
Abstract
Gangliosides are known to be important in many biological processes. However, details concerning the exact function of these glycosphingolipids in cell physiology are poorly understood. In this study, the role of gangliosides present on the surface of rodent mast cells in maintaining cell structure was examined using RBL-2H3 mast cells and two mutant cell lines (E5 and D1) deficient in the gangliosides, GM1 and the α-galactosyl derivatives of the ganglioside GD1b. The two deficient cell lines were morphologically different from each other as well as from the parental RBL-2H3 cells. Actin filaments in RBL-2H3 and E5 cells were under the plasma membrane following the spindle shape of the cells, whereas in D1 cells, they were concentrated in large membrane ruffles. Microtubules in RBL-2H3 and E5 cells radiated from the centrosome and were organized into long, straight bundles. The bundles in D1 cells were thicker and organized circumferentially under the plasma membrane. The endoplasmic reticulum, the Golgi complex, and the secretory granule matrix were also altered in the mutant cell lines. These results suggest that the mast cell- specific α-galactosyl derivatives of ganglioside GD1b and GM1 are important in maintaining normal cell morphology.
G
Another unique feature of gangliosides is their enrichment in plasma membrane microdomains (lipid rafts), along with sphingomyelin, cholesterol, and a number of functionally related proteins (Pike 2003, Ng et al. 2005; Sonnino et al. 2007). There is growing evidence suggesting that lipid rafts play an important role in the regulation of membrane-cytoskeleton interactions and cytoskeleton-dependent cellular processes, particularly in the control of cellular biomechanics (Levitan and Gooch 2007). Lipid rafts are critical for actin organization, membrane trafficking, and cell polarity, as well as cytokinesis (Ng et al. 2005). They are implicated in the functioning of diverse signaling pathways, such as those mediated by growth factors, morphogens, integrins (Simons and Toomre 2000), and antigen receptors on immune cells (Field et al. 1995; Xavier et al. 1998; Cheng et al. 2001; Dráber and Dráberová 2001; Holowka and Baird 2001; Miceli et al. 2001). These membrane microdomains also act as portals of entry for bacterial toxins such as cholera toxin, the B sub-unit of which binds to the lipid raft-enriched GM1 ganglioside (Wolf et al. 2008). Indeed, this property of cholera toxin B subunit has been widely exploited to visualize lipid rafts on a variety of cell types (Gupta and DeFranco 2003; Mitchell et al. 2009).
The α-galactosyl derivatives of the ganglioside GD1b are unique gangliosides present on the surface of rodent mast cells and are specifically recognized by monoclonal antibody (MAb) AA4 (Guo et al. 1989). These gangliosides derived from GD1b have been identified as components of lipid rafts in the plasma membrane of RBL-2H3 cells (Sheets et al. 1999; Silveira e Souza et al. 2008). However, the functional role of these gangliosides, which are close to FcyRI (Basciano et al. 1986), in the lipid rafts is still not clear. It is known that when the α-galactosyl derivatives of ganglioside GD1b are bound by MAb AA4, histamine release is inhibited in a time- and concentration-dependent manner and that morphological and biochemical changes similar to those seen with activation of FcyRI are produced (Basciano et al. 1986; Oliver et al. 1992; Swaim et al. 1994; Stephan et al. 1997). Using two cell lines deficient in the α-galactosyl derivatives of ganglioside GD1b, as well as the parent cell line, RBL-2H3, Silveira e Souza et al. (2008) demonstrated the importance of these gangliosides for the correct assembly of lipid rafts and consequently for FcyRI-mediated degranulation in rodent mast cells.
There is accumulating evidence showing that cellular function is highly affected by gangliosides; however, few studies have evaluated the influence of these lipids on cell structure. Using the mutant cell lines B6A4A2III-E5 and B6A4C1III-D1, which are deficient in α-galactosyl derivatives of ganglioside GD1b (Silveira e Souza et al. 2008), as well as the parent cell line, RBL-2H3, the present study evaluated the role of these gangliosides and GM1 in maintaining cell structure.
Materials and Methods
Cell Lines
Mutant RBL-2H3 cells designated B6A4A2III-E5 (E5) and B6A4C1III-D1 (D1) were generated by exposure to ethyl methane sulfonate, followed by subcloning and identification of sublines deficient in IgE-mediated degranulation as well as in binding of the monoclonal antibody AA4 (Stracke et al. 1987) that is specific for α-galactosyl derivatives of ganglioside GD1b (Guo et al. 1989). The wild-type RBL-2H3 cells (a rat mast cell line) and the mutant cell lines were grown as monolayer, as previously described (Barsumian et al. 1981).
Antibodies
The following primary antibodies were used: mouse MAb anti-β-tubulin (MAB3408, Clone KMX-1, 0.75 μg/ml; Millipore, Billerica, MA) (Birkett et al. 1985); goat anti-GRP 78 [sc-1050, raised against a peptide mapping at the N terminus of the 78-kDa glucose-regulated protein (GRP 78) of human origin, 15 μg/ml, Santa Cruz Biotechnology, Inc.; Santa Cruz, CA] (Hatayama et al. 1992); rabbit anti-γ-tubulin (T3320, raised against a peptide corresponding to the C terminus of Xenopus γ-tubulin, aa 437–451, 1:15,000; Sigma-Aldrich, St. Louis, MO) (Oakley and Oakley 1989); and mouse MAb anti-GM130 (610823, Clone 35, 10 μg/ml; Transduction Laboratories, Lexington, KY) (Nakamura et al. 1997). The following secondary antibodies were used for immunofluorescence: goat anti-mouse IgG F(ab)'2-Alexa 488 or 594, and donkey anti-goat IgG F(ab)'2-Alexa 488 and goat anti-rabbit IgG F(ab)'2-Alexa 594 (1/300 in PBS; Molecular Probes, Eugene, OR).
