
Abstract
Sixteen newly established cell lines with small supernumerary marker chromosomes (sSMC) derived from chromosomes 1, 2, 4, 6, 7, 8, 14, 15, 16, 18, 19, 21, and 22 are reported. Two sSMC are neocentric and derived from 15q24.1-qter and 2q35-q36, respectively. Two further cases each present with two sSMC of different chromosomal origin. sSMC were characterized by multicolor fluorescence in situ hybridization for their chromosomal origin and genetic content. Moreover, uniparental disomy of the sister chromosomes of the sSMC was excluded in all nine cases studied for that reason. The 16 cases provide information to establish a refined genotype-phenotype correlation of sSMC and are available for future studies.
Keywords
S
For de novo sSMC, particularly those ascertained prenatally, Paoloni-Giacobino et al. (1998) stated that they are not easy to correlate with a clinical outcome. It is known that 32% of sSMC are derived from chromosome 15; 11% are i(12p) = Pallister-Killian, ~10% are der(22)-, ~7% are inv dup(22)-cat-eye-, and z6% are i(18p)-syndrome-associated sSMC (Liehr et al. 2004). In general, the risk for an abnormal phenotype in prenatally ascertained de novo cases with sSMC is given as ~13% (Warburton 1991). This has been refined to 7% (for sSMC from chromosome 13, 14, 21, or 22) and 28% (for all non-acrocentric autosomes) (Crolla 1998). Also, generally speaking, sSMC inherited from a normal sSMC carrier to its children are usually not correlated with clinical problems, even though exceptions are described (Liehr et al. 2004). The maternal line is predominantly involved in inheritance (Liehr 2006b).
