
Abstract
Fukutin-related protein (FKRP) is a protein involved in the glycosylation of cell surface molecules. Pathogenic mutations in the FKRP gene cause both the more severe congenital muscular dystrophy Type 1C and the milder Limb-Girdle Type 2I form (LGMD2I). Here we report muscle histological alterations and the analysis of 11 muscle proteins: dystrophin, four sarcoglycans, calpain 3, dysferlin, telethonin, collagen VI, α-DG, and α2-laminin, in muscle biopsies from 13 unrelated LGMD2I patients with 10 different FKRP mutations. In all, a typical dystrophic pattern was observed. In eight patients, a high frequency of rimmed vacuoles was also found. A variable degree of α2-laminin deficiency was detected in 12 patients through immunofluorescence analysis, and 10 patients presented α-DG deficiency on sarcolemmal membranes. Additionally, through Western blot analysis, deficiency of calpain 3 and dystrophin bands was found in four and two patients, respectively. All the remaining proteins showed a similar pattern to normal controls. These results suggest that, in our population of LGMD2I patients, different mutations in the FKRP gene are associated with several secondary muscle protein reductions, and the deficiencies of α2-laminin and α-DG on sections are prevalent, independently of mutation type or clinical severity.
T
Pathogenic mutations in the FKRP gene result in muscular dystrophy (MD) phenotypes, which were identified in congenital MD Type 1C (MDC-1C) and Limb-Girdle MD Type 2I (LGMD2I) (Brockington et al. 2001a, b). Both MDC and LGMD2I are heterogeneous groups of autosomal recessively inherited muscle diseases. MDC is characterized by onset of symptoms within the first few months of life, and in the 1C form, patients never acquire ambulation. LGMD2I has a milder and variable course, with the age at onset varying from the first to the fourth-fifth decade of life and slower progression. In both conditions, serum creatine kinase is elevated and intelligence is preserved, although structural brain changes have been detected in some patients with FKRP mutations (Topaloglu et al. 2003; Mercuri et al. 2006; Quijano-Roy et al. 2006).
Components of the extracellular matrix, such as α2-laminin, integrin α-7, and collagen VI, interact with most defective proteins responsible for MDC, whereas the defects responsible for the LGMD forms are largely associated with sarcolemmal proteins α, β, γ, and δ-sarcoglycans, caveolin, dysferlin, and sarcomeric proteins such as telethonin and myotilin (Brockington et al. 2002; Bushby and Beckmann 2003). Although MDC-1C and LGMD2I are allelic disorders, it became clear that, in addition to the clinical and pathological heterogeneity, they are also genetically diverse. Different types of mutation cause MDC-1C, whereas the majority of the LGMD2I patients already described carry a common C826A missense mutation in the FKRP gene (Mercuri et al. 2003; Wicklund and Hilton-Jones 2003; Brown et al. 2004).
Although the function of FKRP is still unknown, it has been suggested that it might be involved in the glycosylation of α-DG in muscle membrane. The DG complex is important in muscle formation and maintenance and cell adhesion, and it also plays an important role in the function of other tissues, such as brain, kidneys, and peripheral nerves. A single gene DAGI encodes a polypeptide that is post-translationally modified to yield the two glycoproteins referred to as α- and β-DG (Michele et al. 2002; Martin 2003; Cohn 2005). α-DG is a heavily glycosylated peripheral membrane component of the dystrophin-associated-glycoprotein complex (DGC), whereas β-DG is a transmembrane protein that links to dystrophin intracellularly. The disruption of this linkage underlies several forms of MD, underscoring its importance in striated muscle, which contributes to the structural integrity of the sarcolemma (Ervasti and Campbell 1993).
Providing evidence for the glycosyltransferase function of FKRP, patients with MDC-1C typically show abnormalities of α-DG glycosylation, in addition to a secondary reduction in α2-laminin (Brockington et al. 2001b). All studied patients with this severe form had a variable secondary reduction in α2-laminin expression, which was less marked than the reduction in α-DG (Brockington et al. 2002; Brown et al. 2004). Moreover, muscle biopsy findings seem to be less uniform in patients with LGMD2I. Analysis of muscle biopsies has shown variable secondary protein abnormalities, and unlike MDC-1C, detection of abnormal α2-laminin chain on IHC is not so remarkable (Poppe et al. 2003).
