
Abstract
Mutations in the DYSF gene that severely reduce the levels of the protein dysferlin are implicated in muscle-wasting syndromes known as dysferlinopathies. Although studies of its function in skeletal muscle have focused on its potential role in repairing the plasma membrane, dysferlin has also been found, albeit inconsistently, in the sarcoplasm of muscle fibers. The aim of this article is to study the localization of dysferlin in skeletal muscle through optimized immunolabeling methods. We studied the localization of dysferlin in control rat skeletal muscle using several different methods of tissue collection and subsequent immunolabeling. We then applied our optimized immunolabeling methods on human cadaveric muscle, control and dystrophic human muscle biopsies, and control and dysferlin-deficient mouse muscle. Our data suggest that dysferlin is present in a reticulum of the sarcoplasm, similar but not identical to those containing the dihydropyridine receptors and distinct from the distribution of the sarcolemmal protein dystrophin. Our data illustrate the importance of tissue fixation and antigen unmasking for proper immunolocalization of dysferlin. They suggest that dysferlin has an important function in the internal membrane systems of skeletal muscle, involved in calcium homeostasis and excitation-contraction coupling.
Keywords
The DYSF gene encodes dysferlin, a 230-kDa protein that is absent or severely reduced in patients with limb girdle muscular dystrophy type 2B, Miyoshi myopathy, and distal myopathy with anterior tibial onset, skeletal muscle-wasting syndromes collectively referred to as dysferlinopathies (Urtizberea et al. 2008). Inheritance is autosomal recessive, and disease-causing mutations have been identified across the DYSF gene (Nguyen et al. 2005). Since its discovery, dysferlin has been referred to as a plasma membrane protein that is also found in cytoplasmic vesicles (Anderson et al. 1999; Bansal et al. 2003), largely because it appears enriched at the sarcolemma in cross sections of snap-frozen, unfixed muscle. The accumulation of subsarcolemmal vesicles in dysferlinopathic muscle, and studies of muscle fibers cultured from mice lacking dysferlin that involve laser wounding or other damaging treatments, have suggested that dysferlin’s function is to repair disrupted plasma membranes (Bansal and Campbell, 2004; Glover and Brown, 2007; Han and Campbell, 2007).
Recent reports suggest that cytoplasmic dysferlin may be present, at least in part, in the transverse tubules (t-tubules) of skeletal muscle (Ampong et al. 2005; Lostal et al. 2010; Waddell et al. 2011). This location suggests that dysferlin is required for maintaining the integrity of the t-tubules or perhaps for their coupling with the junctional sarcoplasmic reticulum (SR). Here, we report an improved method for immunolocalizing dysferlin in the internal membranes of rat, mouse, and human skeletal muscles.
Our methods rely on the “unmasking” of epitopes on dysferlin by heating fixed cryosections of muscle in mildly acidic citrate buffer. Heat-induced antigen retrieval (AR) in citrate buffers improves the labeling of several muscle proteins (Mundegar et al. 2008), but its effectiveness for dysferlin have not been documented. Using our modification of this method, we show that dysferlin is much more abundant in the intracellular membranes of skeletal muscle fibers than it is at the sarcolemma and that the small amount of dysferlin at the sarcolemma is present where the t-tubules insert, rather than at other sarcolemmal domains that are enriched in dystrophin (Williams and Bloch 1999). We show further that intracellular dysferlin concentrates in a reticulum that flanks the Z-disks of each sarcomere, consistent with its presence in t-tubules, the junctional SR, or both. We conclude that the absence of dysferlin from intracellular membranes, rather than or in addition to its absence from the sarcolemma, contributes to the mechanisms underlying dysferlinopathies.
Methods
We optimized our labeling techniques by testing several modifications of methods for labeling the rat tibialis anterior (TA) muscle with antibodies to dysferlin and then applying them to frozen sections of rat, mouse, and human TA muscles.
Rats and Mice
We used adult male rats and mice (12–16 weeks) for our study. Sprague-Dawley rats (12–14 weeks) were obtained from Harlan Laboratories (Indianapolis, IN). We originally obtained control, A/WySnJ mice, and dysferlin-deficient A/J (Ho et al. 2004) and Bl10.SJL mice (von der Hagen et al. 2005) from the Jackson Laboratories (Bar Harbor, ME: A/WySnJ, A/J) and Dr. A. J. Wagers (Harvard University, Cambridge, MA: Bl10.SJL) and are now breeding them at the University of Maryland School of Medicine, Baltimore. Additional control, C57BL10 mice were from Jackson Laboratories. All protocols for the handling of animals were approved by the Institutional Animal Care and Use Committee (University of Maryland School of Medicine).
Human Muscle
Human TA muscle samples were from cadavers donated to the Department of Anatomy, University of Maryland School of Medicine. Cadavers were embalmed using standard procedures of the State Anatomy Board of Maryland. Briefly, the cadaver was perfused via the brachial artery (in) and femoral artery (out) under pressure (138 g/cm2), with a mixture of methanol (33%), phenol (27%), glycerin (34%), and formaldehyde (6%). Segments of muscle (~20 mm × 5 mm × 5 mm) were snap frozen in a slush of liquid nitrogen and stored at −80C for later study. Our use of cadaveric muscle samples was exempt from review by the Institutional Review Board (IRB), University of Maryland School of Medicine.