It should be emphasized that a comprehensive marker chromosome characterization would be best for carriers of sSMC; however, this is not always possible due to lack of corresponding methods and/or sufficient probe sample. After a complete molecular cytogenetic characterization, sSMC cell lines could also be available for further research, e.g., depending on the modes of sSMC formation, karyotypic evolution of an sSMC, or changed expression profiles of cells due to presence of sSMC.
We recently studied 19 of the ~30 commercially available cell lines from the European Collection of Cell Cultures which, according to this company's catalogue, were positively karyotyped. Surprisingly, six of nine cell lines with sSMC previously characterized for their chromosomal origin by others had to be revised, and no sSMC was detected in five others (Brecevic et al. 2006). Thus, there is an obvious need for well-characterized sSMC cell lines. We present here the beginning of a sSMC cell bank with 16 well-characterized sSMC cases.
Materials and Methods
(Molecular) Cytogenetics
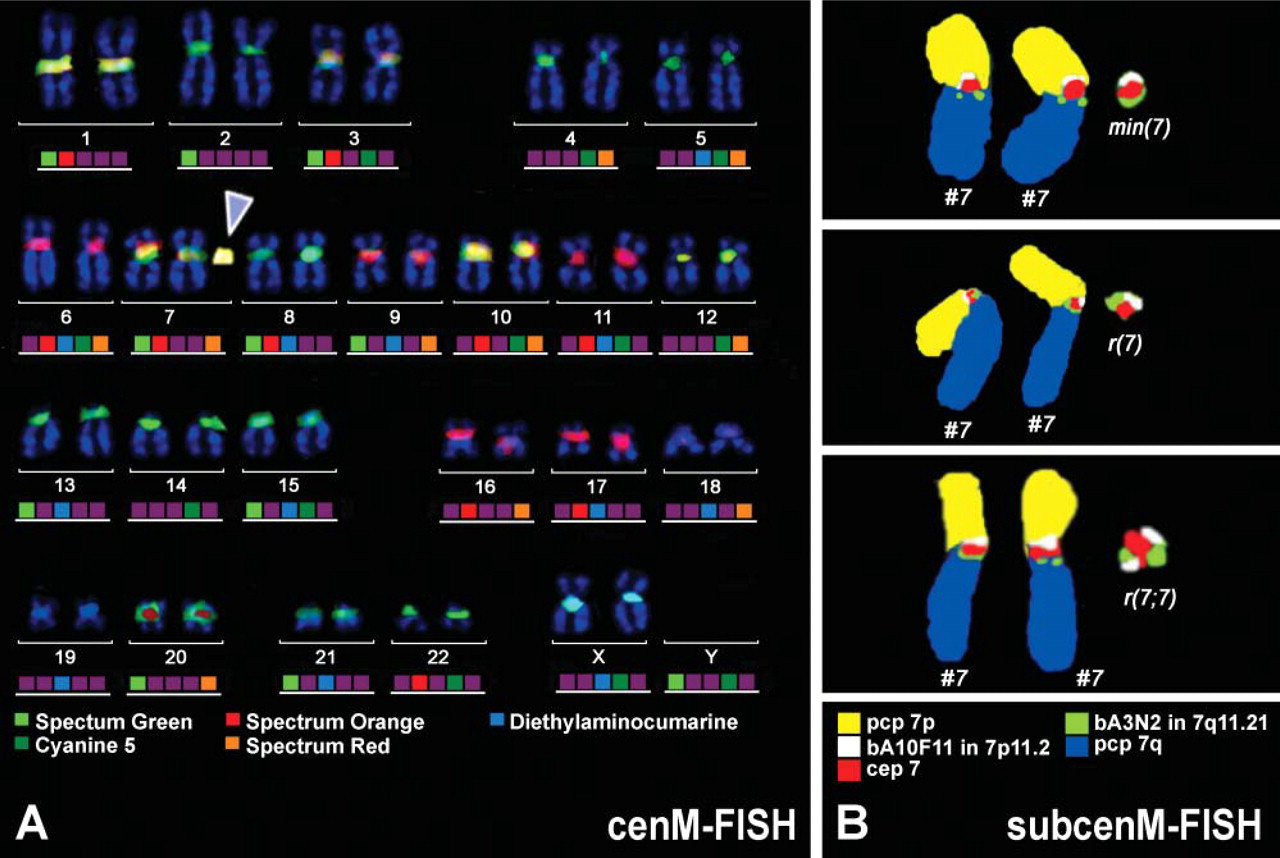
Banding cytogenetics was done according to standard procedures (Seabright 1971). Multicolor FISH (mFISH) approaches applied were also previously described in detail: centromerespecific mFISH (cenM-FISH) (Figure 1A) (Nietzel et al. 2001), multiplex FISH (M-FISH) using whole chromosome-painting probes (Speicher et al. 1996), subcentromere-specific mFISH (subcenM-FISH) (Figure 1B) (Starke et al. 2003), microdissection and reverse painting (Starke et al. 2001), or multicolor banding (MCB) (Liehr et al. 2002) was applied to different extents in the 16 cases (see Table 1).
