
Abstract
Hevin, also known as SC1, MAST 9, SPARC-like 1, RAGS1 and ECM2, is a member of the SPARC-related family of matricellular proteins. Mouse hevin is 53% identical to mouse SPARC, and both proteins share a follistatin-like module and an extracellular Ca2+-binding (E-C) domain. SPARC functions as a modulator of cell-matrix interactions, a regulator of growth factor activity, a de-adhesive protein, and a cell cycle inhibitor. Although the functions of mouse hevin are unknown, its human orthologue has been shown to be de-adhesive for endothelial cells. We now report the production of recombinant mouse hevin in insect cells through the use of a baculoviral expression system and its purification by anion-exchange, size-exclusion chromatography, and isoelectric focusing. Furthermore, we have produced rat anti-hevin monoclonal antibodies (MAbs) that have been characterized by indirect and capture ELISAs, immunoblotting, immunoprecipitation, and immunohistochemistry (IHC). Recombinant hevin, present as a soluble factor or bound to tissue-culture plastic, inhibited the spreading of bovine aortic endothelial cells in vitro. IHC analysis of hevin in normal human and mouse tissues revealed a limited expression pattern in many tissues, with particularly dominant staining in dermis, ducts, vasculature, muscle, and brain. In lung and pancreatic tumor xenografts, we found distinct reactivity with MAbs that were selective for stromal cells, tumor cells, and/or endothelial cells. Although similar to SPARC in its anti-adhesive activities, hevin nevertheless exhibits a distinctive histological distribution that, in certain invasive tumors, is associated with desmoplasia.
M
The function of hevin is unclear. However, analysis of hevin activity in vitro has shown that it can modulate cell shape and cell adhesion to different substrates, including fibronectin (Girard and Springer 1996). Hevin is thought to participate in lymphocyte transendothelial migration in high endothelial venules and might alter vascular permeability, in a manner similar to SPARC (Girard and Springer 1996). Interestingly, the N-terminal portion of hevin augments B-cell lymphopoiesis and clonal proliferation of mature lymphocytes through an undefined mechanism that requires interaction of hevin with B-lymphocytes and is dependent on divalent cations (Oritani et al. 1997). Hambrock et al. (2003) have recently described the production and purification of recombinant hevin, and have reported its binding to collagen I fibrils and to the ECM secreted by osteosarcoma cells in vitro.
Levels of hevin/SC1 expression are apparently regulated differentially in neoplastic tissue and in tissue undergoing repair (McKinnon and Margolskee 1996; Mendis et al. 1996; Nelson et al. 1998; Isler et al. 2001; Peters et al. 2001), data indicating that hevin might modulate cell proliferation. By immunoblotting or Northern analysis, hevin is found in tissues such as brain, lung, kidney, and heart (Hambrock et al. 2003). A band of Mr 55,000 found in extracts of brain, heart, and muscle (Hambrock et al. 2003; and unpublished observations) suggests that hevin is specifically cleaved posttranslationally or that an alternative transcript is present in some cells. In addition to SPARC and other ECM components, hevin has recently been associated with the activated stromal (desmoplastic) response of host tissue to adenocarcinomas that develop in the pancreas (Ryu et al. 2001; Iacobuzio-Donahue et al. 2002,2003). In situ hybridization studies demonstrate mRNA encoding hevin in angiogenic endothelial cells in stromal areas of invasive pancreatic adenocarcinoma (Iacobuzio-Donahue et al. 2002). Other analyses cataloging the level of hevin mRNA in cancerous tissues have shown that levels of hevin decrease in tumor cells (Bendik et al. 1998; Claeskens et al. 2000). The apparent context-dependent expression of hevin is consistent with the regulation of other matricellular proteins (SPARC, thrombospondins, CCN proteins) in tumor tissue (Brekken and Sage 2001). Interestingly, and consistent with results from other mice with targeted deletions of certain matricellular genes, hevin-null mice do not display obvious differences from wild-type littermates (McKinnon et al. 2000). However, responses of hevin-null mice to challenges have demonstrated phenotypes with respect to wound healing and foreign body response that are distinct from those of wild-type and of SPARC-null mice (M. Sullivan et al., unpublished observations). Compensation of hevin function by other SPARC family proteins is also a possibility that should not be overlooked in the interpretation of the hevin-null phenotype.
We have expressed recombinant mouse hevin in insect cells and have developed a protocol for its purification. Recombinant hevin was the antigen for the production of rat anti-hevin monoclonal antibodies (MAbs) that have been used to characterize the synthesis and distribution of hevin in normal and neoplastic tissues. We show that recombinant hevin inhibited the spreading of endothelial cells in vitro in a time- and concentration-dependent manner. These functions are reminiscent of those of SPARC and other matricellular proteins and implicate hevin in a variety of cell–ECM interactions that affect development and response to injury.
Materials and Methods
Production and Purification of Recombinant Mouse Hevin
Mouse hevin (SC1) cDNA (Soderling et al. 1997), minus the signal sequence, was amplified by polymerase chain reaction (PCR) using sequence-specific primers designed to generate a single product with Xba I-5′ and Bgl II-3′ sensitive ends (forward: 5′-GAATTCTCTAGAGACAAGTACA-AGGTTTCTCTTTGACCAC-3′; and reverse 5′-GAAT-TCAGATCTTTTTTTTTTTTTTTGGGGAGGTTTTATAG-3′). The Xba I/Bgl II-digested PCR product was subcloned into the pAcGP67A baculovirus expression vector (Pharmingen; San Diego, CA) in frame with the viral gp67 signal sequence to allow efficient protein secretion of hevin by virally-transfected Spodoptera frugiperda 9 (Sf 9) cells, which were propagated in TMN-FH medium supplemented with 10% fetal bovine serum (FBS; Pharmingen) at 27C. The cloned product was dideoxy-sequenced in both directions to confirm proper insertion of the cDNA into the multiple cloning site of the plasmid. Recombinant baculovirus was generated by co-transfection of the pAcGP67/SC1 vector with linearized baculovirus (AcUW1.lacZ; Pharmingen) into Sf9 cells. Culture supernatants were analyzed for hevin expression by immunoblotting analysis with two separate guinea pig anti-peptide hevin polyclonal IgGs (A. Bradshaw, J. Soderling, and E.H. Sage, unpublished experiments). Transfected cell supernatants were then used to generate high-titer stocks of recombinant virus for later infections of cells grown in protein-free medium (SF900 II SFM; Invitrogen; Carlsbad, CA).
For purification, 1/10 volume of 200 mM 3-(N-morpholino) propane sulfonic acid (Mops, pH 6.5) was added to the starting material and the pH was adjusted to 6.5. The supernatant was applied to a column (1.7 × 20 cm) containing Q-Sepharose Fast Flow anion-exchange resin (Amersham Biosciences; Piscataway, NJ), equilibrated in 200 mM LiCl, 20 mM Mops (pH 6.5). Recombinant hevin was eluted from the column with a continuous salt gradient from 200 to 600 mM LiCl, 20 mM Mops (pH 6.5). The peak fractions were identified by spectrophotometry and were confirmed by SDS-PAGE and staining with Coomassie Brilliant Blue R (Laemmli 1970). Fractions containing recombinant hevin were pooled, dialyzed against Hanks buffered saline solution (HBSS), flash frozen in liquid N2, and stored at −80C.
Further purification was achieved by size-exclusion chromatography and/or isoelectric focusing: For size-exclusion chromatography, peak fractions from the anion-exchange chromatography (described above) were pooled, concentrated in a Centricon Plus-80 centrifugal filter device with 30,000-kD molecular weight cut-off (Millipore; Bedford, MA), and applied to a column (16 × 100 cm) containing Superdex 200 (Amersham Biosciences) equilibrated with HBSS without glucose, 50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH 7.5. The peak fractions were identified by spectrophotometry, SDS-PAGE, and staining with Coomassie Brilliant Blue R. The fractions containing recombinant hevin of highest purity were pooled, dialyzed against HBSS, concentrated, flash-frozen in liquid nitrogen, and stored at −80C. For isoelectric focusing, peak fractions from anion-exchange chromatography were pooled and concentrated as above and were purified by continuous native-PAGE (pH 6.6) on a Model 491 Prep Cell (Bio-Rad Laboratories; Hercules, CA) according to the manufacturer's instructions. Fractions containing recombinant hevin were identified as above, pooled, dialyzed against HBSS, concentrated, flash-frozen in liquid nitrogen, and stored at −80C.
Recombinant hevin, or recombinant hevin digested for 30 min at 37C with trypsin (Worthington Biochemicals; Lakewood, NJ; 1:500 molar ratio of trypsin to hevin) was resolved by SDS-PAGE under reducing conditions, electrophoretically transferred to a PVDF membrane, and subjected to N-terminal sequencing (B. McMullen and E. Davie, Department of Biochemistry, University of Washington, Seattle, WA).