Because FKRP protein cannot be easily measured, secondary effects on muscle protein presence and organization can be used as additional information on the mechanism of the disease and could explain the huge clinical variability observed among LGMD2I patients (de Paula et al. 2003). Here we report muscle biopsy analyses and the expression of muscle proteins of the sarcolemma (dystrophin, four sarcoglycans, dysferlin), extracellular matrix (α2-laminin, α-DG, collagen VI), sarcomere (telethonin), and cytosol (calpain 3) in 13 LGMD2I families with 10 different mutations in the FKRP gene.
Materials and Methods
Patients
We analyzed 13 unrelated LGMD2I patients with different mutations in the FKRP gene (Table 1). These patients were molecularly selected and clinically classified in a previous study of 86 Brazilian families (de Paula et al. 2003), ascertained in the Human Genome Research Center, Department of Biology, IB-USP, and classified as LGMD according to the criteria reported in Bushby (1995) and Bushby and Beckmann (2003). The study was performed after receiving the Institutional Human Subjects Review Board (IRB) approval. All patients signed the informed consent form before any procedure of the study.
Among the 13 studied patients carrying FKRP mutations that were clinically affected, 11 presented a milder LGMD course (ambulant after age 16), 1 (already deceased) had a severe Duchenne-like phenotype (wheelchair bound at age 12), and 1 had an intermediate course (wheelchair bound at age 17) (Table 1).
Patients were classified according to the mutation into three groups: Group 1, four patients carrying the common C828A mutation in the homozygous state; Group 2, five patients homozygous for other mutations; and Group 3, four patients who were compound heterozygous for two different mutations in the FKRP gene (Table 1).
Muscle samples were obtained from biceps biopsies (for diagnostic purposes or after informed consent), frozen in liquid nitrogen immediately after removal, and stored at −70C until use. Routine histological and histochemical procedures were done, with staining for hematoxylin/eosin, modified Gomori trichrome, NADH, ATPase 9.4, 4.3, and acid and alkaline phosphatase (Dubowitz 1999).
Molecular and clinical findings in LGMD2I Brazilian patients