Analysis for Uniparental Disomy
Uniparental disomy (UPD) for the sister chromosomes of the sSMC (see Figure 2) were excluded either by microsatellite analysis (Starke et al. 2003) or by methylation test in SNRPN region (Nietzel et al. 2003). Informative microsatellite markers are listed in Table 1.
Karyotypes of the newly established EBV-transformed cell lines after GTG banding and FISH, UPD result, and clinical signs of the cases a
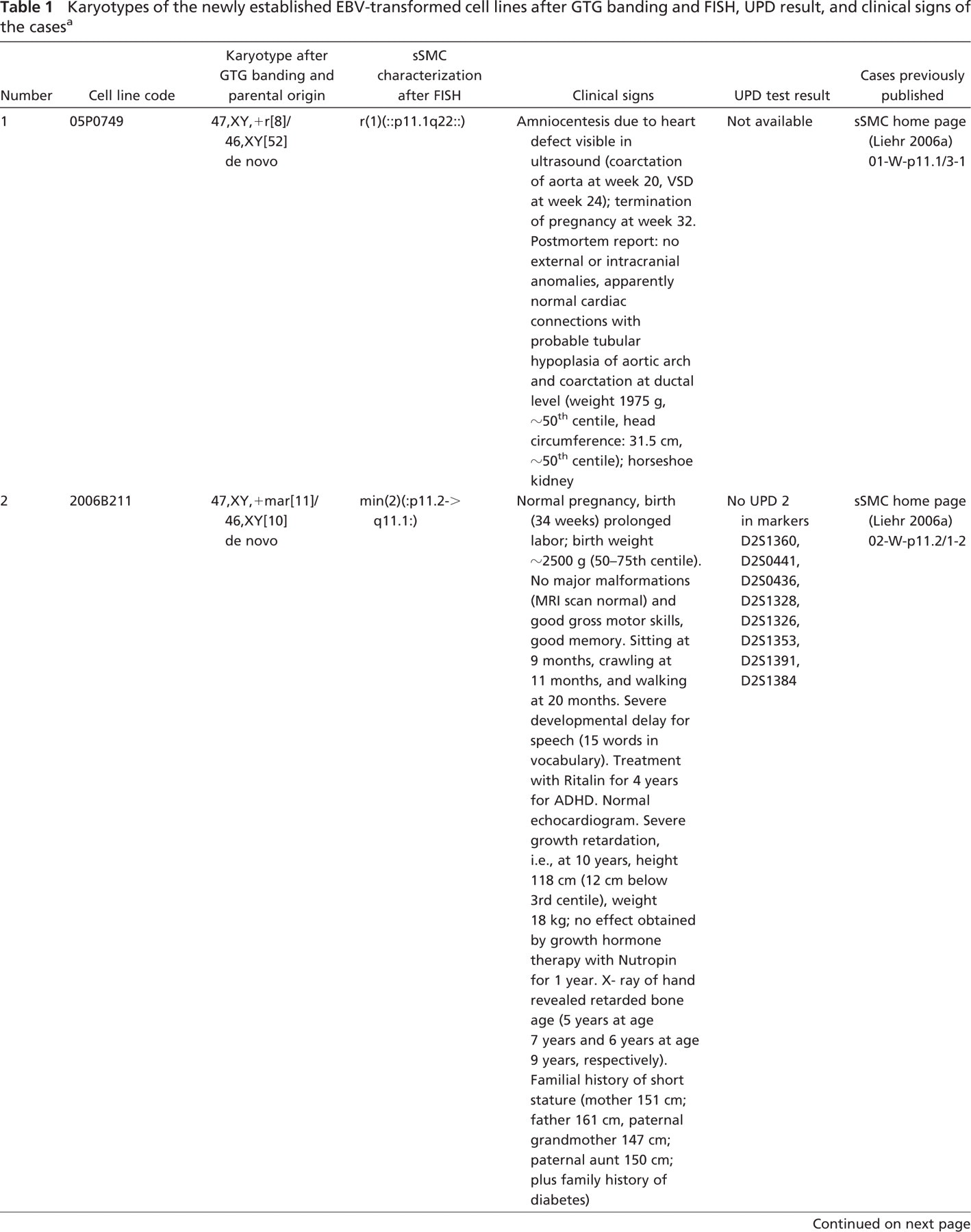
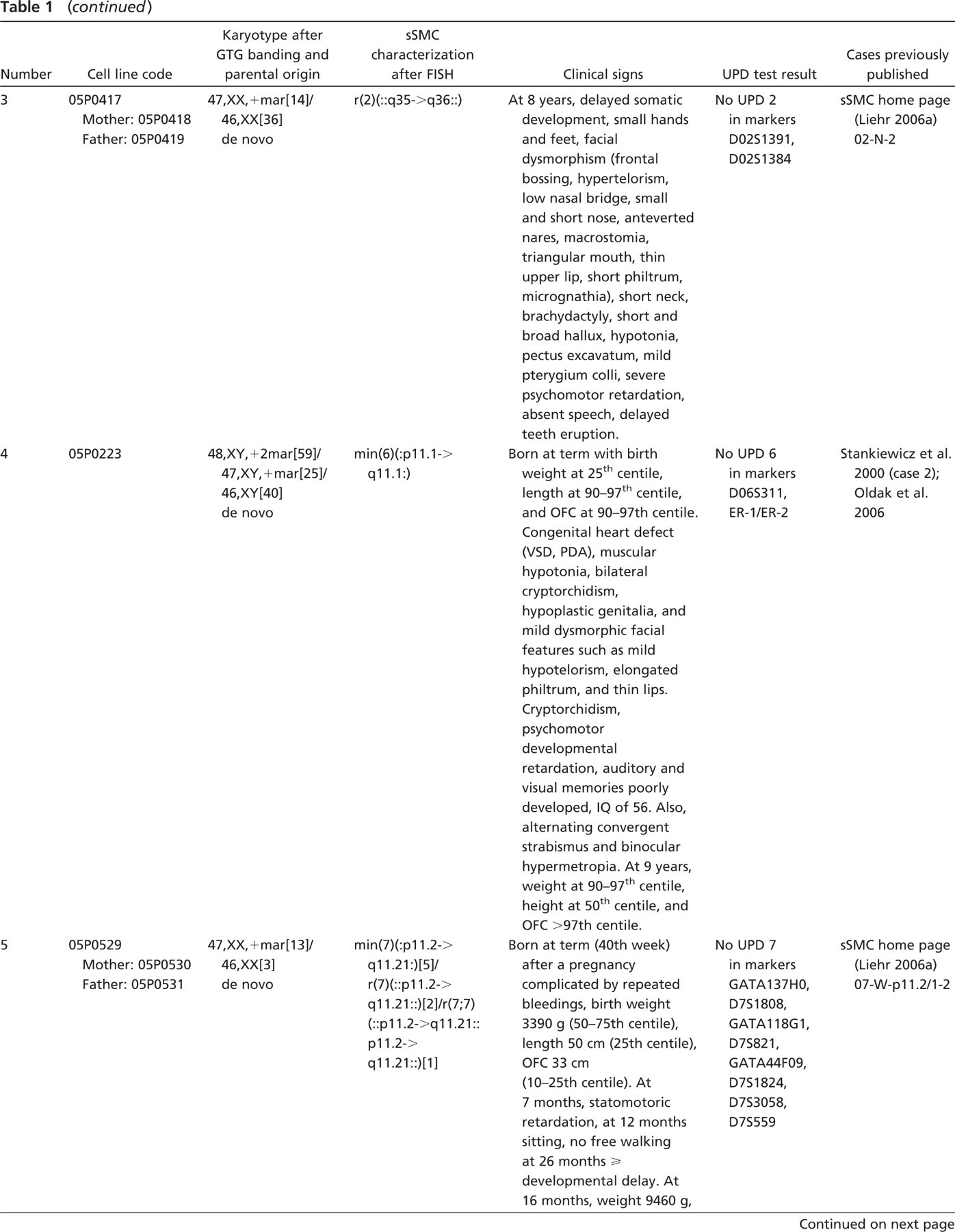
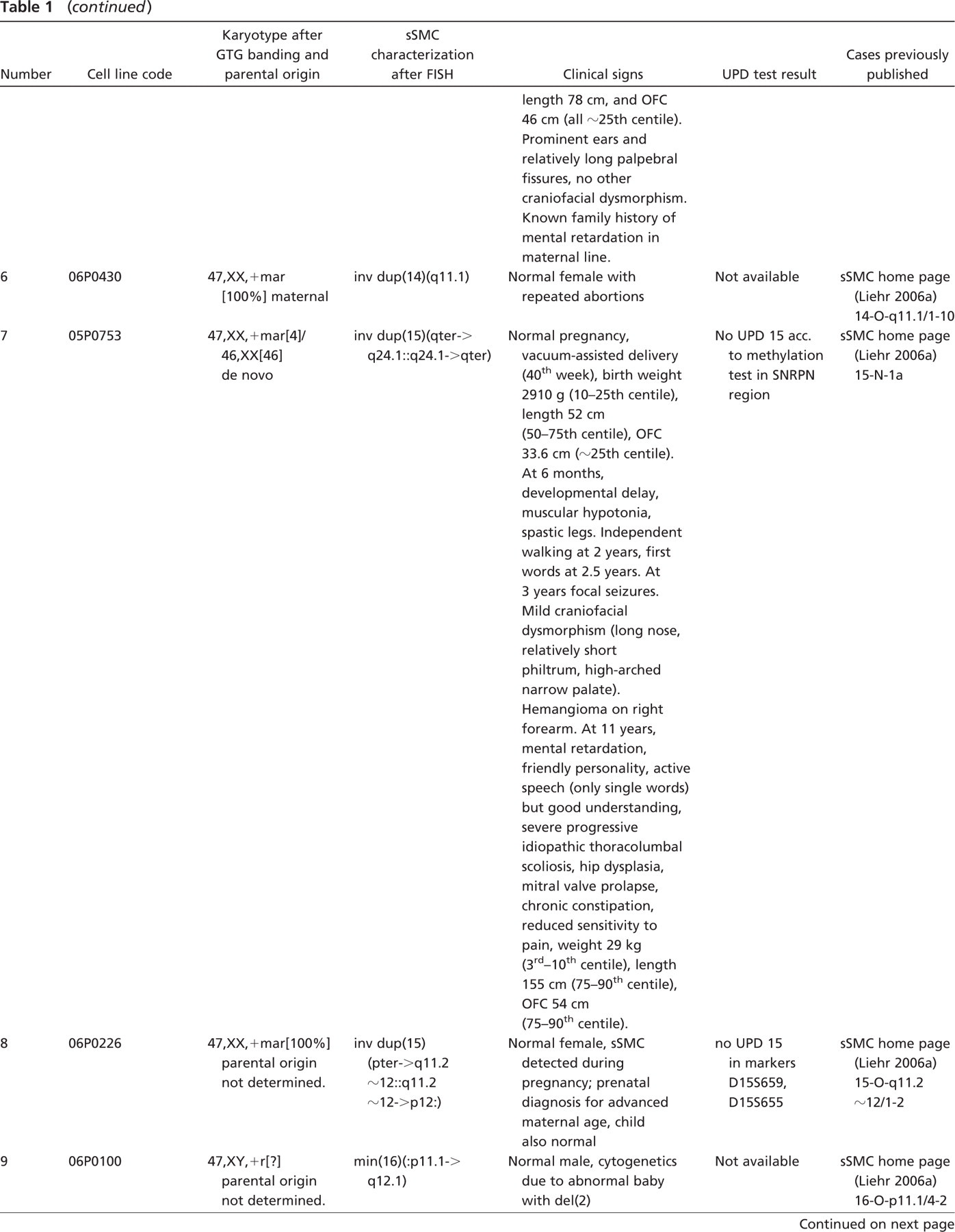
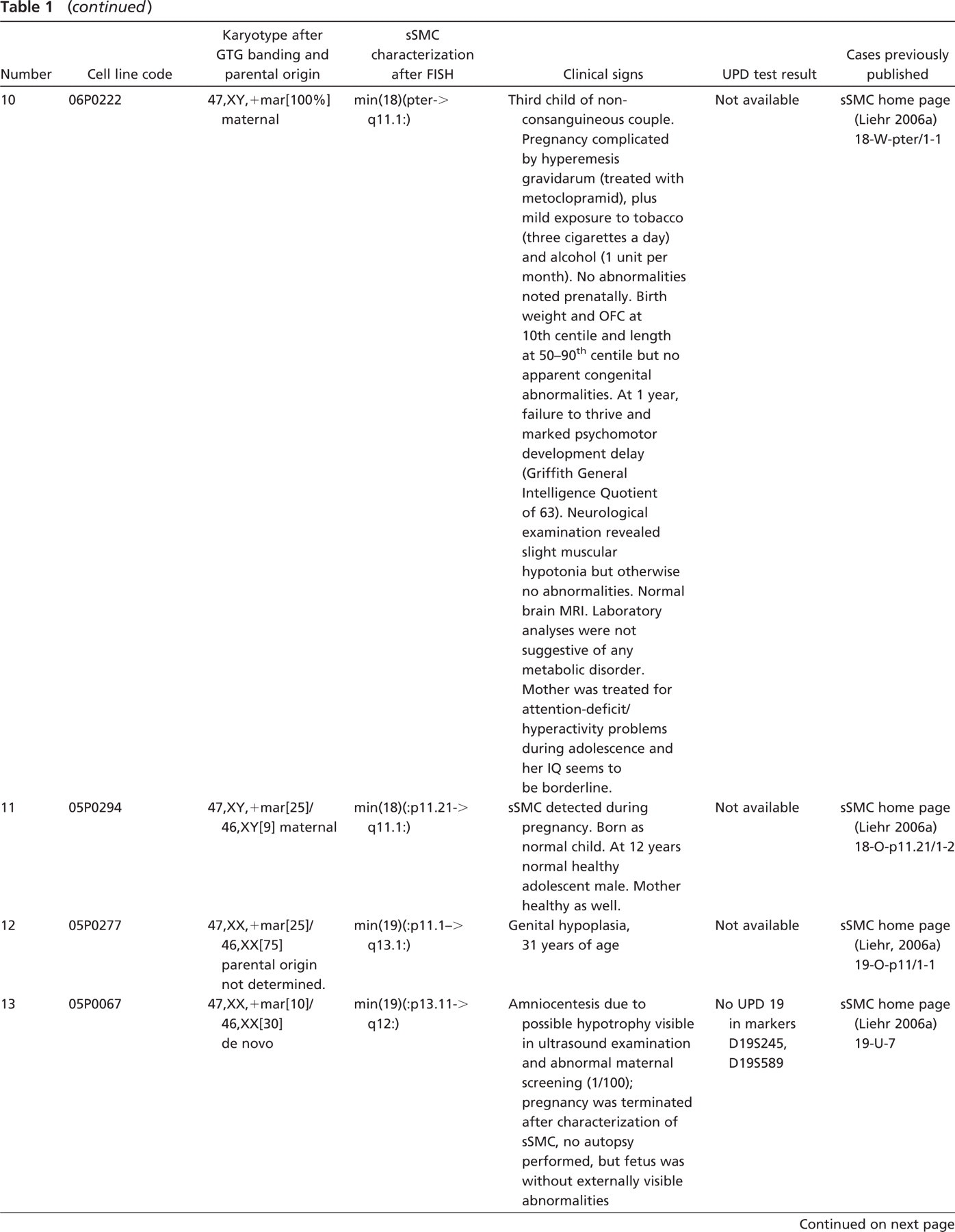
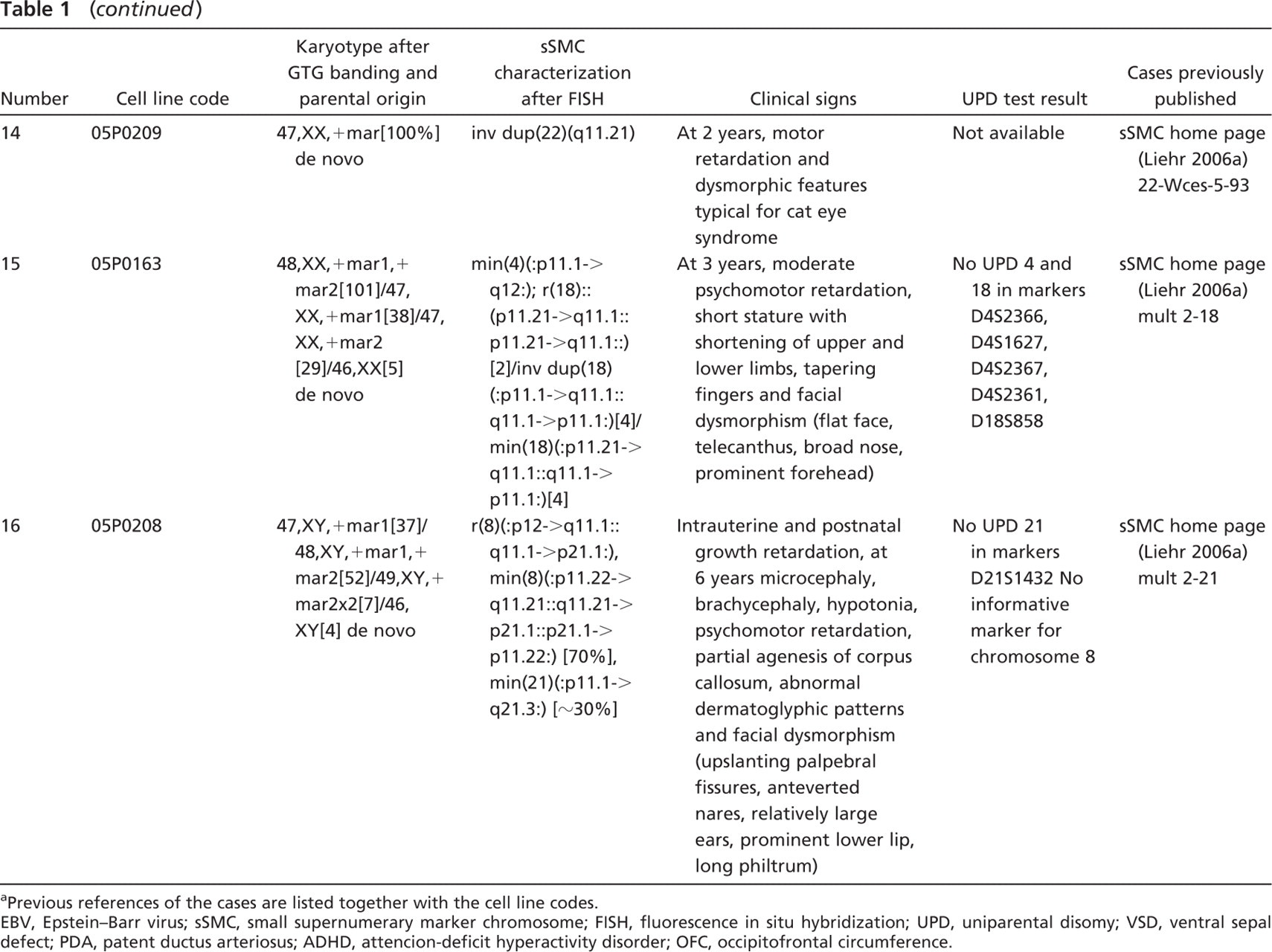
Previous references of the cases are listed together with the cell line codes.