Endotoxin levels were quantified with the Limulus Amebocyte Lysate Pyrochrome kit (Associates of Cape Cod; Falmouth, MA) according to the manufacturer's instructions.
Generation and Screening of Hybridomas
Male Sprague–Dawley rats were immunized IP with 50 μg of recombinant hevin in Ribi R-700 adjuvant (Corixa; Hamilton, MT). After two boosts with the same mixture on days 21 and 42 and a final IP boost without adjuvant on day 63, spleens were collected under sterile conditions on day 66. Splenocytes were fused with P3-X63-Ag8.653 mouse myeloma cells (American Type Culture Collection; Rockville, MD) according to established procedures (Kearney et al. 1979; Lane 1985). The resulting cell mixture was plated into multiple 96-well tissue culture plates in medium consisting of Iscove's modified Dulbecco's medium (Gibco; Grand Island, NY) supplemented with 10% (v/v) Fetal Clone I serum (HyClone Laboratories; Logan, UT), 10% (v/v) BM Condimed H1 (Roche Applied Science; Indianapolis, IN), 2 mM
On day 9, supernatants from the fusion plates were screened by indirect ELISA. Immulon II plates (Thermo Lab-systems; Franklin, MA) were coated with 0.5 μg/ml, 55 ml/well, of recombinant hevin diluted in 10 mM PBS overnight at 4C. Unbound antigen was removed, the wells were washed twice with PBS containing 0.05% Tween-20 (PBST), and the wells were blocked for 1 hr at room temperature with PBST containing 1% (w/v) bovine serum albumin (BSA) (Sigma; St Louis, MO), 200 μl/well. After removal of the blocking solution, the wells were washed twice with PBST, after which culture supernatants from the fusion plates were replicaplated onto ELISA plates at 55 μl/well. The plates were incubated for 1 hr at RT, after which the supernatants were removed and the wells washed four times with PBST. For detection of bound antibody, an HRP-conjugated goat anti-rat IgG (Fc-specific) reagent (Southern Biotechnology Associates; Birmingham, AL) was diluted 1:5000 in Iscove's modified Dulbecco's medium containing 2% Fetal Clone I serum and was added to the wells at 55 μl/well. After a 1-hr incubation at RT, the wells were washed five times with PBST. 3,3′5,5′-Tetramethylbenzidine (TMB) substrate in substrate buffer (Genetic Systems; Redmond, WA) was prepared according to the manufacturer's instructions and was added to the wells (55 μl/well). After a 10–15 min incubation at RT, color development in the wells was terminated by the addition of 1 N H2SO4 and the optical density of the wells was read on an ELISA plate reader (Molecular Devices; Sunnyvale, CA) at 450 nm. All supernatants that were positive in this assay were evaluated in a second indirect ELISA using recombinant human SPARC (Bradshaw et al. 2000) as a negative control protein to ensure that the positive supernatants in the initial assay contained antibody that was specific for hevin.
Hybridomas in wells with anti-hevin specificity were cloned at least twice by culturing the cells at an average of less than one cell per well in Costar 96-well half-area plates (Corning; Acton, MA) and assessing specificity of antibody produced by the clones as described above. MAbs produced by each of the final clones were evaluated for IgG subclass and light chain composition with a rat Monoclonal Antibody Isotyping kit (Roche Applied Science).
Antibody Purification
IgG antibodies were purified from tissue culture supernatant by chromatography on protein G using the Pierce ImmunoPure Binding/Elution buffering system (Pierce; Rockford, IL). IgGs were evaluated for purity by SDS-PAGE and staining with Coomassie Brilliant Blue R and for reactivity with recombinant hevin by indirect ELISA.
Capture ELISA
Recombinant hevin was biotinylated by incubation of biotinamidocaproate NHS-ester (Sigma) with hevin at a 20:1 molar ratio in HBSS (Gibco). The solution was rotated gently at RT for 1.5 hr, after which the reaction was stopped by addition of Tris-HCl/glycine (pH 8) to a final concentration of 10 mM. Biotinylated hevin was separated from excess biotin on a PD-10 column (Amersham Biosciences). For the capture ELISA, microtiter plates coated with protein G (Pierce) or purified IgG (100 ng/well) were blocked with 5% casein acid hydrolysate (CAH) (Sigma) and were incubated with biotinylated hevin at various concentrations. Peroxidase-conjugated NeutrAvidin (NA-Hrp) (Pierce) was added as a second layer, and reactive wells were developed with the peroxidase substrate TMB (BioFx Laboratories; Owings Mills, MD). Reactions were stopped after 15 min with 1 M H3PO4 and were read spectrophotometrically at 450 nM.
Immunoblotting
A total of 250 ng of purified recombinant hevin, 75 μg of whole mouse brain lysate (Brekken et al. 2000), and 75 μg of whole hevin-null mouse brain lysate were resolved by SDS-PAGE on a 4–12% Bis-Tris polyacrylamide gel (Invitrogen) under reducing conditions (100 mM DTT added to protein samples). Proteins were transferred to Immun-Blot PVDF membranes (Bio-Rad) and were blocked for 1 hr at RT with AquaBlock (East Coast Biologics; North Berwick, ME). Each membrane was incubated with an anti-hevin MAb at 1 μg/ml, or antibody tissue culture supernatant at a dilution of 1:10, in PBST + 0.05% CAH for 1 hr at RT or overnight at 4C, followed by washes with PBS + 0.2% Tween-20. The membranes were developed with Super Signal West Pico chemiluminescence substrate (Pierce) after incubation with peroxidase-conjugated goat anti-rat secondary antibodies (Jackson ImmunoResearch Laboratories; West Grove, PA) according to the manufacturers' instructions. Equal transfer of the proteins is routinely confirmed by staining of the transferred gels with Coomassie Brilliant Blue R.
Immunoprecipitation
One ml of conditioned media (CM) from hevin-infected sf9 cultures, or 1 ml of HBSS spiked with 500 ng of purified recombinant mouse hevin, was mixed with 200 μl of anti-hevin MAb 12–18 tissue culture supernatant or 2–5 μg of purified 12–18 and was incubated for 2 hr at 4C. The IgG was precipitated by the addition of protein G-Sepharose beads, and the precipitate was washed three times with PBS/0.5% CAH/0.2% Tween-20. Proteins were resolved after disulfide-bond reduction by SDS-PAGE on a 4–12% Bis-Tris polyacrylamide gel. The presence of hevin in the precipitate was determined by immunoblotting with MAb 155 (1.0 μg/ml).
Immunohistochemistry and Immunocytochemistry
Formalin- and methyl Carnoy's-fixed tissues embedded in paraffin were sectioned by the Histopathology Laboratory at the University of Washington. Methyl Carnoy's-fixed sections were deparaffinized under standard conditions. If necessary, endogenous peroxidases were blocked in methanol with 1.0% H2O2 for 30 min. Some of the sections were subsequently treated with Autozyme (10 μl enzyme concentrate/1 ml buffer for 6 min at RT) (BioMeda; Foster City, CA). The sections were incubated with primary antibody for 1 hr, washed with PBST, incubated for 1 hr with the appropriate peroxidase-labeled secondary antibody (Jackson ImmunoResearch), developed with stable diaminobenzidine (DAB) (ResGen; Huntsville, AL), counterstained with hematoxylin, and cover slipped in Permount (Fisher; Fair Lawn, NJ). Paraffin-embedded sections of mouse brain were evaluated for reactivity as described above except that reactivity was visualized by the use of appropriate fluorescently labeled secondary antibodies (Jackson ImmunoResearch). MAb TUJ1 (BabCO; Berkeley, CA) was used as a neuron marker. Sections of frozen tissue (5–7 μm) were air-dried, fixed in fresh acetone for 5 min, rehydrated in PBST, and blocked in PBST containing 20% Aquablock (East Coast Biologics) for 30 min. The slides were incubated with primary antibodies and were developed as described above.
Acetone-fixed frozen sections of human tumor xenografts grown in SCID mice and tumor specimens from patients obtained from the National Cancer Institute Cooperative Human Tissue Network (Southern Division; Birmingham, AL) were evaluated for reactivity with the anti-hevin MAbs as described above, except that the peroxidase substrate aminoethylcarbazole (AEC) used was as the chromogen.
We screened the MAbs for reactivity with bovine aortic endothelial cells (BAECs) (passage 10; isolated and propagated in our laboratory) (Funk and Sage 1991) plated onto glass coverslips and grown to 70% confluence in DMEM with 10% FBS. Cells were washed once with PBS, incubated at 37C for 30 min in serum-free DMEM (SFM), fixed with 3% formaldehyde/SFM for 20 min at RT, and rinsed with PBS. Cells were blocked with 5% normal goat serum in PBST for 20 min at RT and the anti-hevin MAbs were added at concentrations of 10, 5, and 2.5 μg/ml for 1 hr at RT. Negative controls were normal rat serum and the secondary antibody alone. Reactivity was detected by incubation of the coverslips with the appropriate fluorescein (FITC)-conjugated secondary antibody (Jackson ImmunoResearch). The coverslips were rinsed and nuclei were stained with 20 μg/ml Hoechst 22258 in PBS (Molecular Probes; Eugene, Oregon) for 3 min at RT, washed in PBS, and mounted on slides with Vectashield mounting medium (Vector Laboratories; Burlingame, CA).