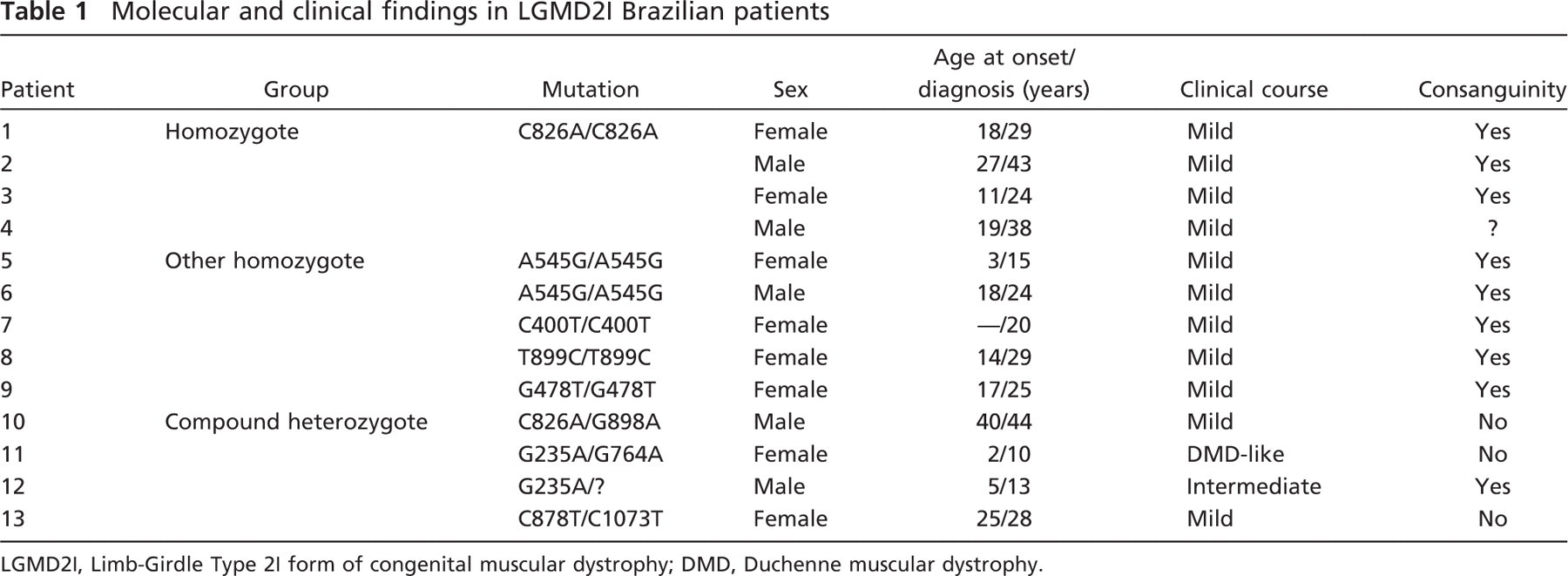
LGMD2I, Limb-Girdle Type 2I form of congenital muscular dystrophy; DMD, Duchenne muscular dystrophy.
Protein Studies
Protein analyses have been done in these patients since 1990 for diagnostic purposes. When sample material was available for analysis, new reactions were performed for this study.
Immunofluorescence Analysis
IHC analyses were performed, using simple or double reactions, as previously described (Vainzof et al. 2003, 2005). Briefly, four to six cryostat sections were thawed on polylysine-coated slides, air dried for 1 hr, and preincubated with PBS containing 10% horse serum (PBSS). The sections were incubated with primary antibodies for 6 hr at room temperature, washed three times with PBS, and incubated with anti-mouse or rabbit IgG-, FITC-, or CY3-conjugated secondary antibodies for 1 hr. After three additional washings, the sections were mounted with Vectashield (Vector Laboratories; London, UK) and analyzed under a Zeiss Axiophot microscope with epifluorescence (Carl Zeiss; Oberkochen, Germany). Image exposure time was standardized using the positive normal control for each reaction/antibody (Figure 1). The same exposure time was used for the images from each analyzed patient. Digital pictures were taken using the Axiovision 4.5 software (Carl Zeiss).
Multiplex Western Blot Analysis
Routine Western blot analysis was performed using 6% SDS-PAGE gels, and proteins were transferred at 150 V for 1 hr. The blots were reacted with a mix of three primary antibodies against dystrophin (rod domain), dysferlin, and calpain 3 (94-kDa band). Incubations with primary antibodies were done overnight, and detection was done using alkaline phosphatase-conjugated secondary antibodies (anti-mouse IgG conjugated to alkaline phosphates) and colorimetric reaction for the enzyme, using nitroblue tetrazolium and 5-bromo-4-chloro-3-indolyl phosphate as substrate (Vainzof et al. 2003). Quantitative analysis was done by comparing each protein band to the other two present in the same blot, and all were normalized to the myosin content of the muscle extract, as detected in the Ponceau S prestained blot (Figure 2). Quantification was done using ID Image Analysis Software (Kodak Digital Science; Rochester, NY).