Light Microscopy
For routine examination by differential interference contrast (DIC), 1–5 × 10 cells were cultured overnight on 13-mm round coverslips. The cells were rinsed twice in PBS and fixed for 20 min with 2% formaldehyde (EM Sciences; Fort Washington, PA) in PBS at room temperature. Some samples were fixed with Carnoy's solution (3% chloroform, 1% acetic acid, and 6% ethanol) for 15 min at room temperature and stained with Alcian Blue (1% Alcian Blue in 120 mM hydrochloric acid, pH 1.0) for 15 min at room temperature; then the samples were rinsed twice in 70% ethanol and once in milli-Q water. The samples were counterstained with Weigert's Fucsin-Resorcin (1% basic fucsin, 2% resorcin, 90% ethanol, 240 mM hydrochloric acid, and 30% FeCl3) for 15 min at room temperature, dehydrated in a graded series of ethanol (50, 70, 80, 90, and 100%), cleared in xylol:ethanol, xylol, and mounted with Permount (Fischer Scientific; Hanover Park, IL). To stain F-actin, 1–5 × 104 cells were cultured overnight on 13-mm round coverslips. Cells were fixed with 4% formaldehyde in PBS for 20 min at room temperature, rinsed in PBS, briefly washed with 0.1 M glycine in PBS, permeablized with 0.3% Triton X-100 (Sigma-Aldrich) for 10 min, rinsed in PBS, and then incubated for 45 min with 2.6 U/ml Phalloidin-Alexa 488 (Molecular Probes). To visualize the gangliosides GM1 (Molecular Probes), 1–5 × 104 cells were cultured overnight on 13-mm round coverslips. The cells were rinsed in PBS, fixed for 20 min with 4% formaldehyde (EM Sciences) in PBS, rinsed in PBS, briefly washed with 0.1 M glycine in PBS and incubated with cholera toxin B conjugated to Alexa 488 (6 μg/ml) for 1 hr at room temperature. The cells were then rinsed in PBS, and coverslips were mounted with Fluoromount-G (EM Sciences). For immunostaining of the endoplasmic reticulum (ER; goat anti-GRP 78) and the Golgi complex (GM130), 1–5 × 104 cells were rinsed in PBS, fixed for 20 min with 2% formaldehyde (EM Sciences) in PBS, rinsed in PBS, briefly washed with 0.1 M glycine in PBS, and permeabilized with acetone at −20C for 4 min. To visualize the microtubules, the cells were fixed with 4% formaldehyde, 50 μM taxol (Sigma-Aldrich), and 50 mM EGTA (Sigma-Aldrich) in PBS for 20 min at 37C, and permeabilized with 0.3% Triton X-100 in PBS for 10 min at room temperature. The cells were then processed as above. After fixation and permeabilization, the cells were rinsed in PBS, briefly washed with 0.1 M glycine in PBS, and blocked for 30 min at room temperature in PBS containing 1% BSA and 5 μg/ml donkey IgG. The cells were then labeled with the primary antibody diluted in PBS +1% BSA for 1 hr at room temperature. After incubation, the cells were rinsed thoroughly in PBS, and the samples were incubated for 45 min at room temperature with the secondary antibody diluted in PBS. All samples were then rinsed in PBS, and coverslips were mounted with Fluoromount-G (EM Sciences). For nuclear staining, after incubation with secondary antibodies, the cells were incubated for 15 min at room temperature with 4',6-diamidino-2-phenylindole (DAPI) (Molecular Probes) at a concentration of 0.2 μg/ml in PBS. Controls consisted of omitting the primary antibody or substituting normal mouse or rabbit IgG for the primary antibody. All controls were negative.
The cells were examined with a Nikon microscope (Nikon E 800; Nikon Instruments, Inc., Melville, NY) equipped with a digital camera (DXM 1200; Nikon), or a LEICA TCS-NT scanning confocal microscope (Leica Microsystems; Heidelberg, Germany) or an Olympus BX50 (Olympus; Melville, NY) microscope equipped with a Nikon DXM 1200 digital camera.
Flow Cytometry
The cells were harvested with trypsin-EDTA (Invitrogen Life Technologies; Carlsbad, CA) and washed by centrifugation in PBS (2000 rpm/5 min) and blocked for 30 min at room temperature in PBS containing 1% BSA and 5 μg/ml donkey IgG. Then the cells were incubated with cholera toxin B conjugated to Alexa 488 (6 μg/ml) for 1 hr at room temperature. The cells were then washed 5 times in PBS (2000 rpm/5 min). After rinsing, the cells were analyzed with a BD FACS Calibur flow cytometer, using the CellQuest program (Becton-Dickinson Labware; San Jose, CA).
Scanning Electron Microscopy (SEM)
Cells (3 × 104/coverslip) were plated on 13-mm round coverslips. Cells were rinsed in warm PBS (37C) and fixed in 2% glutaraldehyde (Ladd Research Industries; Burlington, VT) in warm PBS for 2 hr at room temperature. Cells were postfixed in 1% OsO4 (EM Sciences) for 2 hr, rinsed in Milli-Q water, incubated with a saturated solution of thiocarbohydrazide (EM Sciences), followed by 1% OsO4. This step was repeated once. The cells were dehydrated in a graded series of ethanol and critically point-dried with liquid CO2 in a BAL-TEC-CPD 030 Critical-Point Dryer (BAL-TEC; Liechtenstein, AG), mounted on aluminum stubs with silver paint (EM Sciences), and coated with gold in a BAL-TEC SCD 050 Sputter Coater (BAL-TEC). Samples were examined with a JEOL JSM-5200 scanning electron microscope (Jeol, Ltd.; Tokyo, Japan).
Transmission Electron Microscopy (TEM) Cells were plated at 2 × 105 cells/well in 6-well tissue culture plates and cultured for 3 days before fixation. Media was changed 16–24 hr before fixation. Cells were fixed in 2% glutaraldehyde (Ladd Research) plus 2% formaldehyde (EM Sciences) in 0.1 M cacodylate buffer, pH 7.4, containing 0.05% CaCl2 for 1 hr at room temperature. Cells were postfixed in 1% OsO4 (EM Sciences) in 0.1 M cacodylate buffer, pH 7.4, for 2 hr, rinsed in milli-Q water, dehydrated in a graded series of ethanol. Cells were removed from the tissue culture plates with propylene oxide and embedded in EMBED 812 (EM Sciences). Thin sections were cut with a diamond knife, mounted on copper grids, and stained for 10 min each in Reynolds lead citrate (Reynolds 1963) and 0.5% aqueous uranyl acetate and examined with a Philips EM 208 transmission electron microscope (Fei Company; Eindhoven, The Netherlands).
Identification and Quantification of Glycosaminoglycans
Synthesis of [35S]-sulfated Glycosaminoglycans. Post-confluent RBL-2H3, E5, and D1 cells were grown in 35-mm culture dishes in DMEM (Invitrogen) containing 150 μCi/ml of [35S]-sulfate for 24 hr. The conditioned medium was removed, and the cells were washed three times with 0.1 M PBS and then scraped from the dishes using 3.5 M urea in 0.05 M Tris-HCl, pH 7.4. Protein-free glycosaminoglycan chains were prepared from the cells and conditioned medium after incubation with maxatase (0.1 mg/ml in 0.05 M Tris-HCl, pH 8.0, containing 0.15 M NaCl, 4 hr, 60C, final volume 100 μl) and identified and quantified using agarose gel electrophoresis and enzymatic degradation with glycosaminoglycan lyases (chondroitinases AC and ABC, heparinase, and heparitinases I and II) as previously described (Nader et al. 1989; Trindade et al. 2008).
