
Abstract
Pallister-Killian syndrome (PKS) is characterized cytogenetically by mosaic tetrasomy of chromosome 12p. Routine prenatal diagnosis of PKS is still complicated because of the difficulties of discriminating between the supernumerary isochromosome 12p and the duplication 21q and because of the variable level of mosaicism. The frequency of cells with an extra metacentric chromosome i(12)(p10) is usually determined by tissue-limited or tissue-specific mosaicism. We demonstrated a decrease of the abnormal clone with extra i(12p) in the amniotic fluid cells of the PKS fetus during amniocyte subculturing. The rapid loss of the i(12p) in the course of amniocyte subculturing should be the focus of attention during prenatal karyotyping. This is especially necessary for cultures with slow growth, which require further interpretation of the result during cytogenetic diagnosis of PKS.
P
PKS is cytogenetically characterized by a tissue-limited mosaicism. Most fibroblasts have 47 chromosomes, with an extra small metacentric chromosome, whereas the karyotype of lymphocytes is usually normal. The extra metacentric chromosome is an isochromosome of the short arm of chromosome 12: i(12)(p10) (Peltomaki et al. 1987; Warburton et al. 1987). Rarely, it contains a neocentromere (Dufke et al. 2001). Clinical findings in PKS include profound mental retardation, seizures, streaks of hypo- or hyperpigmentation, and facial anomalies (“coarse” face), including prominent forehead with sparse anterior scalp hair, flat occiput, hypertelorism, short nose, flat nasal bridge, and short neck (OMIM #601803; http://www.ncbi.nlm.nih.gov/Omim/).
The main ultrasound indicators of PKS are hydramnios, congenital diaphragmatic hernia, and micromelia of a predominantly rhizomelic type. The hydrops fetalis, hygroma colli, or increased nuchal translucency, fetal overgrowth, ventriculomegaly, dilatation of cavum pellicidum, absence of stomach visualization, and presence of a sacral appendix are less common (Doray et al. 2002).
Prezygotic origin of the additional chromosome is suggested in a number of studies. Maternal and paternal discrepancies of meiosis have been demonstrated, and several mechanisms have been proposed for i(12p) formation (Hunter et al. 1985; Rivera et al. 1986; Van Dyke et al. 1987).
Routine prenatal diagnosis of PKS can still be complicated: on the one hand, in the discrimination of the supernumerary 12p isochromosome from the inv dup 21q; and on the other, because of the variable level of mosaicism. The frequency of cells with an extra metacentric chromosome i(12)(p10) usually is determined by tissue-limited or tissue-specific mosaicism.
We report the first case of PKS in Belarus. In this prenatal case, we describe a rapidly decreasing number of metaphase cells with i(12p) in cultured amniocytes in vitro in the course of several subculturings.
Materials and Methods
Case Report
A 39-year-old woman presented to the Belorussian Republic Genetic Service Center at 20 weeks' gestation for amniocentesis because of advanced maternal age. The pregnancy was uncomplicated. The first child (16 years old) in the family had mental retardation and developmental delay (cytogenetic examination was not carried out). The amniocentesis of the current pregnancy was performed at 20 weeks of gestation.
Cytogenetics
The first non-mosaic karyotype was obtained using a 21-day culture of amniotic fluid cells. Cell culture time was more than 50 days between the start of the culturing and the final fourth amniocyte subculture because of poor and slow culture growth.
For cytogenetic study, fluorescence in situ hybridization (FISH) analysis of the extra chromosome at the second to fourth amniocyte subcultures was used.
Results
The conventional GTG banding study of cultures of amniocytes showed the additional metacentric marker chromosome in 20 metaphases studied. The karyotype was interpreted as 47,XX, + mar (Figure 1A). The parental karyotypes were both normal.
First, FISH investigation using a commercially available whole-chromosome paint (WCP) probe for chromosome 12 was performed. Hybridization with WCP#12 revealed the origin of the extra chromosome and the mosaic status of the isochromosome: (WCP#12; Vysis, Baar, Switzerland) (Figure 1B). That provided the diagnosis of 47,XX, + mar.ish i(12)(p10)(wcp12+)[53]/46,XX[6].