Adhesion Assays In Vitro
BAECs (passages 5–12) were plated in DMEM into 24-well tissue culture plates (Corning) at 104 cells/well. Recombinant hevin (10 or 40 μg/ml) was added immediately to cells in suspension and the cells were monitored hourly by phase-contrast microscopy, up to 24 hr. Alternatively, tissue culture wells were precoated overnight at 4C with 10 μg/ml fibronectin (Biomed-Tech; Ancona, Italy) or hevin and were washed once with Dulbecco's PBS. BAEC were subsequently plated in serum-free DMEM at 104 cells/well, and cells were monitored by phase-contrast microscopy from 30 min to 24 hr. Quantification of cell spreading was performed by calculation of a Rounding Index, as described by Lane and Sage (1990).
Results
Expression and Purification of Recombinant Murine Hevin
We had originally cloned hevin (as SC1) from mouse brain and had shown moderate expression of a 3.2-kb hevin mRNA in mouse heart, adrenal gland, and lung, and lower levels in other organs, relative to the high levels characteristic of brain (Soderling et al. 1997). In contrast to SPARC, which is abundant in cultured cells, hevin mRNA was not evident in endothelial cells, smooth muscle cells, or fibroblasts cultured from rat, bovine, mouse, and human tissue. By ISH, we noted a consistent distribution of hevin mRNA in vessels of all organs that we examined, although representatives of both micro- and macrovasculature were negative (Soderling et al. 1997). Although the expression patterns of hevin and SPARC mRNA are not coincident, there is appreciable overlap (Soderling et al. 1997). Indeed, at the protein level, secreted hevin could be available to endothelial or stromal cells, e.g., in instances where SPARC is diminished or absent, as a compensatory matricellular protein. For these reasons it became imperative to study hevin at the protein level and to generate reagents, such as recombinant hevin and anti-hevin MAbs, to facilitate this inquiry. Accordingly, in the following tables and figures we show (a) expression and purification of the murine recombinant protein, (b) its biological activities on cultured cells, (c) a panel of anti-hevin MAbs and their characteristics, and (d) distribution of hevin protein in selected normal and malignant tissues.
Figures 1 and 2 illustrate the purification of hevin from the culture medium of Sf9 cells transfected with the baculoviral expression vector described in Materials and Methods. Chromatography on Q-Sepharose (Figure 1) produced hevin of Mr 120,000–130,000 as estimated by SDS-PAGE on 10% polyacrylamide gels (Figure 1C, Lane 2). Despite the calculated peptide mass of hevin (70,900 D), this size range is consistent with published results (Johnston et al. 1990; Girard and Springer 1995,1996; Mendis et al. 1996) and with the apparent Mr of hevin from mouse brain extract (Figure 1C, Lane 3). The apparent Mr of recombinant hevin was only slightly altered in the presence of DTT (Figure 1B, Lanes 3 and 6). The sequence in the expression vector encoding hevin has been verified by dideoxy sequencing of multiple overlapping products. In addition, protein sequence analysis of the N-terminus and of trypsin-derived peptides of the purified product has validated the protein as hevin, with a 14-residue N-terminal leader derived from the pAcGP67A vector. Recombinant hevin is glycosylated by Sf9 cells (not shown), but the presence of other carbohydrate modifications, coupled with its low pI, account for the low mobility of hevin on SDS-PAGE gels (Figure 1C; and Hambrock et al. 2003).
Purification of hevin to near-homogeneity was achieved by size-exclusion chromatography on Superdex 200 (Figure 2A). Further purification of the protein in Figure 2A (Lane 2) has been performed by re-chromatography of the peak fractions on Superdex 200. This procedure, although producing highly purified hevin, results in sizable losses of the protein. As an alternative protocol, we subjected partially purified recombinant hevin to isoelectric focusing, as shown in Figure 2B. The purified full-length protein (fractions/ lanes 40–45), as well as some of the bands of higher mobility on SDS-PAGE, were reactive with anti-hevin MAbs (not shown), indicating possible proteolysis or heterogeneity in posttranslational modification of the full-length protein.
Recoveries of recombinant hevin are shown in Table 1. Based on scanning densitometry of SDS-poly-acrylamide gels, which tends to overestimate the contribution of impurities, preparations of recombinant hevin were ∼80% pure after elution from an isoelectric focusing column. Greater yields, however, with less purity, were obtained from size-exclusion chromatography compared with isoelectric focusing.
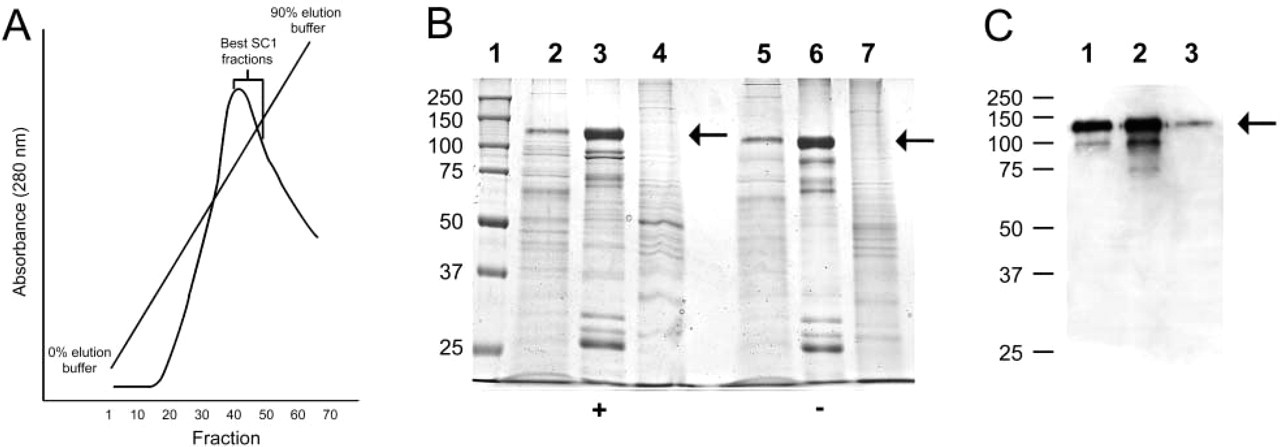
Purification of recombinant mouse hevin from Sf9 cell conditioned media by ion-exchange chromatography. (
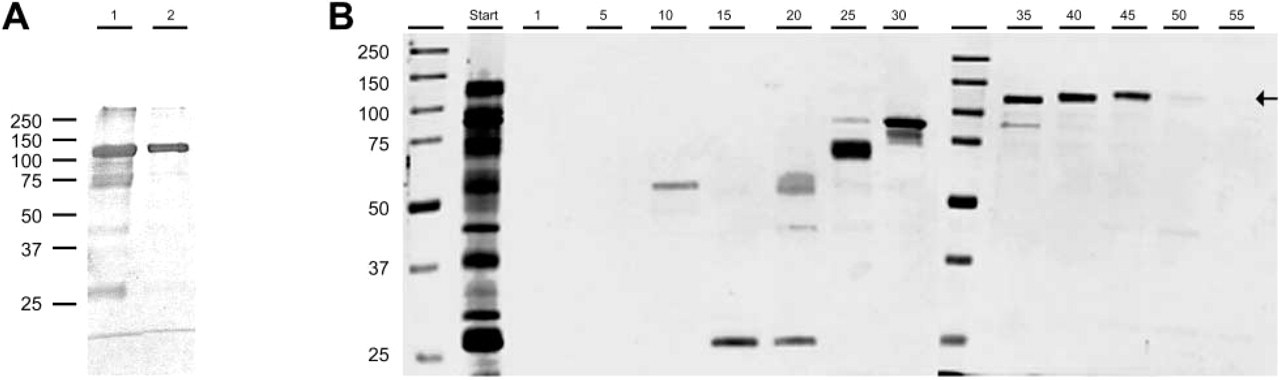
Purification of recombinant mouse hevin by Superdex 200 chromatography and isoelectric focusing. (
Bioactivity of rHevin In Vitro
Figures 3 and 4 confirm that recombinant hevin (rhevin), expressed and purified as described above, is biologically active. Human hevin has been described as anti-adhesive for cultured human umbilical vein endothelial cells (HUVECs) (Girard and Springer 1996). We have confirmed these data and have extended the findings to define an anti-adhesive activity of mouse recombinant hevin on BAECs in vitro. As shown in Figures 3A and 3B, purified hevin, added at 10 μg/ml to cells in suspension, inhibited the spreading of BAECs on tissue culture plastic (i.e., was anti-adhesive) in the presence of 0% FBS. This effect was apparent up to 6 hr after plating, after which hevin-treated BAECs completed adhesion and resembled non-treated or BSA-treated controls. Quantification of these data as Rounding Indices is shown in Figure 4. These results were also obtained in the presence of 1% FBS (not shown).