Results
Expression of GM1 Is Lower in D1 Cells
As previously reported (Silveira e Souza et al. 2008), RBL-2H3 cells highly express the derivatives of GD1b ganglioside on their surface, whereas the E5 cells express 35% and the D1 cells express <1% of these gangliosides. To verify whether the expression of another ganglioside was also affected in the E5 and D1 cells, RBL-2H3 cells and the mutant cell lines were incubated with cholera toxin B subunit, which recognizes GM1. Similar to that observed for the ganglioside derivatives of GD1b, RBL-2H3 cells highly express the ganglioside GM1 on their surface (Figure 1A), whereas in the E5 cell line, the level of expression of GM1 is only slightly reduced when compared with RBL-2H3 cells (Figure 1B). However, the D1 cells show only a minimum level of expression of these gangliosides (Figure 1C). By flow cytometry, the E5 and D1 cell lines express 89.2% and <1%, respectively, of GM1 relative to the amount present on RBL-2H3 cells (Figure 1D). Therefore, the level of expression of gangliosides in the E5 mutants varies with the ganglioside type, whereas in the D1 cell line, these two types of gangliosides are virtually absent.
Lack of Gangliosides Affects Cell Structure
Because gangliosides are known to play a role in the structure of the plasma membrane, the possibility that the decreased expression of GM1 and the α-galactosyl derivatives of gangliosides GD1b (Silveira e Souza et al. 2008) may produce morphological alterations in the mutant cell lines was investigated. DIC microscopy (Figure 2) shows that the cell lines RBL-2H3 and E5 are primarily spindle-shaped, whereas the D1 cells are round, somewhat smaller, and grow as small colonies, indicating that the lack of gangliosides affects the structural characteristics of these cells. These morphological differences were confirmed by scanning electron microscopy (Figure 2). Similar to that seen by DIC, by scanning electron microscopy, RBL-2H3 cells are elongated and spindle-shaped, and their surface is covered with small, short microvilli. E5 cells have morphology similar to that of RBL-2H3 cells. The majority of D1 cells are round and somewhat smaller, and their surface is dramatically altered, possessing a few large ruffles instead of fine microvilli (Figure 2, arrows).
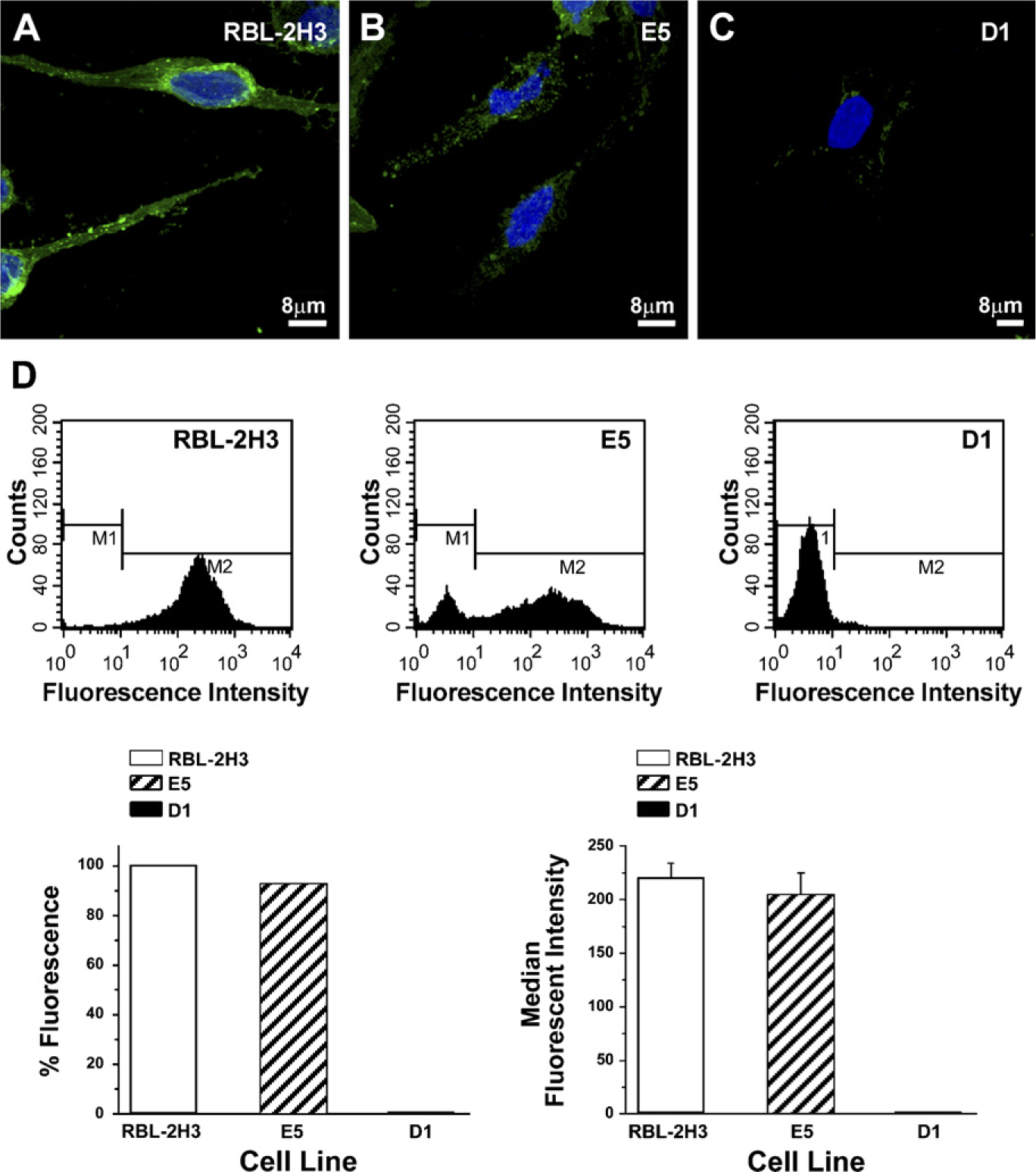
Expression of GM1 gangliosides. GM1 gangliosides were labeled with the B subunit of cholera toxin conjugated to Alexa 488. All of the RBL-2H3 cells express GM1 gangliosides on their surface (
Because the D1 cells have a distinct morphology, including extensive membrane ruffles, it was of interest to compare the distribution of the cytoskeleton in the three cell lines. In RBL-2H3 and E5 cells (Figure 3), the actin filaments are under the plasma membrane following the spindle shape of the cells and in association with microvilli. In D1 cells, the actin filaments are concentrated in large membrane ruffles (Figure 3, arrows), and extend into the cytoplasmic projections marking the entire cell. The distribution of the microtubules is also altered in the D1 cells (Figure 3). Microtubules in RBL-2H3 and E5 cells radiate from the centrosome and are organized into long, straight bundles that give a fusiform shape to the cells. In D1 cells, the microtubules are also associated with a centrosome, but the bundles in D1 cells appear thicker and are organized circumferentially in a basket shape and are largely restricted to the cell body.