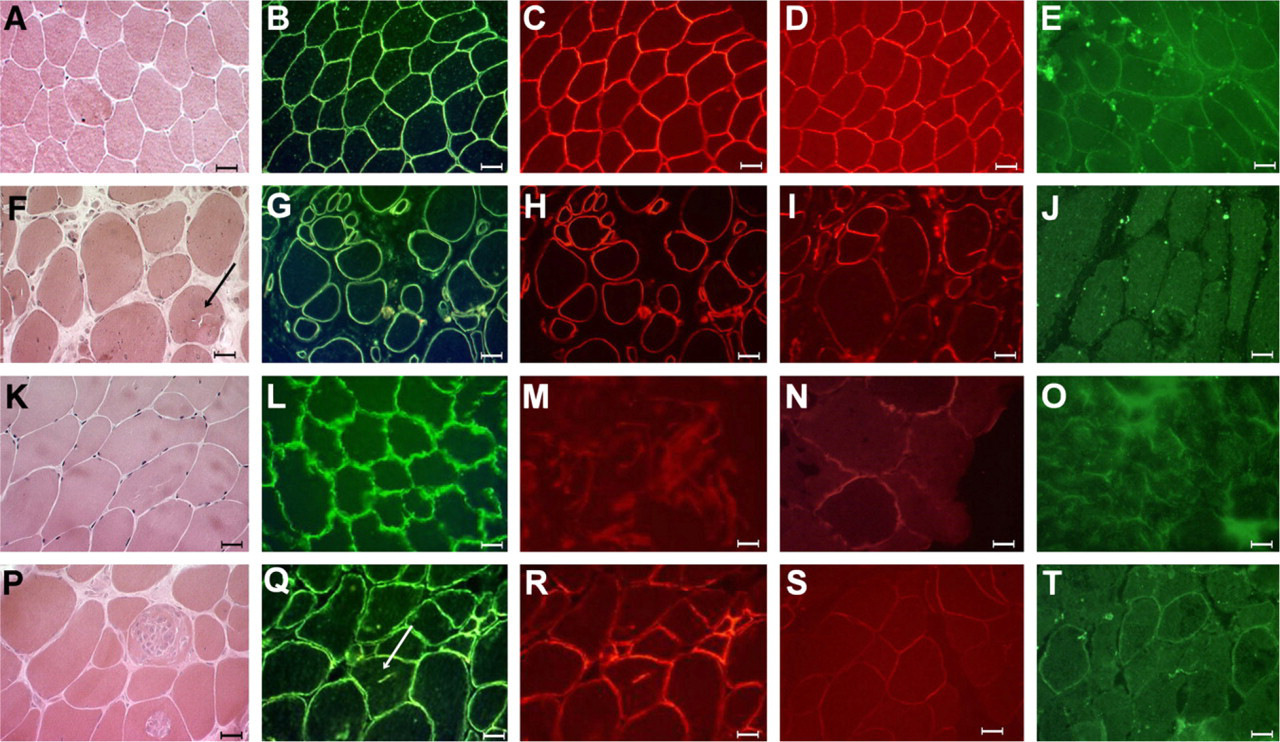
Histological and protein finding in one patient from each group of mutations: normal control
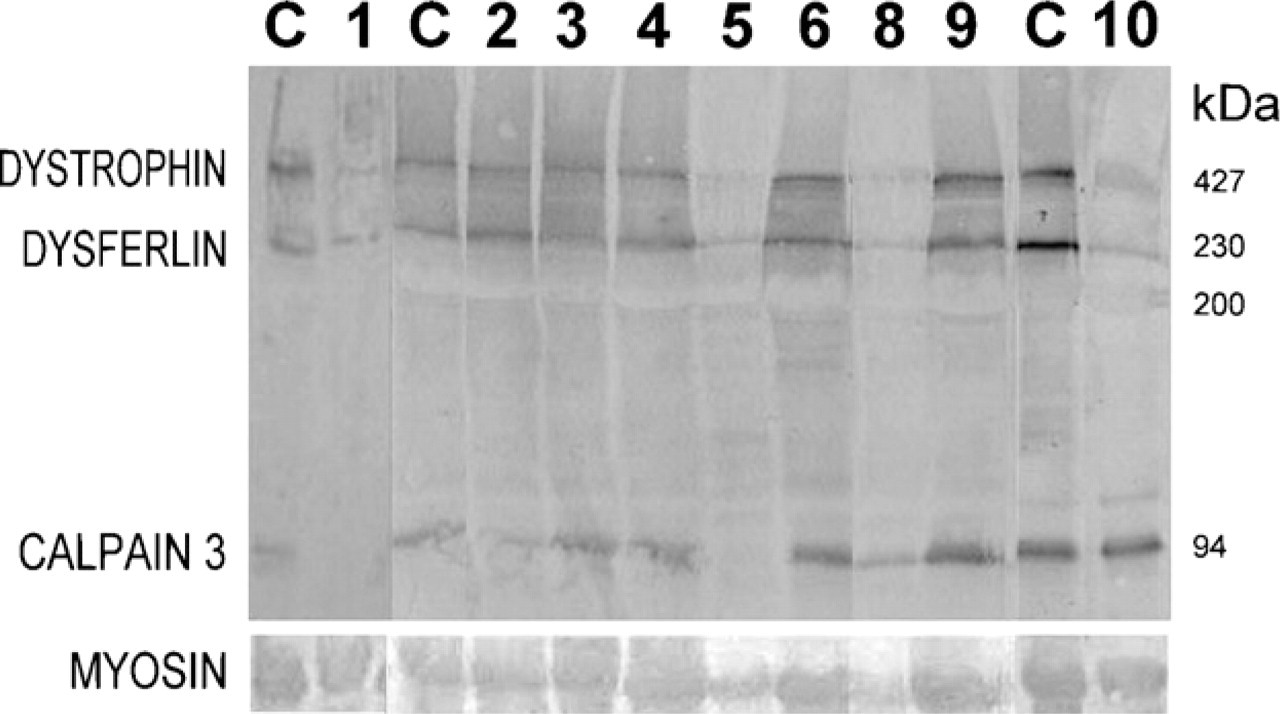
Multiplex Western blot analysis for dystrophin, dysferlin, and calpain 3 in nine of the patients with Limb-Girdle Type 2I form of congenital muscular dystrophy. MYOSIN, myosin band detected in the Ponceau prestained blot for the evaluation of loaded muscle proteins.
Antibodies
The antibodies listed in Table 2 were used for immunofluorescence and/or Western blot analyses.
Results
Histological Findings
Muscle biopsy taken from one individual from each family showed a typical dystrophic histopathological pattern in all samples. Most biopsies showed significant variations in fiber size, very large hypertrophic fibers, hyaline fibers, rounded and necrotic fibers, split fibers, centrally nucleated and whorled, lobulated, and “moth-eaten” fibers. There was intense endomysial and perimysial connective tissue replacement. Small groups of atrophic fibers were detected, suggesting multiple splitting of large fibers or secondary neurogenic features. ATPase reactions showed a mosaic pattern, with some patients showing variable degrees of Type II or Type I predominance. Interestingly, in 8 of the 13 cases, a variable number of rimmed vacuoles were observed inside muscle fibers (Figure 1, arrow).
Protein IHC Analysis
Immunofluorescence analysis of the skeletal muscle section showed no abnormalities for the expression of the proteins dystrophin, dysferlin, telethonin, α, β, γ, and δ-sarcoglycans, or collagen VI in all patients.
A variable degree of deficiency in membrane labeling was observed for α2-laminin, mainly with the most sensitive N-terminal 300-kDa antibody, which showed a patchy and weaker pattern in 12 of the 13 studied patients. Labeling for α-DG showed a total deficiency in 5 of 11 patients, a partial deficiency in 5 of 11 patients, and a normal pattern in 1 of the patients (Figure 1; Table 3).