We also measured the anti-adhesive activity of recombinant hevin presented as a substrate to trypsin-released BAECs. As shown in Figure 5, BAECs in the absence of FBS adhered to substrates coated with fibronectin (Figures 5E and 5H), but were rounded and clumped on hevin-coated wells (Figures 5F and 5I). This effect was maintained over ∼12 hr, after which most of the cells began to spread. Intermediate levels of adhesion that were time-dependent were seen with substrates coated with 10% FBS, and rounding similar to that shown in Figures 5C, 5F, 5I, and 5L was observed with substrates coated with recombinant SPARC (not shown). Cell viability was maintained under all conditions.
Purification of recombinant murine hevin
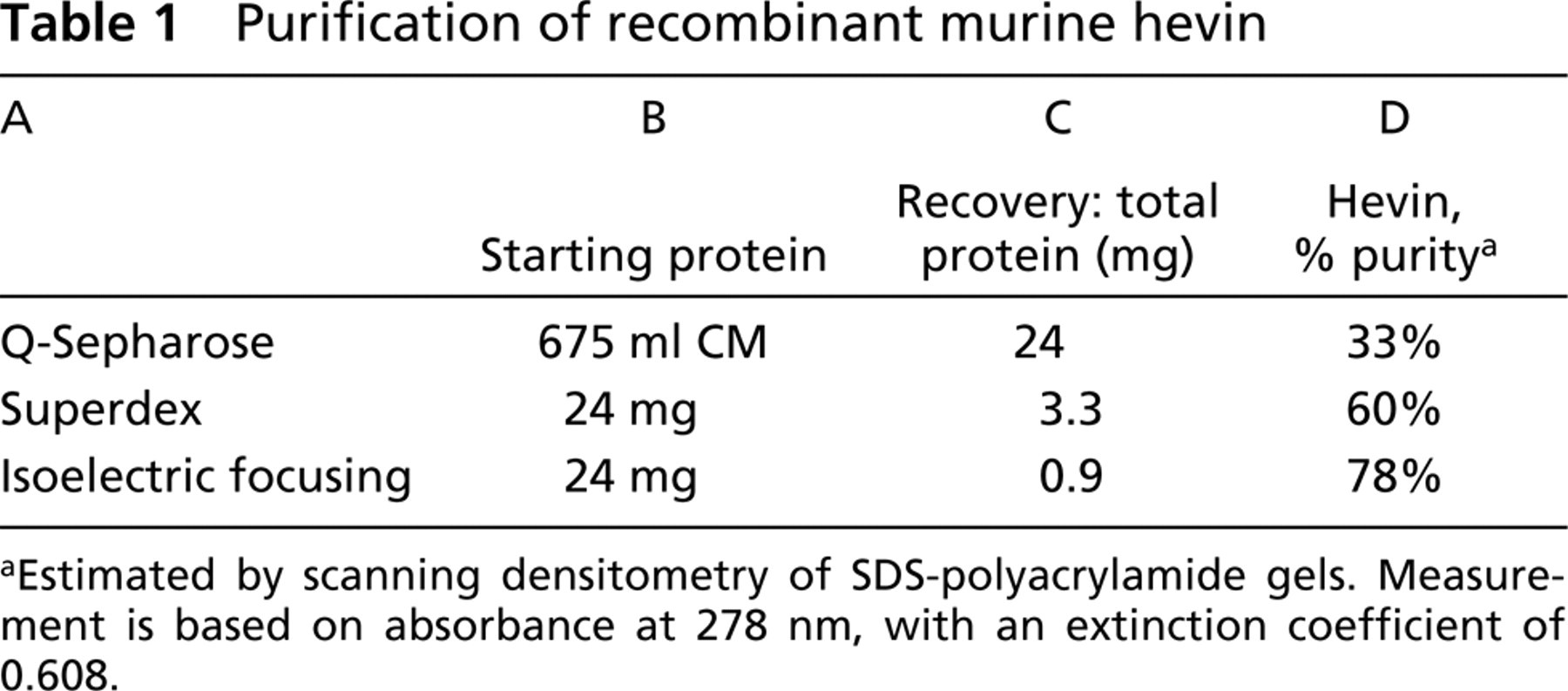
Estimated by scanning densitometry of SDS-polyacrylamide gels. Measurement is based on absorbance at 278 nm, with an extinction coefficient of 0.608.
Production and Characterization of Anti-hevin MAbs
Partially purified hevin was used to immunize rats to produce MAbs specific for mouse hevin. This protocol resulted in over 200 hybridoma clones that bound strongly to hevin by ELISA (data not shown). These reagents were acutely needed because there were no anti-hevin antibodies that reacted reliably with mouse tissues or with paraformaldehyde-fixed tissue, that blocked any activity of native hevin, or that could immunoprecipitate native antigen. For selection of hybridomas, tissue culture supernatants from the clones were screened for reactivity with recombinant hevin in a variety of assays. A partial list of the most extensively characterized rat anti-hevin hybridomas is shown in Table 2. The majority of MAbs are IgG2a, and each clone reacts with mouse hevin by indirect ELISA. In contrast, many MAbs failed to react with hevin in solution, as determined by a capture ELISA (Table 2).
Several of the MAbs described in Table 2 were used to identify hevin in mouse brain lysates (Figure 6A, Lane 1). In each lysate, a band of Mr 105,000–110,000 was the predominant immunoreactive species detected by three different MAbs. Minor bands most likely reflect differences in posttranslational modification of the hevin chain (Hambrock et al. 2003) because tissue lysates contain secreted as well as cellular (endoplasmic reticulum and Golgi-associated) hevin, with differing degrees of glycosylation. Immunoprecipitation of hevin from extracts of mouse brain by MAb 12–18 also showed a protein of Mr 105,000–110,000 by SDS-PAGE (Table 2; Figure 6B, Lane 2). In no cases was immunoreactivity obtained with proteins in lysates from hevin-null brains (Figure 6A, Lanes 2). The disparity in Mr of hevin observed in Figures 1 and 2 vs Figure 6 is likely due to the differences in polyacrylamide gel composition (10% in Figures 1 and 2 vs 4–12% gradient in Figure 6), as well as in buffer conditions (Tris-HC1 vs Bis-Tris).