Because organization of the cytoskeleton was altered in the mutant cells, especially in D1 cells, it was of interest to examine the structure of two organelles, the Golgi apparatus and the rough ER, which are dependent on the cytoskeleton for their organization. Both the ER and the Golgi complex (Figure 4) are altered in the mutant cell lines. The ER in RBL-2H3 cells has a filamentous distribution throughout the cytoplasm. In contrast, in the E5 and D1 cells, the ER is more vesicular, especially in the perinuclear region. By TEM, the ER in the RBL-2H3 cells appears filamentous with long, thin cisternae. The E5 cells have numerous short cisternae, whereas in the D1 cells, the cisternae appear slightly dilated. The Golgi complex in all three cell lines, RBL-2H3, E5, and D1, is located in a perinuclear position (Figure 4). By TEM, the RBL-2H3 cells have a well-organized Golgi complex, with their cisternae showing the typical arrangement of flattened saccules. In the E5 cells, although the Golgi complex is well-developed, the cisternae are not as well-organized as in the RBL-2H3 cells and appear slightly dilated. The Golgi complex of the D1 cells is smaller, and the cisternae are shorter and disorganized. Therefore, the fact that these organelles are altered in the mutant cell lines could affect ganglioside synthesis and/or transport.
Because both the ER and the Golgi apparatus are altered in the mutant cells, the matrix of the secretory granules, produced by these organelles, was also examined. The cells were stained with Alcian Blue, which labels the glycosaminoglycans, to visualize the granules and to view their distribution. In RBL-2H3 cells, the granules are present throughout the cytoplasm (Figure 5, arrows). In E5 cells, the granules appear to be smaller (Figure 5, arrow), and the D1 cells contain fewer but larger granules than do the RBL-2H3 cells (Figure 5, black arrows). Clear vacuoles (Figure 5, green arrow) are also present in the cytoplasm of the D1 cells. The difference in the cytoplasmic granules was even more apparent when the cells were examined by TEM. The granules in the RBL-2H3 cells contain electron-dense material (Figure 5, arrows), most likely granule matrix. In contrast, the cytoplasmic granules in the E5 (Figure 5, arrows) and D1 (Figure 5, arrows) cells are largely electron-lucid and the granule matrix appears different.
Because sulfated glycosaminoglycans are a major component of the mast cell granule matrix, the content of sulfated glycosaminoglycans from the three cell lines was analyzed by agarose gel electrophoresis. All three cell lines have chondroitin sulfate and heparin. Analysis of metabolically [35S]-sulfate-labeled lysates from the E5 and D1 cells (Figure 6) showed that they contained, respectively, 19% and 56% of the amount of chondroitin sulfate seen in the RBL-2H3 cells. However, the E5, and especially the D1 cells (206% and 549%, respectively), contain much more heparin than do the RBL-2H3 cells, suggesting that the synthesis of glycosaminoglycans and the reduction in the amount of gangliosides in the mutant cell lines may be related.
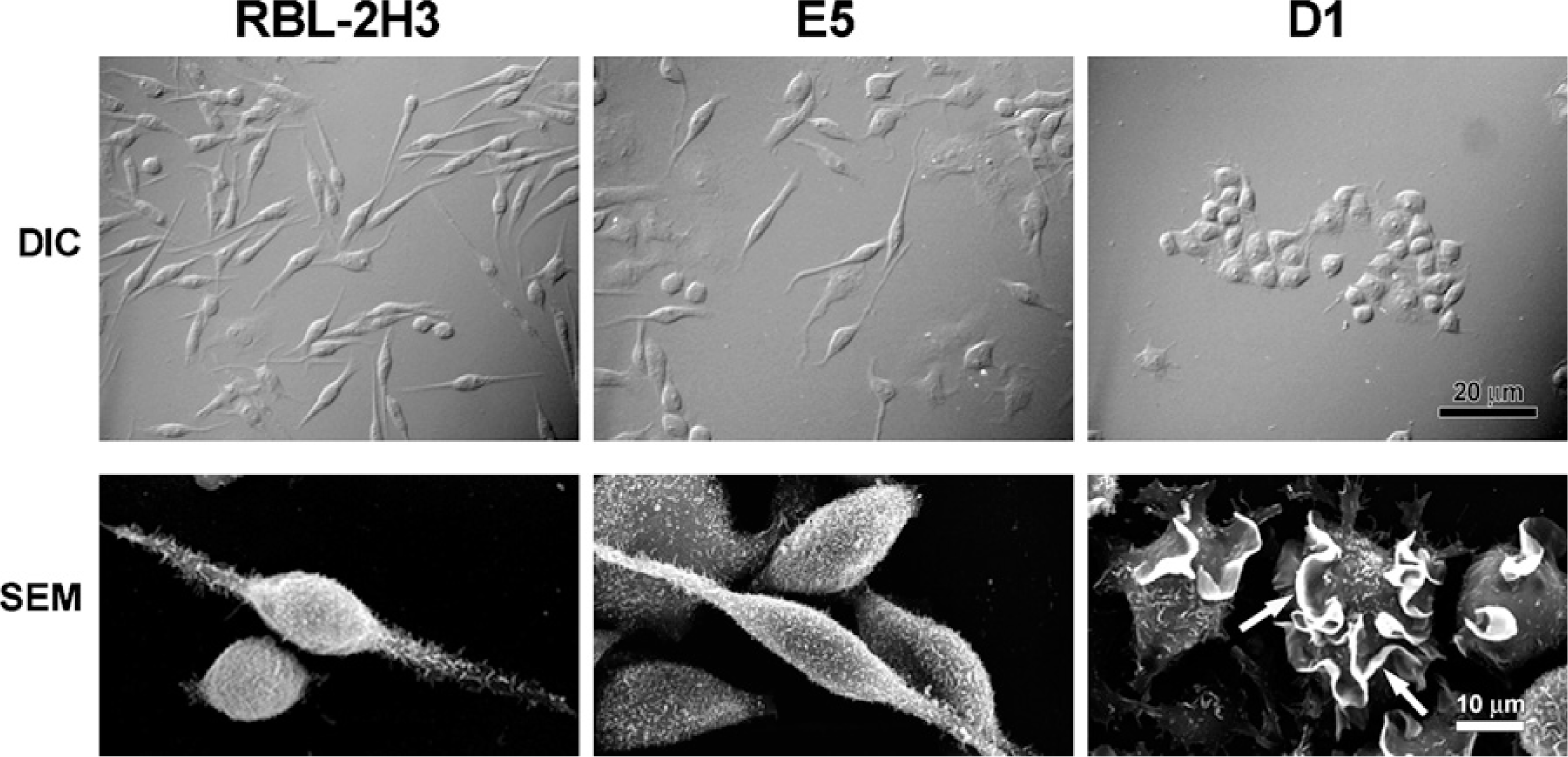
D1 cells are morphologically distinct from RBL-2H3 and E5 cell lines. By differential interference contrast (DIC), the RBL-2H3 and E5 cells are spindle-shaped. D1 cells are round and grow close to each other as small colonies. By scanning electron microscopy (SEM), RBL-2H3 cells are spindle-shaped and their surface is covered with short microvilli. E5 cells have morphology similar to that of RBL-2H3 cells. The majority of D1 cells are round, and their surface is dramatically altered possessing only a few large ruffles (arrows).