EBV, Epstein-Barr virus; sSMC, small supernumerary marker chromosome; FISH, fluorescence in situ hybridization; UPD, uniparental disomy; VSD, ventral sepal defect; PDA, patent ductus arteriosus; ADHD, attencion-deficit hyperactivity disorder; OFC, occipitofrontal circumference.
Epstein-Barr Virus Transformation of Peripheral Blood B-lymphocytes
Epstein-Barr virus (EBV) was done as previously described using 2–10 ml of heparinized peripheral blood (Pattengale et al. 1973; Neitzel 1986; Xie et al. 2005). This study was approved by the Ethical Commission of the Friedrich Schiller University, Jena, Germany (internal code 1457-12/04).
Results
To date we have established for our sSMC cell bank 16 new sSMC cell lines (see Table 1) by EBV transformation of peripheral blood B-lymphocytes. sSMC were characterized by molecular cytogenetics for their chromosomal origin and genetic content (Table 1). An example for the applied FISH methods is given for case 05P0529 in Figure 3.
Cell lines presented here contain sSMC derived from chromosomes 1 (05P0749), 2 (2006B211, 05P0417), 4 (05P0163), 6 (05P0223), 7 (05P0529), 8 (05P0208), 14 (06P0430), 15 (05P0753, 06P0226), 16 (06P0100), 18 (06P0222, 05P0294, 05P0163), 19 (05P0277, 05P0067), 21 (05P0208), and 22 (05P0209). Two neocentric sSMC cell lines derived from 2q35–q36 and 15 q24.1-qter, respectively, were established (05P0417, 05P0753). In two further cases, two sSMC of different chromosomal origin each were present (05P0163, 05P0208).
Parental DNA was available for UPD analysis in 9/16 cases, and a UPD was excluded in all (2006B211, 05P0417, 05P0223, 05P0529, 05P0753, 06P0226, 05P0067, 05P0163, 05P0208; Table 1).
Discussion
As mentioned above, there is a lack of sSMC cell lines; thus, 16 new EBV-transformed sSMC cell lines are presented here (see Table 1). EBV transformation was chosen for immortalization because it is a well-established and, in our hands, easily performed approach. It has been taken into account that the mechanism of EBV transformation is still not completely understood (Yamashita et al. 2006), and problems were also reported concerning chromosomal (Okubo et al. 2001), genetic, and proteomic effects of the procedure (Toda et al. 2000). However, for the 16 reported immortalized cell lines, future studies will now be possible without the need for taking further blood samples. However, for case 05P0749, this would not be possible due to termination of pregnancy.