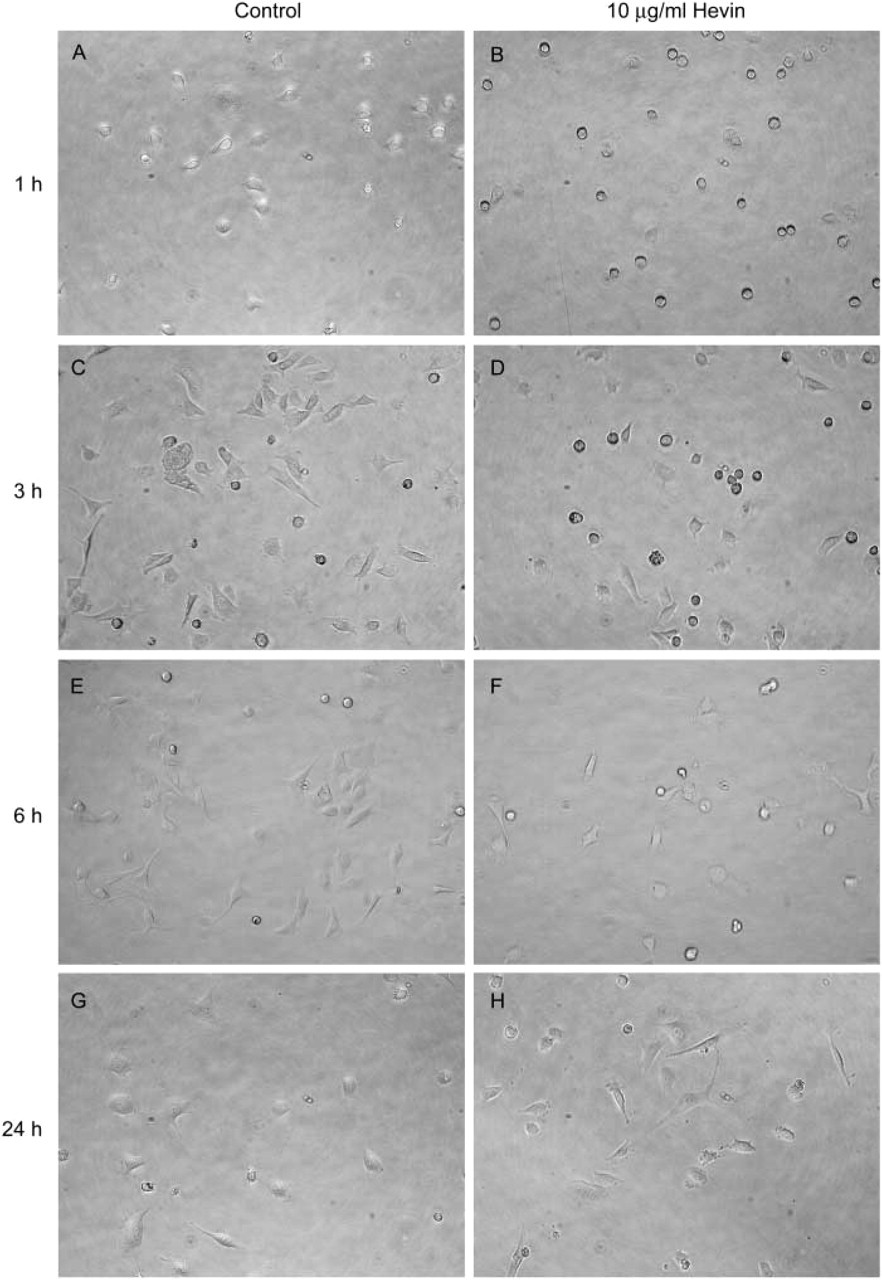
Soluble hevin is anti-adhesive for BAEC in vitro. BAECs (passages 5–12) in serum-free DMEM were plated in triplicate into 96-well tissue culture plates at 2 × 103 cells/well. Recombinant hevin (10 μg/ml) was added immediately to cells in suspension, and the cells were monitored hourly by phase-contrast microscopy. (
Expression of Hevin in Normal and Malignant Tissue
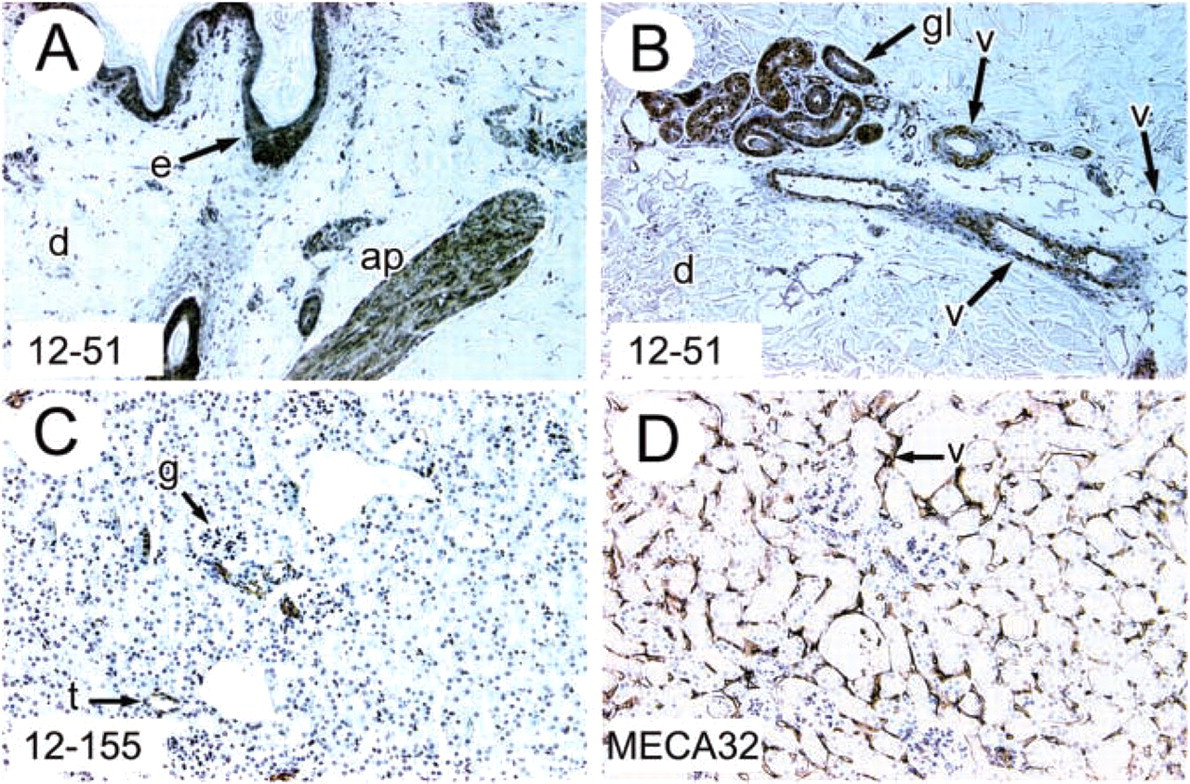
We found that MAb 12–51 was particularly useful for IHC localization of hevin in frozen or paraffin-embedded tissue because it also crossreacts with human hevin. Figure 7 shows the reactivity of 12–51 with human skin and mouse kidney. Strong reactivity was apparent in the dermal layers of the skin, as well as in muscle and the epithelia of glands (Figures 7A and 7B). In the brain, 12–51 highlighted specific cells in the parenchyma of the cerebellum and also vessels in other regions (not shown). Arterioles, venules, and capillaries in the dermis were also reactive with MAb 12–51 (Figure 7B). In paraffin-embedded mouse kidney, hevin was localized by MAb 12–155 to certain glomeruli and tubule epithelial cells (Figure 7C). These data are consistent with previous results in which hevin mRNA was localized to vessels, epidermis, and selected populations of renal tubules by ISH (Soderling et al. 1997).
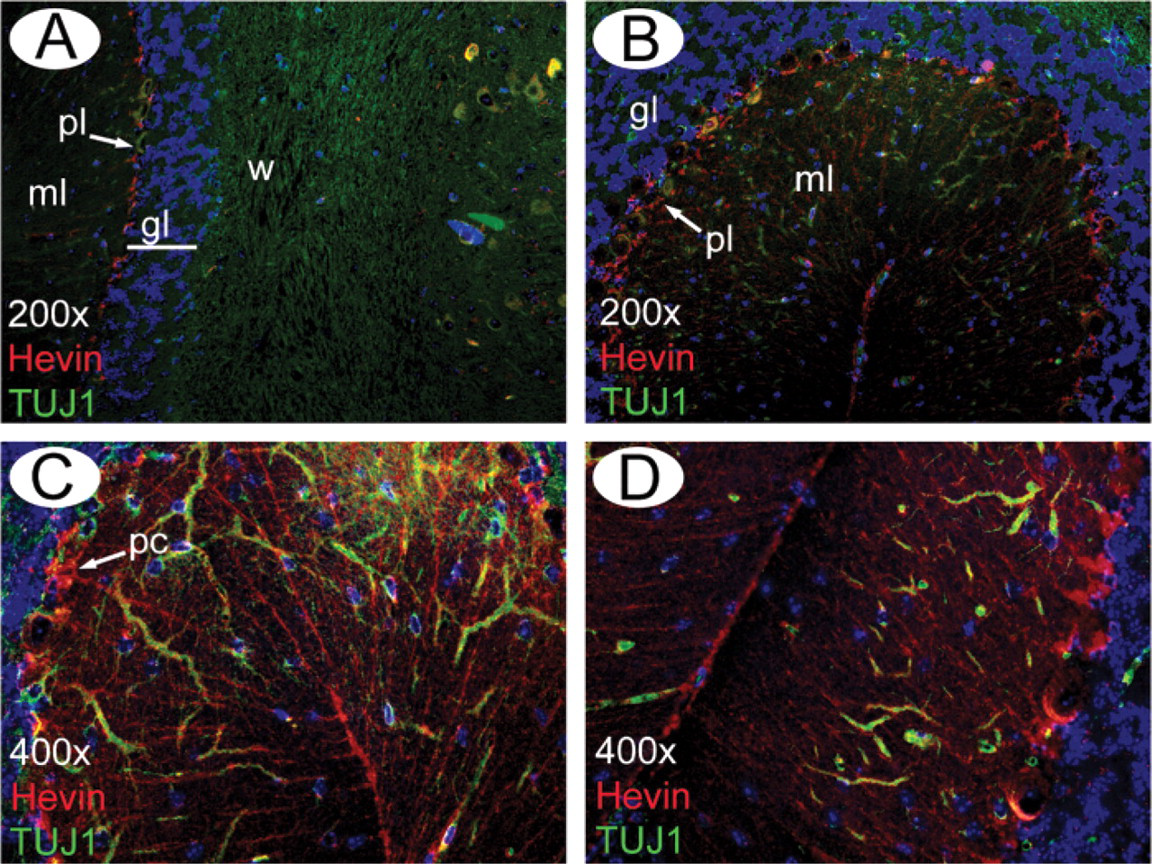
Staining for hevin in mouse cerebellum with MAb 12–155 revealed patterns as shown in Figure 8. Hevin, shown in red, localized conspicuously to the Purkinje cell layer (Figures 8A and 8B) and to the Bergmann glia and Purkinje cells throughout the molecular layer (Figures 8C and 8D). Neurons, shown in green, were localized with MAb TUJ1, which is specific for neuronal class III β-tubulin; nuclei, shown in blue, were stained with Hoechst 22258. These results extend earlier reports of hevin mRNA expression in the Purkinje cell layer (McKinnon and Margolskee 1996; Sullivan and Sage 2004).