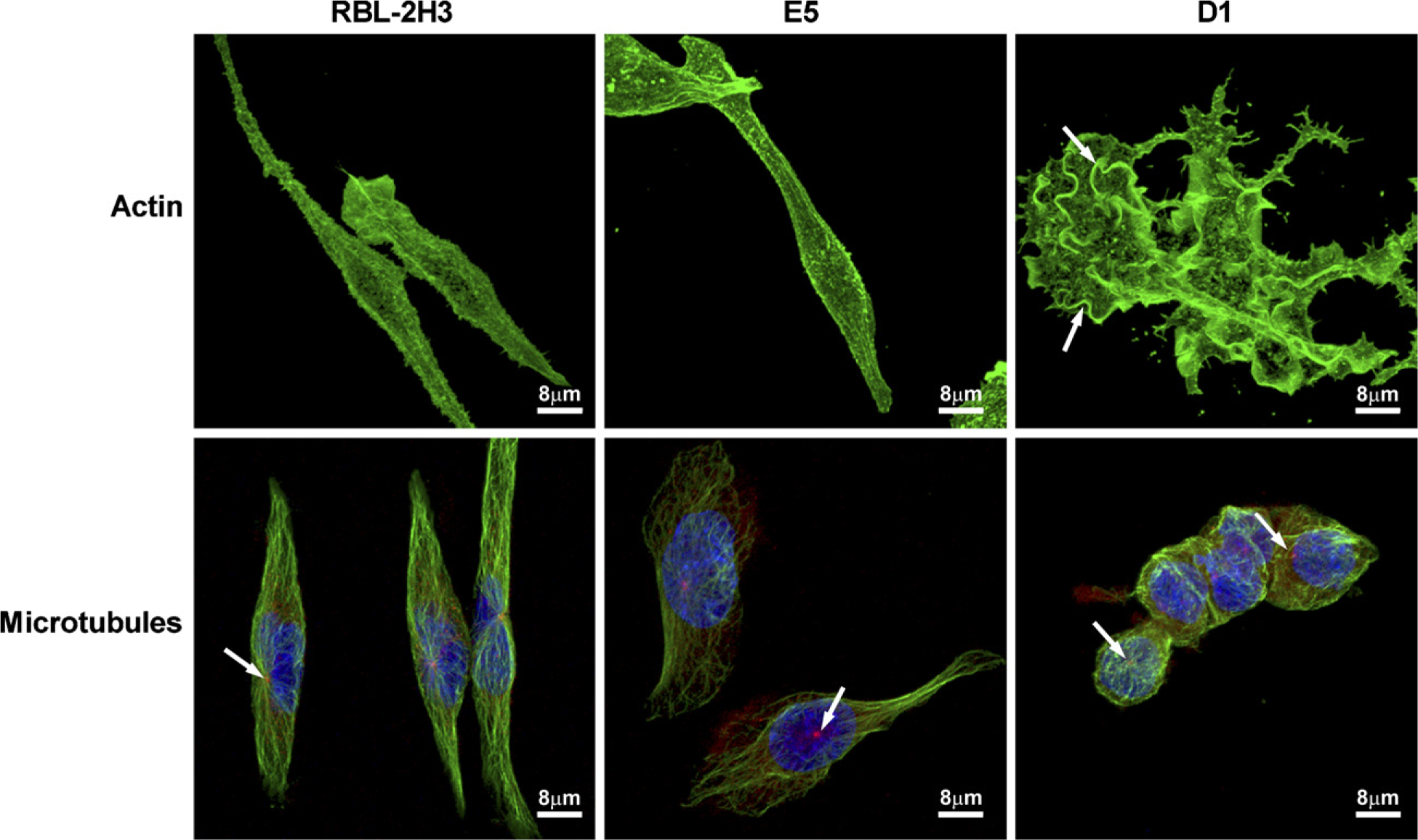
Cytoskeleton is altered in the D1 cells. Actin filaments in RBL-2H3 and E5 cells lie under the plasma membrane following the spindle shape of the cells and in association with microvilli. In D1 cells, actin filaments are concentrated in large membrane ruffles (arrows) and extend into the cytoplasmic projections. F-actin was labeled with phaloidin-Alexa 488. In RBL-2H3 cells, microtubules labeled with anti-β-tubulin are organized into bundles that followed the fusiform shape of the cells, whereas in E5 cells, they appear less organized, and are completely disorganized in D1 cells. In D1 cells, microtubules appear as thick bundles that are restricted to the cell body and are not present in the cytoplasmic extensions. Centrosomes (arrows) were labeled with anti-γ-tubulin. Nuclei are marked in blue with DAPI.
Discussion
The importance of the α-galactosyl derivatives of the ganglioside GD1b for mast cell degranulation via FcyRI and for the correct assembly of lipid rafts was demonstrated by Silveira e Souza et al. (2008). Gangliosides are components of all animal cell membranes, and the plasma membrane is intimately related to cell morphology. In this study, the influence of gangliosides on cell structure and organization was examined using the ganglioside-deficient cell lines E5 and D1 (Silveira e Souza et al. 2008) and the parent cell line, RBL-2H3.
The morphological analysis by DIC and SEM showed that the mutant cell line D1 has a cellular morphology distinct from that of the mutant E5 and RBL-2H3 cell lines, suggesting that the gangliosides are important in the maintenance of normal cell morphology. This role for gangliosides was also observed by Yates et al. (1989) and Ikami et al. (2000). The morphological changes observed in D1 cells could be related to the lipid composition of these cells. In this cell line, there is a large decrease in glycosphingolipids that may affect many physicochemical properties of the plasma membrane (Kato et al. 2003) as well as the anchorage of the cytoskeleton to the plasma membrane (Palestini et al. 2000).
Meivar-Levy et al. (1997) demonstrated in 3T3 fibroblasts that long-term incubation with mycotoxin fumonisin B1 (FB1), a specific inhibitor of sphingolipid synthesis, resulted in a decrease in synthesis of ganglioside GM3 and caused profound changes in a number of processes associated with the actin cytoskeleton, including cell spreading, microvilli formation, cytokinesis, formation of long processes, and disruption of DNA synthesis and cell proliferation. Remarkably, all of the morphological effects of FB1 could be reversed by the addition of low concentrations of GM3.