Western Blot Analysis
Multiplex Western blot analysis showed a quantitative reduction in dystrophin in 2 of 13 patients and in cal-pain 3 in 4 of 13 patients (Figure 2; Table 3).
Discussion
Several recent studies have highlighted the role of the extensive O-linked glycan on α-DG, mediating the binding of extracellular matrix proteins in skeletal muscle (Henry and Campbell 1999; Hewitt and Grewal 2003; Martin 2003). Studies from the Campbell group showed that α-DG is a heavily glycosylated protein that migrates in a heterogeneous pattern on gels (Ibraghimov-Beskrovnaya et al. 1992). The protein is a dumbbell-shaped molecule, with two globular domains separated by a central rod thought to be the mucin-like domain, containing >50 potential sites for O-linked glycosylation on serine or threonine. The dystroglycan gene encodes an ∼70-kDa α-chain polypeptide that migrates at ∼156 kDa in skeletal muscle because of extensive glycosylations. (Martin 2003). O-linked glycans from the mucin region have been sequenced from different sources, and an O-linked α-mannose structure was found, which was elaborated with a sialyl-N-acetylglucosamine to yield NeuAcα2, 3Galβ1, 4GlcNAcβ1, and 2Manα-O-Ser/Th. In the skeletal muscle, the link of α-DG to the basal lamina is done by the G domain of the α2 chain of laminin 2, which interacts with the glycans structures present on α-DG. B-DG interacts with the C-terminal region of α-DG at the membrane periphery and with dystrophin, utrophin, caveolin, actin, and Grb2 in the cytoplasm, thereby linking the extracellular matrix with cytoplasmic and signaling components of the muscle fibers (Henry and Campbell 1999).