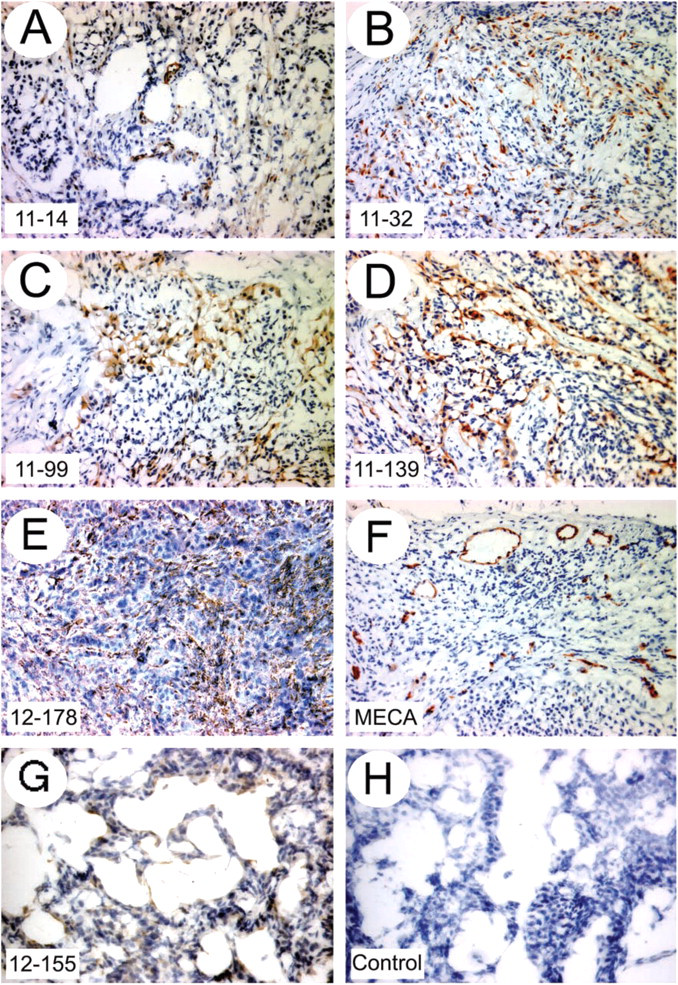
We also evaluated the production of hevin in a limited panel of cancerous tissues (Figure 9). Of the tumor tissues that we screened (human colorectal, lung, breast, pancreatic, and endometrial adenocarcinoma), reactivity was apparent only in endometrial carcinoma associated with the luminal duct epithelial layer (Figure 9G). Although there was no reactivity in two samples of human pancreatic adenocarcinoma, we found tumor as well as stromal reactivity with MAbs 12–51 and 12–155 in human pancreatic tumor xenografts grown in immunocompromised mice (not shown). Xenografts of human non-small-cell lung carcinoma from immunocompromised mice are shown in Figures 9A–9F), after incubation with several additional anti-hevin MAbs (identified on each panel) that have not been listed in Table 2. Each of these staining patterns reflects a stromal/vascular component that was identified by the respective anti-hevin MAb. These data provide evidence for hevin as a stroma-associated protein of neoplastic tissue, an observation consistent with findings by other investigators from serial analysis of gene expression (SAGE) analysis of invasive pancreatic carcinomas (Ryu et al. 2001; Iacobuzio-Donahue et al. 2003).
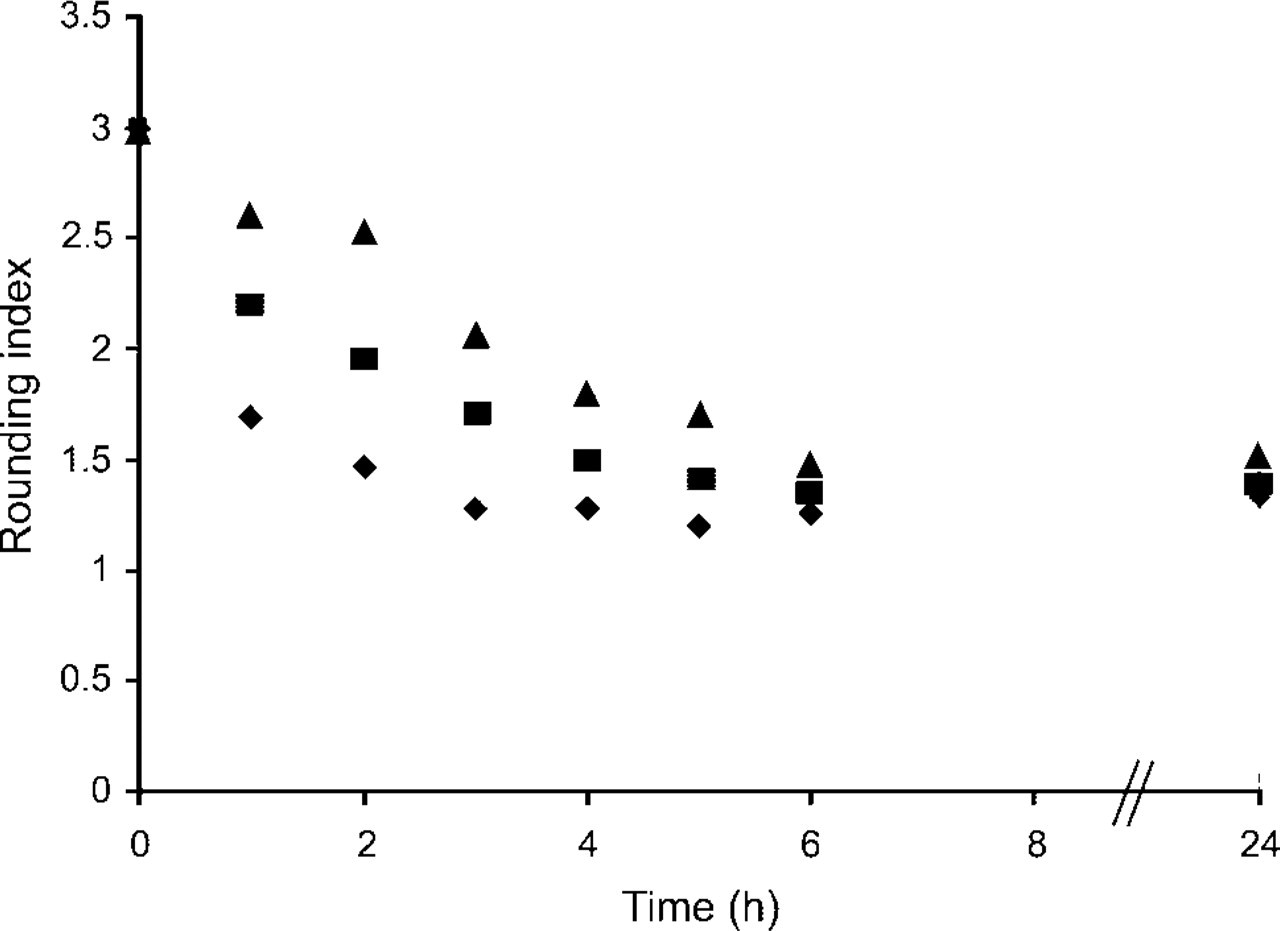
Inhibition of cell adhesion by hevin. Cells (images from experiments shown in Figure 3) were scored for degree of cell spreading according to Lane and Sage (1990). A score of 3 indicates rounded cells that have not initiated spreading, whereas a score of 1 indicates fully spread cells. Diamonds, control cells plated without exogenous hevin. Squares, cells plated with 10 μg/ml hevin. Triangles, cells plated with 40 μg/ml hevin.