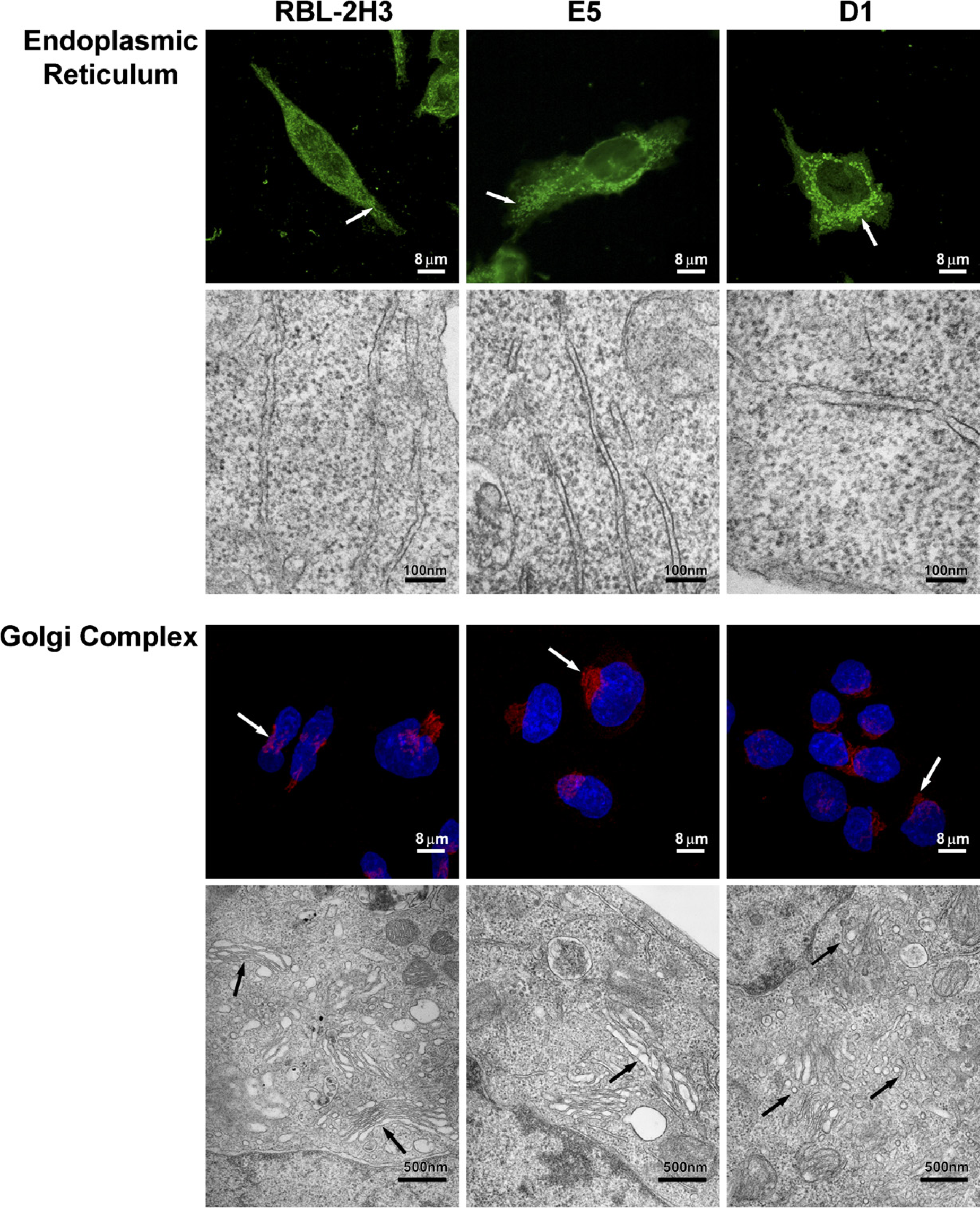
Lack of gangliosides also affects the endoplasmic reticulum (ER) and the Golgi complex. ER was labeled with anti-GRP-78, and alterations were assessed in the three cell lines. ER in RBL-2H3 cells is filamentous in form (arrow). In the E5 and D1 cell lines, ER appears vesiculated (arrows). By transmission electron microscopy (TEM), ER in the RBL-2H3 cells is filamentous with long, thin cisternae. E5 cells have numerous short cisternae, and in D1 cells, the cisternae seem slightly dilated. Golgi complex in the RBL-2H3, E5, and D1 cell lines has perinuclear localization (arrows). The size of the Golgi complex is reduced in D1 cells. Cis-Golgi cisternae were labeled with anti-GM130, and nuclei were stained with DAPI. By TEM, the RBL-2H3 cells have a well-organized Golgi complex, with their cisternae showing the typical arrangement of flattened saccules (arrows). In E5 cells, the Golgi cisternae (arrow) are not as well-organized and appear slightly dilated. Golgi complex of the D1 cells is smaller and the cisternae are disorganized (arrows).
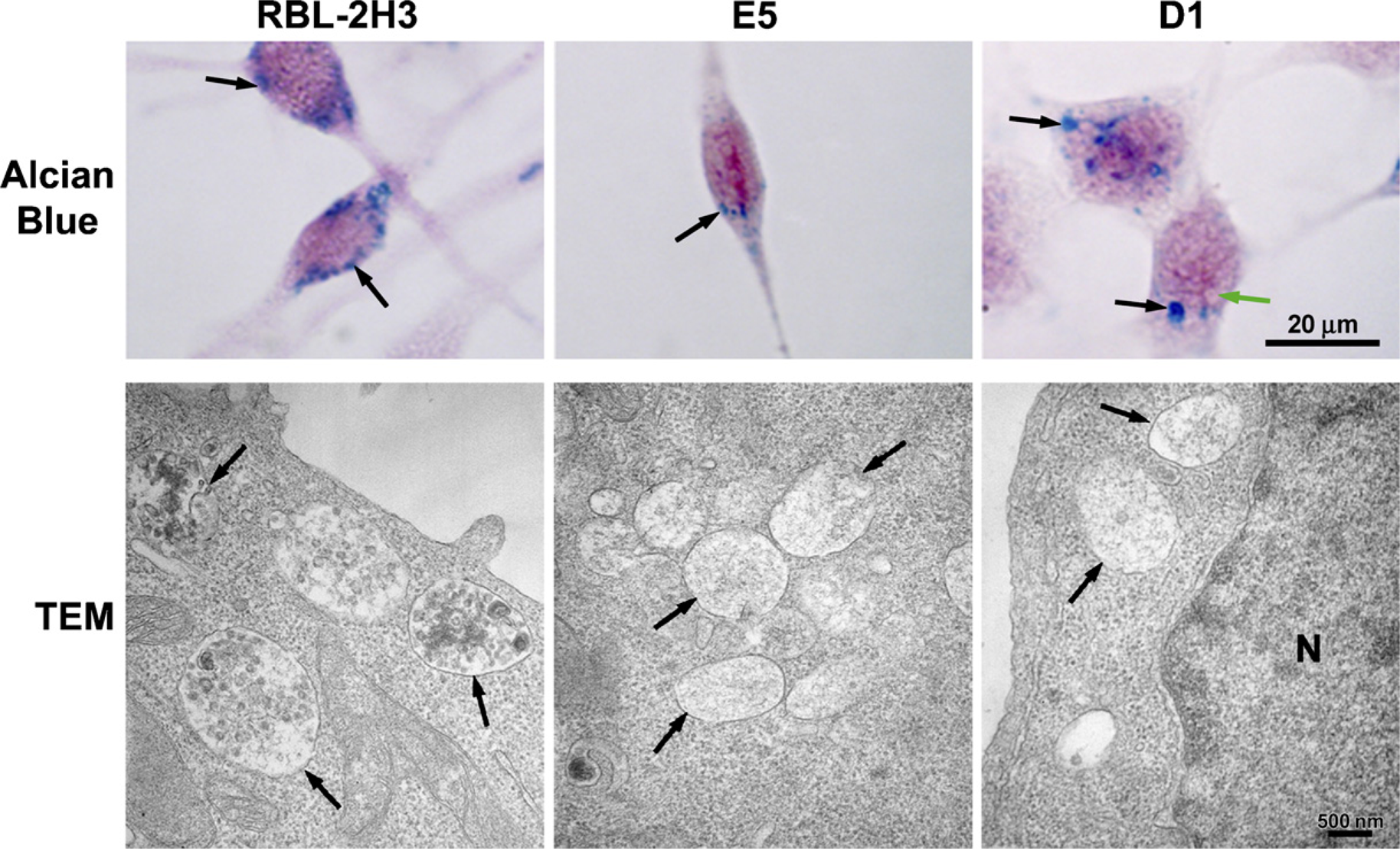
Secretory granules are altered in the mutant cell lines. In RBL-2H3 cells, the granules are distributed through the cytoplasm (arrows). In E5 cells, the granules are smaller (arrow) and stain less intensely. D1 cells have a few large granules (black arrows), and their cytoplasm appears to be filled with clear vacuoles (green arrow). Alcian Blue staining counterstained with Weigert's Fucsin-Resorcin. By TEM, RBL-2H3 cells contain electron-dense granules (arrows). However, in the E5 and D1 cells, the granules are electron-lucid (arrows). N, nucleus.