List of the used antibodies
Histological and protein findings in the 13 LGMD2I Brazilian patients
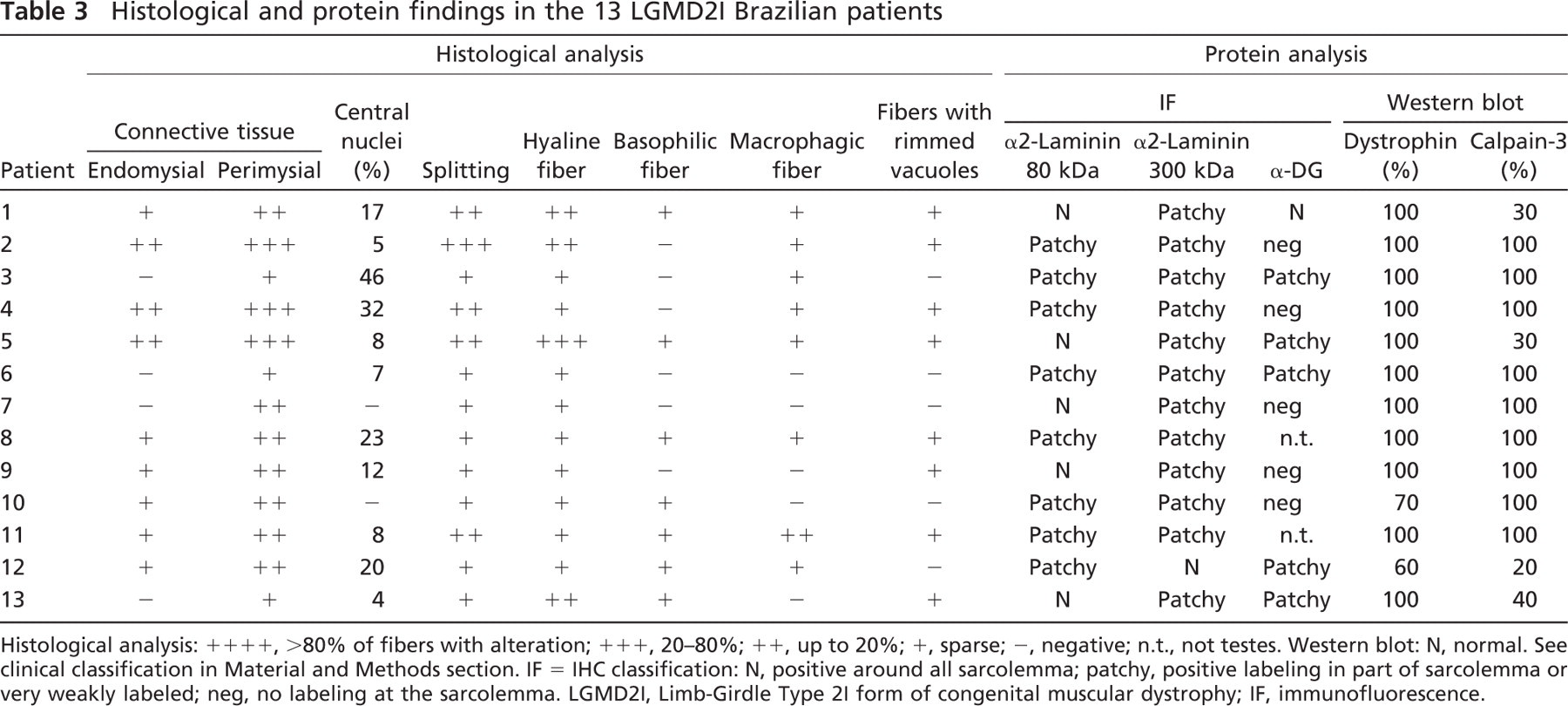
Histological analysis: ++++, >80% of fibers with alteration; +++, 20–80%; ++, up to 20%; +, sparse; −, negative; n.t., not testes. Western blot: N, normal. See clinical classification in Material and Methods section. IF = IHC classification: N, positive around all sarcolemma; patchy, positive labeling in part of sarcolemma or very weakly labeled; neg, no labeling at the sarcolemma. LGMD2I, Limb-Girdle Type 2I form of congenital muscular dystrophy; IF, immunofluorescence.