Monoclonal rat anti-hevin antibodies
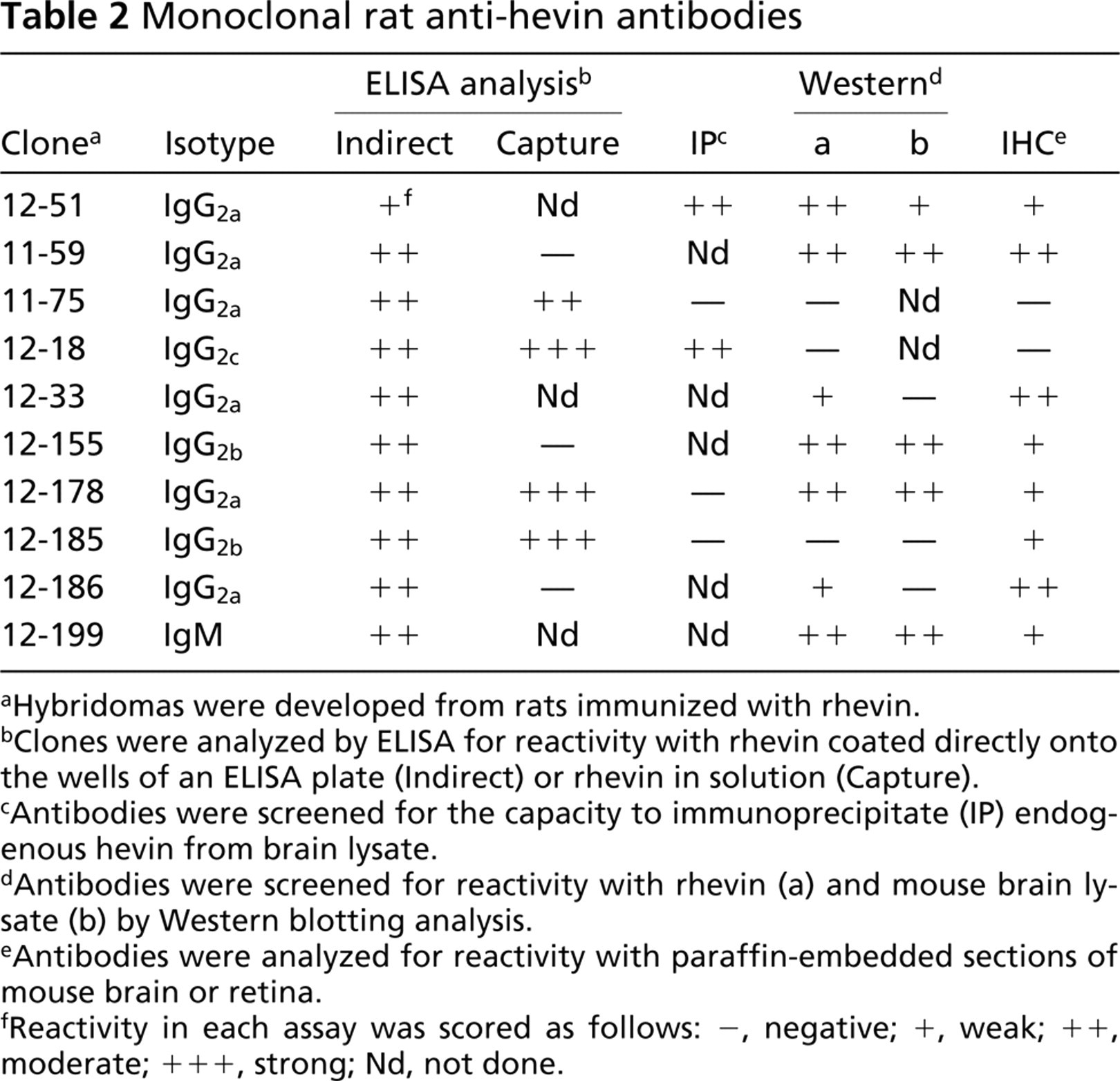
Hybridomas were developed from rats immunized with rhevin.
Clones were analyzed by ELISA for reactivity with rhevin coated directly onto the wells of an ELISA plate (Indirect) or rhevin in solution (Capture).
Antibodies were screened for the capacity to immunoprecipitate (IP) endogenous hevin from brain lysate.
Antibodies were screened for reactivity with rhevin (a) and mouse brain lysate (b) by Western blotting analysis.
Antibodies were analyzed for reactivity with paraffin-embedded sections of mouse brain or retina.
Reactivity in each assay was scored as follows: −, negative; +, weak; ++, moderate; + + +, strong; Nd, not done.
Discussion
Matricellular proteins regulate the interaction of cells with the ECM, a critical function in the maintenance of tissue homeostasis. Hevin, originally described in rodents as SC-1, is a matricellular protein belonging to the SPARC family. In the present study we have produced recombinant mouse hevin and have purified it to near homogeneity, developed MAbs specific for hevin, and identified a function of hevin in vitro. The MAbs described herein will be useful for elucidating the functions of hevin in vivo. For example, hevin has recently been shown to be identical to a neuronal guidance protein, the antigen for the MAb RAGS1 (Gongidi et al. 2004). RAGS1 (Radial Glial Stop Signal molecule 1) recognizes an antigen on the surface of radial glial cells that is distributed where neurons terminate their migration. Gongidi et al. (2004) found that the MAbs RAGS1 and 12–155 react on immunoblots with the same protein. Moreover, inhibition of hevin with RAGS1 allowed neurons to migrate past the normal termination point in the cerebral cortex, thereby demonstrating that hevin is critical for appropriate neuron migration.
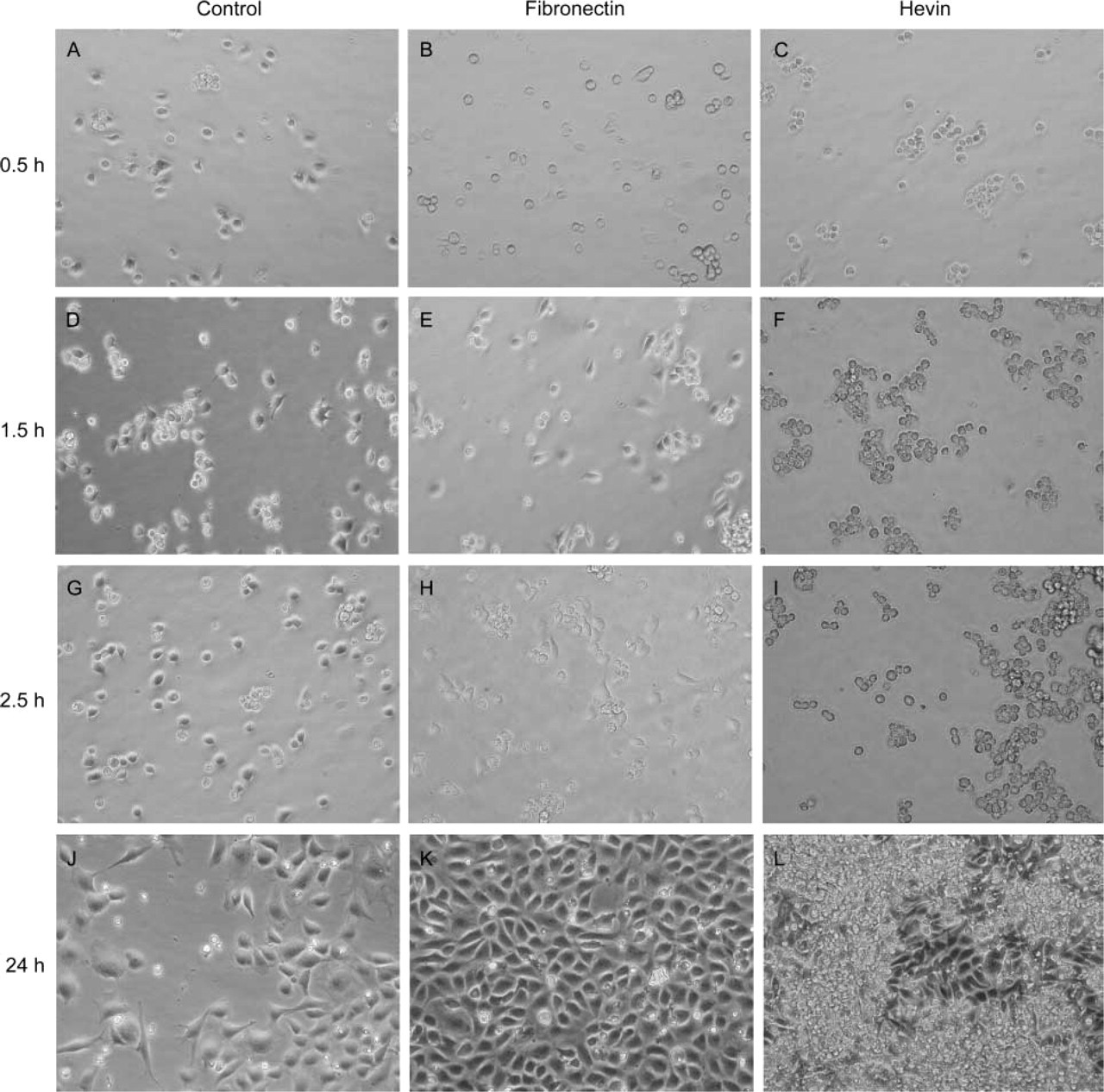
Substrate-adsorbed hevin also inhibits BAEC adhesion. Individual wells of 24-well tissue culture plates were coated with 10 μg/ml protein overnight at 4C. BAECs were plated in serum-free DMEM at 104 cells/well. Cells were monitored hourly by phase-contrast microscopy. (
Among the many cell lines that have been tested for the production of hevin, the few that produce hevin at detectable levels by immunoblotting are tumor lines. Lysates of hevin-producing tumor cells yield a primary immunoreactive band of Mr 130,000 on SDS-PAGE using 10% polyacrylamide gels (not shown). We found hevin of similar Mr in lysates of human and mouse brain tissue, or of Mr 105,000 on 4–12% Bis-Tris gradient gels. The major immunoreactive band of recombinant mouse hevin migrates slightly faster than the Mr 130,000 band characteristic of cell and tissue lysates, a possible consequence of the inability of insect cells to produce N-glycoproteins containing a sialic acid cap, a modification of many complex carbohydrate chains of secreted proteins synthesized by mammalian cells. Note that the apparent Mr of recombinant hevin is 105,000 when estimated by separation on a NuPAGE 4–12% Bis-Tris gel with NuPAGE MOPS SDS running buffer (Invitrogen) (Figure 6). Estimates of the Mr of human hevin in the literature vary from 95,000 (Hambrock et al. 2003) to 150,000 (Bendik et al. 1998) by SDS-PAGE. Therefore, we consider acceptable the apparent size variation that we have observed in recombinant murine hevin and explain it as a possible consequence of the gel resolution system and/or interaction of the acidic N-terminal domain with buffers of varying pH and/or ionic strength.