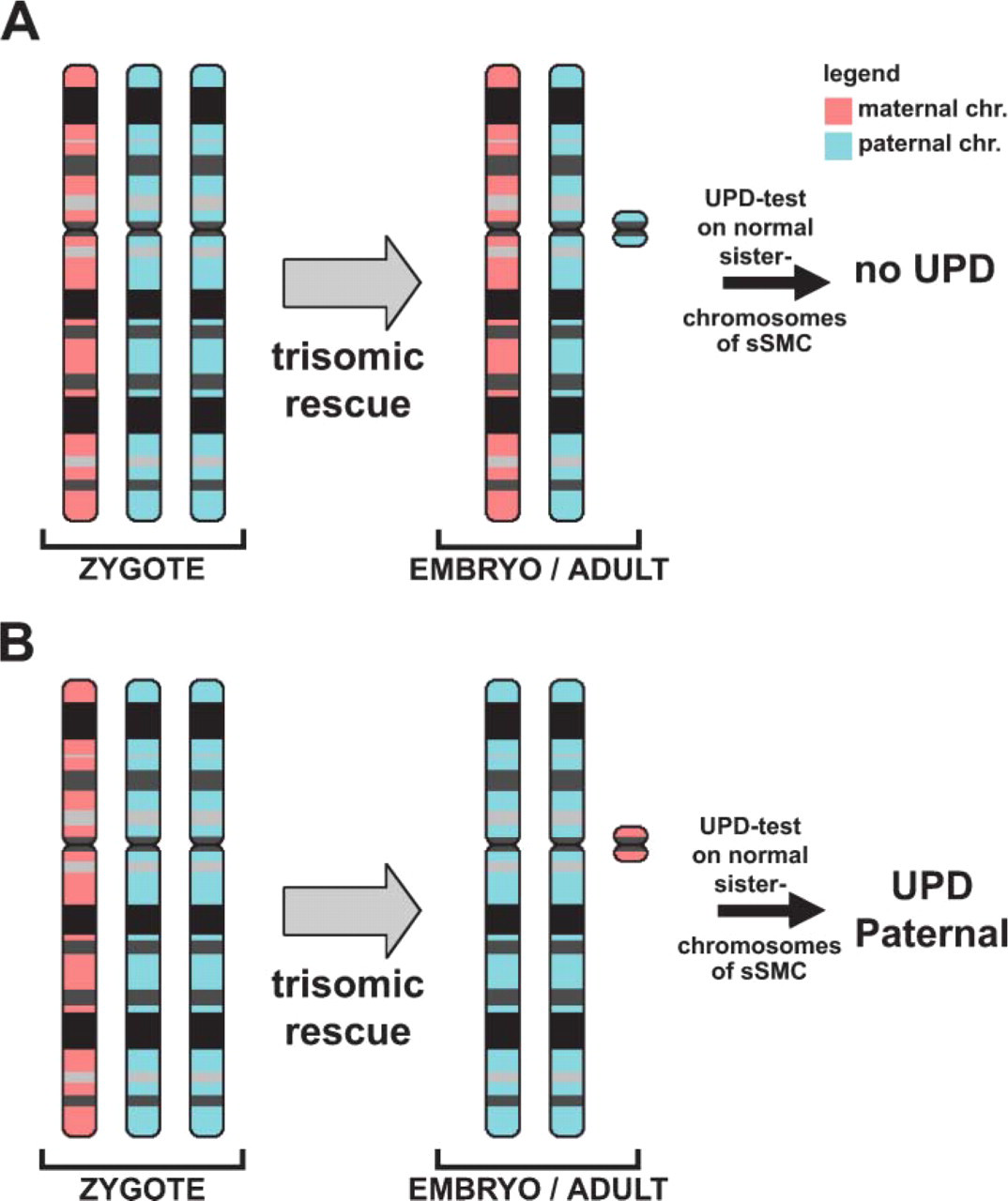
Schematic drawing showing how an sSMC derived from chromosome 7 can occur. This kind of development has already been observed (Bartels et al. 2003): initially there is a trisomy for one chromosome in the zygote, one of the three chromosomes is transformed to an sSMC by trisomic rescue, by uniparental disomy (UPD) analysis of the sister chromosomes of the sSMC it can be clarified whether there is a normal (A) or abnormal (B) parental origin of the cytogenetically normal chromosomes 7. (B) An example of a paternal UPD is depicted.
For all 16 cell lines, clinical data could be collected, and the mosaic state of the sSMC could be determined, as well as the chromosomal origin and genetic content present on the sSMC (see Table 1). Six sSMC carriers were clinically normal, and five of those six had a euchromatic imbalance due to sSMC presence. Thus, those cases could not only provide the genotype-phenotype correlation of sSMC (see below and Figure 3) but are also important for future studies on the question of if and how these genetically relevant regions are silenced, e.g., by heterochromatization.
Ten of the cases included in our sSMC cell bank provided information about the genotype-phenotype correlation as suggested in Liehr et al. (2006) (Table 1). This correlation is based on the idea that, if no UPD of the sister chromosome sSMC is present (Figure 2), the genetic imbalance induced by the presence of the sSMC is causative for clinical symptoms. It was possible to exclude UPD for 9/16 cases. However, as also shown by Barber (2005), there are regions within the human genome that do not lead to clinical abnormalities if present in three or more copies instead of the two regular ones. Similar observations can be made in sSMC cases, and critical regions for clinical abnormalities were suggested for the pericentric regions of all human chromosomes (Liehr et al. 2006). As summarized in Figure 3, cases 05P0749, 2006B211, 05P0529, 06P0430, 06P0226, 06P0222, 05P0294, and 05P0209 confirmed the previously suggested borders of the aforementioned critical regions for chromosomes 1, 2, 7, 14, 15, 18, and 22. Case 06P0100 not only confirmed but even enlarged the region on chromosome 16, which does not lead to clinical problems originally from 16p11.1–q11.2 to 16p11.1–16q12.1. This observation was not only made for case 06P0100 but also for at least three additional cases summarized in Liehr (2006a). Case 05P0277 indicated a relatively uncritical region of 19p11.1–q13.1, which does not lead to serious clinical problems. This suggestion is also supported by three additional cases (Liehr 2006a). The remaining six sSMC cases reported here were not suited to contribute to genotype-phenotype correlations of their corresponding chromosome due to different reasons. Cases 05P0417 and 06P0226 are neocentric sSMC derived from non-pericentric regions of chromosome 2 and 15, respectively, and cases 05P0163 and 05P0208 have two sSMC derived from chromosomes 4 and 18 or 8 and 21, respectively. Thus, it is impossible to know which chromosomal region contributes to what extent to the clinical outcome. Case 05P0223 has in a subset of cells two identical, seemingly heterochromatic sSMC(6) but no UPD 6; however, a severe clinical picture is described for this patient. Thus, either a cryptic chromosomal imbalance in connection with the sSMC or a reason for the phenotype completely independent of the sSMC should be suggested here. In any case, at present, 05P0223 cannot be included in a genotype-phenotype correlation, as with case 05P0067, because clinical information is lacking.