The primary intracellular determinant of cell morphology is the cytoskeleton. Pfeiffer et al. (1985) and Sahara et al. (1990) have previously shown that polymerized actin in RBL-2H3 cells is mainly associated with membrane ruffles after stimulation similar to that seen in unstimulated D1 cells. However, unlike activated RBL-2H3 cells, the D1 cells are not spread over substrate. The actin filaments are important for de-granulation and serve to transport the granules for short distances at the cellular periphery (Kuznetsov et al. 1992). In addition, in resting cells, subcortical actin acts as a physical barrier, preventing the secretion of granules. Frigeri and Apgar (1999) demonstrated that RBL-2H3 cells exposed to latrunculin, an inhibitor of actin polymerization, showed a decrease in F-actin and an increase in degranulation after cell activation.
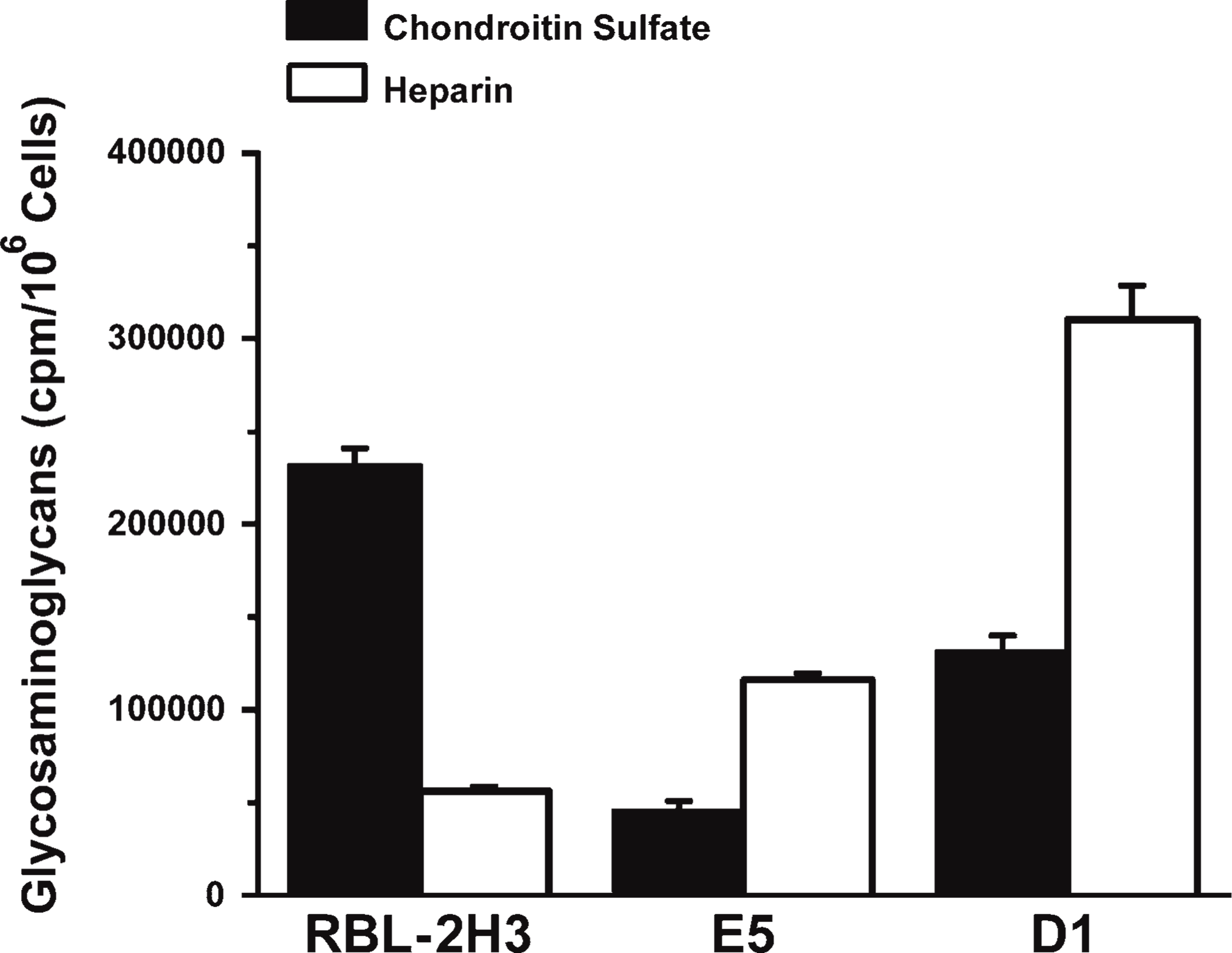
Secretory granule matrix is also altered in the mutant cell lines. The amount of chondroitin sulfate is reduced in D1 and E5 cells in comparison to RBL-2H3 cells, but there is more heparin in the mutant cells (E5 and D1).