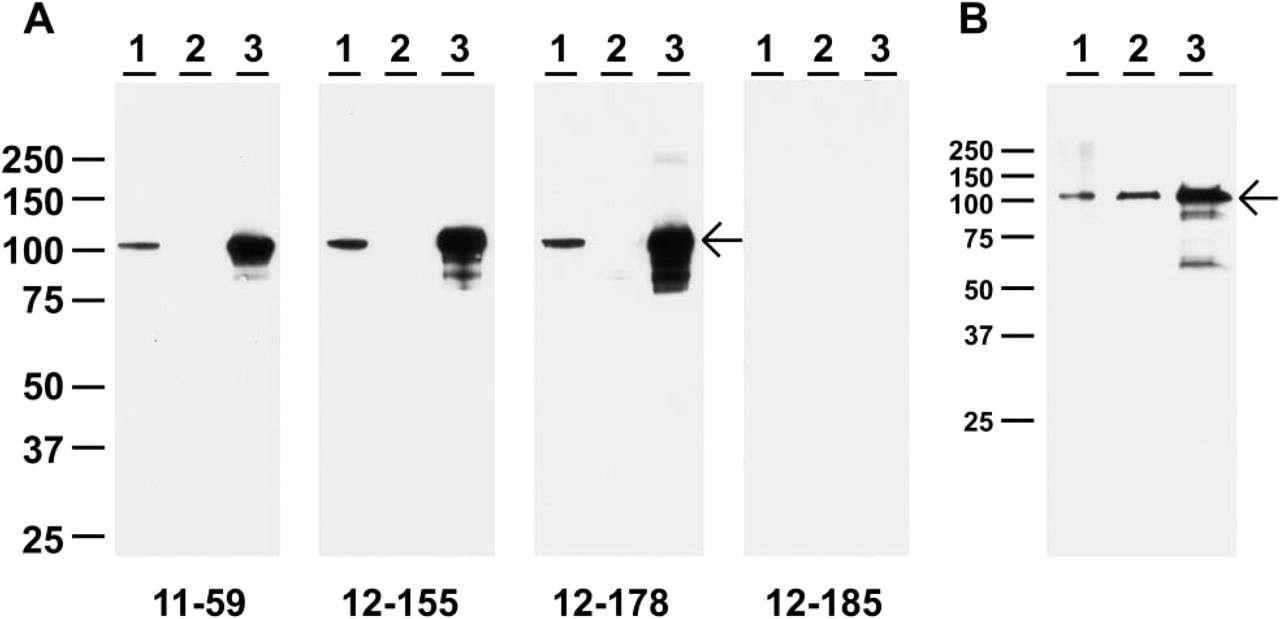
Immunoblotting and immunoprecipitation of murine hevin. (
The de-adhesive effects of hevin that we report are consistent with a prior study. Girard and Springer (1996) found that hevin (in contrast to fibronectin, thrombospondin 1, and tenascin C) was non-permissive for attachment and spreading by HUVECs and BAECs. Furthermore, it diminished adhesion to fibronectin, an effect correlated with reduced formation of focal adhesions. Initially, it was suggested that hevin supported lymphocyte extravasion through high endothelial venules via the diminishment of cell–cell and/or cell–ECM contacts in a dynamic, chemokine-responsive manner (Girard and Springer 1995,1996). We speculate that hevin might play a comparable role in the desmoplastic endothelium associated with tumor growth and metastasis.
The expression and function of hevin in neoplastic tissue appear to be dependent upon the microenvironment, consistent with the expression and function of the hevin homologue SPARC in tumors. For example, SPARC promotes invasiveness in brain tumors (astrocytomas and glioblastomas) (Golembieski et al. 1999; Menon et al. 2000) but induces tumor cell apoptosis in ovarian carcinomas (Yiu et al. 2001). Hevin has been suggested as a potential tumor suppressor due to its downregulation in multiple cancers and its effects on cell proliferation in vitro (Bendik et al. 1998; Claeskens et al. 2000; Isler et al. 2001). However, hevin is also upregulated in the desmoplastic response (Ryu et al. 2001; Iacobuzio-Donahue et al. 2002). Pancreatic adenocarcinomas and renal cell carcinomas, in comparison to normal pancreas or kidney, have been shown to express increased levels of hevin, as determined by SAGE and gene expression profiling (Peale and Gerritsen 2001; Ryu et al. 2001; Iacobuzio-Donahue et al. 2002), and ISH indicated that endothelial cells in the desmoplastic regions of the tumors were primarily responsible for the production of hevin (Ryu et al. 2001; Iacobuzio-Donahue et al. 2002). In contrast, we found that hevin was expressed in or on luminal duct epithelial cells in frozen sections of human endometrial adenocarcinoma (Figure 8), but we did not find hevin in two examples of human pancreatic adenocarcinoma. These discrepancies might be resolved by time-course studies of tumor development and by analysis of the type and degree of desmoplasia associated with each tumor. Hevin was also evident in human pancreatic tumor xenografts grown in immunocompromised mice, although there were different patterns of anti-hevin immunoreactivity in these tissues. Some MAbs reacted clearly with stromal components that included endothelial cells and ECM, whereas other MAbs reacted with distinct stromal cells as well as tumor cells.
Immunolocalization of hevin in normal tissues. IHC analysis of formalin-fixed, paraffin-embedded sections of human skin (
Immunolocalization of hevin in normal mouse cerebellum. IHC analysis of methyl Carnoy's-fixed, paraffin-embedded sections of mouse brain with MAb 12–155 (red). Two ×200 magnification fields (
The function of hevin in the regulation of tissue growth, reaction to injury, and remodeling is not clear, especially with the claim that hevin-null mice lack a discernable phenotype and display no alterations in SPARC levels, in comparison to wild-type mice (McKinnon et al. 2000). SPARC-null mice also do not show major alterations in hevin, as determined by IHC (RAB, unpublished observation). Therefore, the speculation that SPARC and hevin might functionally compensate for each other, although possible, is thus far unsupported. Mice lacking both SPARC and hevin are viable, and studies assessing the responses of these mice to injury are under way (Puolakkainen et al. unpublished observations). Like SPARC, hevin has recently been shown to associate with collagen I fibers in vitro (Hambrock et al. 2003), and IHC analysis of different tissues, particularly subcutaneous tumor xenografts, indicates that hevin is associated with ECM components. Therefore, consistent with a major function of SPARC, hevin might regulate the production, deposition, and/or assembly of ECM proteins or proteoglycans, such as collagen or decorin (Bradshaw et al. 2003; Brekken et al. 2003). Although this idea has not been tested directly, it is important to note that the tools for a thorough examination of the function of hevin are now available. Recombinant hevin, MAbs specific for hevin, hevin-null mice, and hevin/SPARC double-null mice will enable us to probe into the mechanisms by which hevin regulates cell adhesion in vitro and possibly desmoplasia in vivo.
Immunolocalization of hevin in neoplastic tissues. Acetone-fixed frozen sections or human tumor (non-small-cell lung carcinoma) xenografts grown in immunocompromised mice (
Footnotes
Acknowledgements
Funding for monoclonal antibody production was provided by The Hope Heart Institute. Rolf A. Brekken was supported by National Institutes of Health grant F32 HL10352, and E. Helene Sage by R01 GM40711 and by the National Science Foundation–Engineering Research Center (EEC-9529161).
We thank members of the Sage Laboratory and Dr D. Graves for helpful comments, Dr. E. Davie and B. Mc-Mullen for sequencing, and Eileen Neligan for assistance with the manuscript.