Several glycosyltransferases are involved in this process of O-mannosyl oligosaccharide synthesis, such as POMT1 and POMGnT1, and mutations in the respective genes cause severe muscle diseases such as muscle-eye-brain disease and Walker-Warburg syndrome (Schachter et al. 2004). Other enzymes are putative glycosyltransferases. Among them, fukutin is involved in FMD, and mutations in the Large gene cause myodystrophy in mice and congenital MD Type 1D in humans (Hewitt and Grewal 2003; Schachter et al. 2004).
FKRP is also a putative glycosyltransferase, and its activity could play a significant role in the mechanism of glycosylation of α-DG. In this case, different mutations in the FKRP gene could be associated with a diverse pattern of glycosylation of α-DG. Beedle et al. (2007) recently showed that, in the muscle, the FKRP protein is associated with the sarcolemmal dystrophin glycoprotein complex (DGC) forming a stable mature complex. This study also reported that, in mouse models of MD in which DG is destabilized or lost, the FKRP is disrupted. Therefore, different mutations in the FKRP gene could result in the codification of enzymes with variable deficient glycosyltransferase activity. Consequently, the mucin domain of α-DG would be diverse, resulting in variation in the link of the laminin molecule. This modulation in the organization of the dystrophin-glycoprotein complex could result in diverse patterns of disruption of this protein complex in the muscle of LGMD2I patients and could be correlated with their clinical course.
This was particularly observed in patients with the severe congenital MDC-1C form, in which the type of mutation in the FKRP gene was highly correlated with the disruption of membrane proteins associated with α-DG (Matsumoto et al. 2005; MacLeod et al. 2007). All affected patients showed significant reduction of α-DG and other membrane-associated proteins. In contrast, all LGMD2I patients that carry the most common C826A mutation have shown a less abnormal expression of proteins involved with α-DG. The main findings were the association of a reduction of α-DG with a marked reduction of α2-laminin on Western blot analysis, but this was not as clearly detectable by IHC (Brockington et al. 2001b; Poppe et al. 2003; Brown et al. 2004).
In our sample of 13 patients with a molecular diagnosis of LGMD2I, almost all showed a deficiency of α2-laminin in sections, mainly with the more sensitive N-terminal 300-kDa antibody. The LAMA2 gene codes a protein of 390 kDa, and under reducing conditions, the laminin α2 chain migrates as an N-terminal fragment of ∼300 kDa and a C-terminal fragment of 80 kDa. IHC studies have shown that partial deficiencies of the laminin α2 chain are more easily detected with the 300-kDa antibody than with the antibody against the 80-kDa fragment (Sewry et al. 1997). The use of more than one antibody is therefore more informative for this analysis.
Four patients were compound heterozygous for three different mutations, five were homozygous for four different mutations, and the remaining four were homozygous for the most common C826A mutation. Because we found a deficiency of laminin α2 in all these patients, this deficiency may not depend on the type of the mutation in the FKRP gene.
In contrast, comparing our four homozygous patients with the most common mutation, although all showed deficiency of α2-laminin on section, variability was observed for α-DG expression. Because the used antibody for α-DG detects the glycosylated part of the protein, the lack of detection of α-DG on sarcolemma is a signal of defective glycosylation of the protein and not its total absence (Michele et al. 2002). In our sample, two patients were completely deficient, one was partially deficient, and one showed a positive IHC pattern for α-DG in the muscle (comparable to normal control). This suggests no correlation between secondary reductions in these two extracellular matrix proteins, even under the effect of the same mutation.