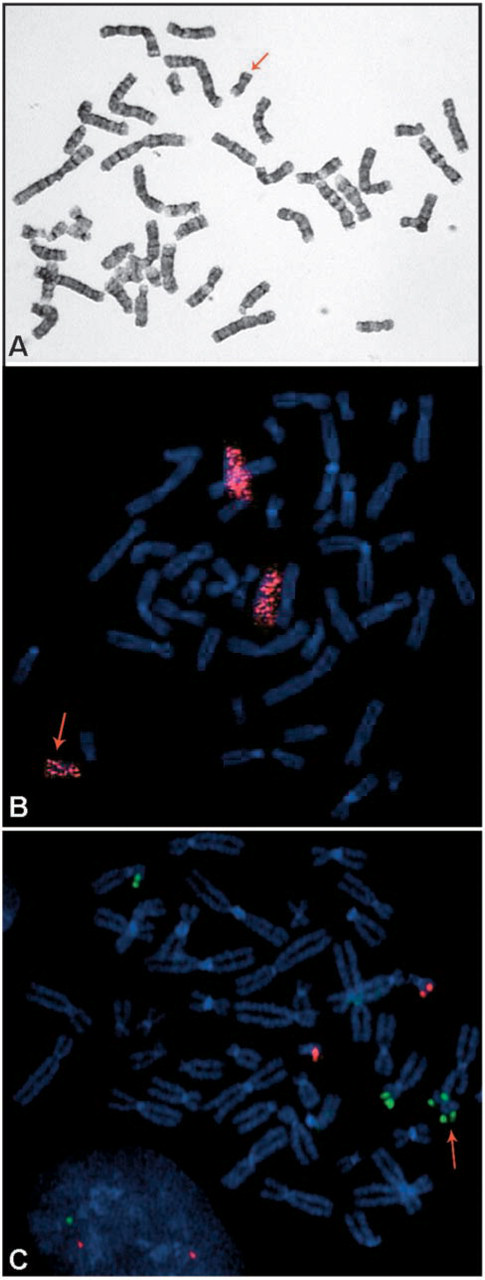
Later, the next FISH detection, with an LSI TEL/AML1 ES dual-color translocation probe (Vysis), was performed using the next subcultures of amniocytes, and the diagnosis was confirmed: 47,XX, + mar.ish i(12)(p10)(12p13×2)[3]/46,XX[48] (Figure 1C). During these molecular cytogenetic tests, a significantly smaller number of cells with i(12p) was discovered. The second FISH investigation showed a decrease in isochromosome 12p frequency in late subcultures of cells.
The diagnosis of PKS was established for this fetus. Pregnancy was terminated at 22 weeks' gestation with the consent of the spouses.
Autopsy verified the following multiple congenital anomalies of fetus: craniofacial dysmorphies were oxycephaly, wide, flat nasal bridge, low-set malformed ears, high-arched palate, short webbed neck, brachydactyly, clinodactyly of five fingers, single palmar crease, bronchopulmonary dysplasia, annular pancreas, and agyria. The diagnosis of PKS was confirmed.
Discussion
Variable tissue-specific mosaicism of i(12p) is the characteristic cytogenetic feature of tetrasomy 12p syndrome. The variation of isochromosome 12p frequency does not correlate with the severity of congenital abnormalities in the PKS fetus (Schinzel 1991).
Isochromosome 12p is seen mainly in skin fibroblast cultures and in chorionic villus and amniotic fluid cell samples. In spite of such tissue-limited mosaicism, the extra chromosome can rarely be identified in blood lymphocytes during postnatal examination (Schubert et al. 1997; Leube et al. 2003) or prenatal studies using cordocentesis (Chiesa et al. 1998).
Therefore, there is no strict limitation on the presence of extra isochromosome 12p in cell tissues, which are generally used in prenatal and postnatal cytogenetic examinations.
The morphology of the extra chromosome also varies. Cases in which two abnormal cell clones were present are described in the literature. One of the clones contained i(12p), giving the tetrasomy of 12p; another had an additional 12p and trisomy (Dong et al. 2003; Leube et al. 2003). In addition, the rare phenomenon of neocentromere formation has been observed in the supernumerary chromosome i(12p) (Dufke et al. 2001). These findings suggest a complicated, multi-stage origin for this chromosomal disorder.
Culture of amniotic fluid cells is the optimal method, in which extra chromosome 12p mosaicism is detectable with high reliability. Nevertheless, our findings, as well as the literature data concerning PKS, demonstrate a significant variation in the amount of i(12p) in this type of cell. We demonstrated a decrease in the abnormal clone with extra i(12p) in the PKS patient during several amniocyte subcultures in which there was pure cellular growth: from 100% (20 metaphases) in the first harvest of 21-day culture, to 89.8% (53/59 metaphases) in the second subculture, and to 5.9% (3/51 metaphases) in the fourth subculture.
Taking into account the literature data concerning a prezygotic (Cormier-Daire et al. 1997) meiotic origin of the extra chromosome i(12p), whether maternal or paternal, and the early zygotic tissue-mosaicism formation, the common mechanisms that appear to explain specific tissue-limited mosaicism and the in vitro decrease in the number of tetrasomic cells can both be identified.
Probably, the mosaicism takes place because of some advantages of the normal cell clone in vivo during the critical stage of early embryogenesis. Consequently, the growth and/or proliferative advantages of the normal clone are the similar selection factor in cell culture in vitro.
The rapid loss of the i(12p) in the course of amniocyte subculturing must be the focus of attention during prenatal karyotyping. This is especially necessary for cultures with slow growth, which require further interpretation of the results during the cytogenetic diagnosis of PKS.
The mechanisms of trisomy/tetrasomy rescue in vivo have not yet been discovered. Therefore, further observations in vitro and the analysis of mosaicism changes could clarify these mechanisms.
Footnotes
Acknowledgements
This work was supported by the Laboratory of Genetics, CH Mulhouse Hospital, by Dr Eric Jeandidier, France, and by DAAD.