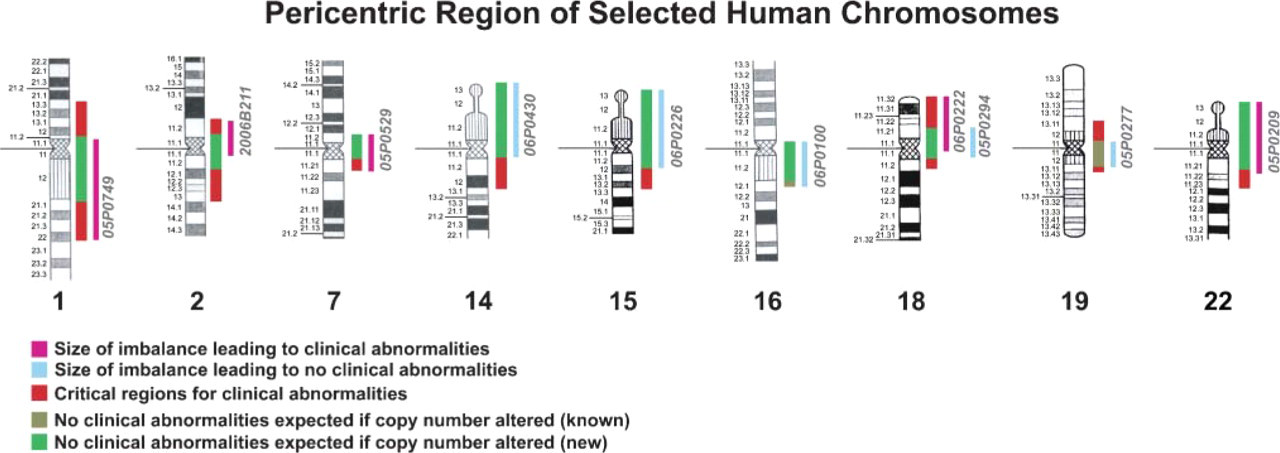
The pericentric region of nine selected chromosomes is shown. According to Liehr et al. (2006), the genotype-phenotype correlation for sSMC in connection with the caused imbalance is depicted. This suggested correlation was confirmed by the sSMC cases presented here for all chromosomes. In the case of chromosomes 16 and 19, the correlation could even be refined, and more non-critical regions were characterized.
In summary, a cell bank of sSMC has been established, and 16 cases have already been included here. In the future, this cell bank will be crucial for an exact genotype-phenotype correlation in sSMC. It will be possible, for example, to select in a chromosome-specific manner for sSMC cases and align the clinical outcome with the genetic and proteomic profiles, e.g., by array CGH (Shaffer and Bejjani 2006) or ProteinChip technology (Ernst et al. 2006). No lack of material will hamper this analysis as presently is the case. The sSMC cell bank also provides, for the first time, cellular model systems in which, e.g., karyotypic evolution of the sSMC can be studied. This is interesting because we recently showed that cryptic mosaicism of sSMC exists and evolves, at least in a subset of cases (Starke et al. 2003; this study, cases 05P0529, 05P0163, and 05P0208). In addition, e.g., studies on the breakpoint regions of sSMC, on trisomic rescue (Figure 2), heterochromatization, etc. can be performed when a sSMC cell bank with some dozens to hundreds of samples will be available.
Footnotes
Acknowledgements
This study was supported in part by the DFG (436 ARM 17/2/04, 436 RUS 17/109/04, 436 RUS 17/22/06, WE 3617/2-1, LI820/11-1), the Schering Foundation, the Boehringer Ingelheim Fonds, the Ernst-Abbe-Stiftung, and the Evangelische Studienwerk e.V. Villigst.
The support of all families with incidences of sSMC and their willingness to support this study by providing blood samples is gratefully acknowledged. Drs. A. Torres and A. Lara from the Hospital San Juan de la Cruz (Úbeda) Jaen, Spain are acknowledged for providing two of the sSMC cases.