A growing amount of evidence indicates that lipid rafts are essential for membrane-cytoskeleton coupling (Levitan and Gooch 2007; Pike 2009). The factors that govern the formation of lipid rafts continue to be elucidated, but lipid raft formation often requires actin filaments. The connection between lipid raft proteins and actin filaments can affect the lateral distribution and mobility of these membrane proteins (Rodgers and Glaser 1993; Lenne et al. 2006). The presence of actin and other cytoskeletal proteins in lipid raft fractions has been known since the earliest description of these structures (Jordan and Rodgers 2003; Badizadegan et al. 2004), and proteomic analyses have documented the presence of numerous cytoskeletal proteins in these fractions (Nebl et al. 2002). Furthermore, cholesterol depletion, which is often regarded as a functional test of dependence on lipid rafts, is associated with loss of phosphatidylinositol 4,5-biphosphate from the plasma membrane and a global reorganization of the actin cytoskeleton (Kwik et al. 2003), suggesting a possible role for actin in the organization and/or function of lipid rafts. However, the extent to which the actin cytoskeleton participates in establishing membrane rafts is not yet established. Importantly, the actin cytoskeleton is a dynamic structure that changes in response to extracellular signals, and it may therefore represent one mechanism for governing the establishment and distribution of membrane rafts in the plasma membrane (Chichili and Rodgers 2007). In a recent review, Chichili and Rodgers (2009) show that there is evidence of structuring of rafts by a synergistic interaction between the cortical actin cytoskeleton and lipid rafts, and that many of the structural and functional properties of rafts require an intact actin cytoskeleton. The data of Fujita et al. (2009) indicate that the GM1 and GM3 clusters in the cell membrane are regulated in different ways and that segregation of the two gangliosides depends on an intact actin cytoskeleton.
It is possible that the abnormal distribution of actin filaments observed in D1 cells is a result of the disorganized lipid rafts in these cells (Silveira e Souza et al. 2008), and that the disorganization of both lipid rafts and actin filaments leads to impaired degranulation in the mutant mast cell lines. A physical association between an outer-membrane lipid and the actin cytoskeleton requires the presence of other structural signaling or regulatory proteins or lipids, or both (Badizadegan et al. 2004). The results obtained previously (Silveira e Souza et al. 2008) and in this study suggest an association between the gangliosides in the outer leaflet of the plasma membrane and actin filaments on the cytoplasmic surface of the plasma membrane. Therefore, lipid rafts might be involved in regulating cytoskeleton organization, thus regulating cytoskeleton-dependent cellular processes. Holowka et al. (2000) demonstrated that functionally important interactions between the high-affinity Fcy receptor in activated mast cells and lipid rafts are dependent on the actin cytoskeleton. A similar role for the actin cytoskeleton in the biology of lipid rafts has been suggested in other systems, such T-cell signaling (Badizadegan et al. 2004). Disruption of lipid rafts by cholesterol depletion in T-cells inhibits the inducible tyrosine phosphorylation of the T-cell receptor (TCR) and prevents its association with actin (Moran and Miceli 1998). The idea that actin plays a central role in T-cell signaling via lipid rafts is further supported by the observation that activation of TCR induces reorganization of actin in a cholesterol-dependent manner (Valensin et al. 2002). Mitchell et al. (2009) found that clustering GM1 lipid rafts in T-cells can regulate b1 integrin function and activate adhesion-strengthening processes that are accompanied by a remodeling of the contact area with the adhesive plane, the induction of high-affinity integrin states, and the rearrangement of integrin-cytoskeleton networks.
Microtubules are one of the major determinants of cell shape and polarity (Ilschner and Brandt 1996; Conde and Cáceres 2009). The ganglioside-deficient D1 cells are round, and the arrangement of their microtubules is completely disorganized. Several studies have shown a relationship between microtubules and gangliosides. Microtubules are essential for ganglioside-mediated neurite elongation (Spero and Roisen 1984; Spero et al. 1986), and exogenously added GM1 induced typical ramified morphological changes in microglia in a dose-dependent manner (Park et al. 2008).
The abnormal distribution of microtubules in D1 cells may be one of the causes of the disorganized arrangement of the Golgi complex observed by TEM in this cell line. Both the structure and positioning of the Golgi complex are highly dependent on the microtubule cytoskeleton. Rivero et al. (2009) observed that when the microtubules are disorganized, the Golgi complex fragments, giving rise to discrete Golgi elements, which are dispersed throughout the cell, exactly as observed in the D1 mutant cell line. This suggests that microtubules are required to link stacks into a single organelle and to ensure its pericentrosomal location (Thyberg and Moskalewski 1999). Additionally, the specific glycosyltransferases that synthesize the oligosaccharide moiety of the gangliosides (Gillard et al. 1994; Crespo et al. 2004) are located in this organelle. Therefore, disruption of the Golgi complex could also interfere with ganglioside synthesis.
The disorganization of the ER, Golgi apparatus, and microtubules may explain the alterations in the secretory granule matrix seen in D1 cells. The ER, and especially the Golgi complex, are involved in the biosynthesis of heparan sulfate/heparin and chondroitin sulfate (Silbert and Sugumaran 2002; Gorsi and Stringer 2007). The distribution of glycosyltransferases along the Golgi cisternae dictates the overall structure of glycosylated compounds (de Graffenried and Bertozzi 2004). A disruption in the orderly passage of glycosylated molecules through the Golgi complex could lead to the production of alternate products, such as heparin instead of chondroitin sulfate.
The results of this study demonstrate that decreased ganglioside expression in the mutant cells, E5 and D1, affects the normal cell structure of the RBL-2H3 mast cells. Various cell processes, such as signal transduction, vesicular transport, endocytosis, and cell adhesion, could be compromised by the decrease in gangliosides that leads to lipid raft disorganization.
Footnotes
Acknowledgements
We thank Coordenação de Aperfeiçoamento de Pessoal de Nível Superior, Fundação de Apoio ao Ensino, Pesquisa e Assistěncia, and Fundação de Amparo à Pesquisa do Estado de São Paulo (individual grant 01/10752-2 to CO) for financial support.
We thank Reuben P. Siraganian, MD, PhD, National Institutes of Health, Bethesda, MD, who generously provided the cell lines; Márcia Sirlene Graeff for assistance with the confocal microscopy; Maria Dolores S. Ferreira, Tereza P. Maglia, and José Augusto Moulin for assistance with the scanning and transmission electron microscopy; and Anderson Roberto de Souza for technical assistance; all from the Department of Cell and Molecular Biology and Pathogenic Bioagents, FMRP-USP, Ribeirão Preto, SP. We also thank Patrícia Vianna Bonini Palma, Fabiana Rossetto, and Camila Cristina Branquinho de Oliveira Menezes, Hemocentro- Faculdade de Medicina de Ribeirão Preto-Universidade de São Paulo, Ribeirão Preto, Brazil, for assistance with the flow cytometry.