Because none of the studied patients showed altered immunofluorescence pattern for dystrophin, dysferlin, telethonin, α-, β-, γ-, δ-sarcoglycans, and collagen VI, glycosylation defects apparently do not perturb the organization of these proteins in the respective compartment in muscle fibers. However, a quantitative deficiency of dystrophin was observed in two patients, without correlation with the mutation or clinical course (Patients 10 and 12). As already observed in some Becker MD patients, a small quantitative reduction in the amount of dystrophin is not easily detectable by immunofluorescence analysis and can be more evident using Western blot methodology (Vainzof et al. 1992). Calpain 3 deficiency could be correlated with muscle degradation, because some samples of muscle biopsies have been maintained frozen for >15 years. In accordance with this fact, the four muscles that showed calpain 3 deficiencies were the oldest studied samples, and one of them showed several degraded bands in Western blot analysis (Patient 12). However, two patients carrying the same A545G mutation, biopsied in the same stage, were concordant for the partial deficiency of α2-laminin and α-DG but discordant for calpain 3 amount. Calpain 3 deficiency was observed in only one of them, again showing no total correlation between the type of the mutation and secondary deficiencies of the studied proteins.
After the molecular classification of our patients, we decided to meticulously verify if any histopathological characteristic could be associated with this LGMD form. Histological findings showed typical dystrophic features in the majority of the patients, with the presence of moth-eaten and whorled fibers. Interestingly, we found a high frequency of patients with many fibers containing rimmed vacuoles, which were also visible using antibodies for sarcolemmal proteins. This alteration is not pathognomonic, because it can be observed in several other forms of MD, but its high frequency among our LGMD2I patients called our attention to the possible physiopathological mechanism of the disease. No correlation with the type of the mutation was observed, because patients presenting rimmed vacuoles were homozygous and heterozygous for different mutations, as well as carriers of the common mutation in the FKRP gene. No correlations were observed with secondary protein alterations as well, because immunofluorescence or Western blot analyses showed the same deficiencies of the α2-laminin associated to variation in the expression of the α-DG and/or in calpain-3 amount in these patients. Additionally, rimmed vacuoles were observed in affected patients of different ages and stages of the disease, suggesting no direct correlation with the progression of the disease. We could therefore infer that the mechanism of muscle degeneration in LGMD2I could have started with a defective glycosylation of extracellular matrix proteins, followed by injury of the muscle, possibly involving necrosis of myofibers with loss of muscle internal architecture, in all stages of the disease.
In conclusion, the analysis of muscle proteins in patients with LGMD2I with different mutations in the FKRP gene suggests that there is no correlation between α2-laminin and α-DG deficiencies in the muscle and the type of mutation in the FKRP gene. Secondary protein deficiencies thus are probably not the cause of the variability in the clinical course observed in LGMD2I patients.
Footnotes
Acknowledgements
This work was supported by Fundação de Amparo a Pesquisa do Estado de São Paulo-Centro de Pesquisa, Inovação e Difusão (FAPESP-CEPID), Conselho Nacional de Desenvolvimento Científico e Tecnologico (CNPq), and Associação Brasileira de Distrofia Muscular (ABDIM).
The authors thank Dr. Rita Pavanello, Lucas S. Maia, Marta Canovas, Patrícia Kossugue, Telma Gouveia, Danielle Ayub, Poliana Martins, Paula Onofre, Dinorah Zilberztajn, Vanessa Lopes, and Viviane Muniz for scientific and technical help and the following researchers for kindly supplying us with antibodies: Dr. L.V.B. Anderson (in memoria), Dr. K. Campbell, Dr. J. Chamberlain, Dr. V. Nigro, Dr. G. Faulkner, Dr. E. Hoffmann, Dr. Nguyen Thi Man, Dr. G. Morris, and the Developmental Studies Hybridoma Bank, University of Iowa. We also thank the patients and their families.