We also obtained frozen cross sections of muscle biopsies (10-µm thick) from the Tissue/Cell Repository of the Wellstone Muscular Dystrophy Cooperative Research Center at the University of Iowa through Dr. S. A. Moore, director of the Wellstone Center and professor of neuropathology. Sections were collected on 4 × 2 grid slides, air dried, and shipped in a slide mailer to our laboratory on dry ice. Our use of these slides was also IRB exempt.
Antibodies Used for Immunofluorescence
For immunofluorescence, we used the following antibodies, diluted in PBS containing 3% (w/v) bovine serum albumin (BSA) and 0.01% (v:v) Triton X-100 (primary antibody dilution buffer, PBS/0.01%Tx/BSA): mouse monoclonal antibody to dysferlin (1:20, NCL Hamlet, Novocastra, Newcastle upon Tyne, UK), rabbit polyclonal antibody to dysferlin (1:200, LS B802, Lifespan, Seattle, WA), rabbit monoclonal antibody to dysferlin (1:20, 2191-1; Epitomics, Burlingame, CA), rabbit polyclonal antibodies to dystrophin and desmin (1:200, RB-9024-P and RB-9014–P, respectively; Labvision Thermo Scientific, Fremont, CA), rabbit polyclonal antibody to dihydropyridine receptor (L-type Ca2+ channel α1C; dihydropyridine receptors [DHPR], 1:100, sc-25686, Santa Cruz Biotechnology, Santa Cruz, CA), and a mouse monoclonal antibody to ryanodine receptors (RyR; 1:200, MA3-925, Thermo Scientific, Fremont, CA). The amino acids in the human sequence of dysferlin targeted by the antibodies were residues 1999 to 2016 (Hamlet), 2004 to 2021 (Lifespan), and ~2050 to 2080 (Epitomics). We used appropriate combinations of the following secondary antibodies, diluted 1:200 in PBS containing 0.01% Triton X-100 (secondary antibody dilution buffer, PBS/0.01%Tx): biotin-conjugated goat anti-mouse or goat anti-rabbit IgG (1:200, B2763 and B2770, respectively, Invitrogen, Carlsbad, CA), Alexa488 conjugated goat anti-rabbit IgG, and Alexa488 conjugated goat anti-mouse IgG (A11008 and A11001, respectively; Invitrogen). Equimolar concentrations of nonspecific mouse or rabbit IgG were used as negative controls.
Sarcolemmal and Nuclear Staining for Immunofluorescence
Where applicable, sections were stained with Alexa555-conjugated wheat germ agglutinin (WGA, W32464, 1:500 in PBS/0.01%Tx; Invitrogen, Burlingame, CA) to label the sarcolemma, and 4′,6′-diamidino-2-phenylindole (DAPI, 71-03-01, 1:1000 in PBS; KPL, Gaithersburg, MD) to label nuclei.
Confocal Microscopy and Image Analysis
All images except those of human biopsies were obtained with a confocal laser scanning microscope (LSM 510 Duo; Carl Zeiss, Poughkeepsie, NY), with a 63× oil immersion lens with the pinhole adjusted to generate a 1.5-µm-thick optical section in each channel. Sections of human biopsies were imaged with a 20× air objective at an optical zoom of 2×. Images were saved as .lsm files and imported into Volocity software (PerkinElmer, Waltham, MA). Plot profiles were obtained with Image J (National Institutes of Health, Bethesda, MD).
Collecting Fixed Rodent Tissue
Fixed rodent muscles were harvested after perfusing animals through the left ventricle with ice-cold relaxing buffer (pH = 7.0) containing 7.28 g/L KCl, 1.12 g/L MgCl2, 3.84 g/L EGTA, 4.76 g/L HEPES, 0.22 g/L disodium ATP, and 2.32 g/L maleic acid in de-ionized water, followed by perfusion with ice-cold 4% paraformaledhyde in PBS (pH = 7.2). Proper fixation was verified by twitching followed by stiffening of the limbs and tail. TA muscles of both hindlimbs were collected, placed on strips of aluminum foil, and snap frozen by quickly submerging them in a slush of liquid nitrogen. Two min later, muscles were transferred into cryostorage vials, precooled in liquid nitrogen, and stored at −80C.
Collecting Sections of Muscle
Cross sections of snap-frozen rodent muscle and human cadaveric muscle were cut at a thickness of 16 µm. Longitudinal sections of rodent muscle and human cadaveric muscle were cut at thicknesses of 20 µm and 5 µm, respectively. All sections of rodent muscle, as well as cross sections of cadaveric muscle, were collected onto glass slides (Superfrost plus, 48311-703; VWR Scientific, Bridgeport, NJ). Cadaveric longitudinal sections were collected with modifications to methods previously described (Reed et al. 2006) by picking up a ribbon of longitudinal sections, floating them on cold collection buffer (a 1:1 mixture of ethanol and a solution of 4% paraformaldehyde, 10 mM EDTA in PBS) on glass slides, and then allowing them to dry at room temperature. Alternatively, collecting longitudinal cadaveric sections directly onto slides and immediately treating them with collection buffer also gave satisfactory morphology in many regions, although this was not the case with longitudinal sections of unfixed rodent muscle or human biopsies.
Cryosections of perfusion-fixed rodent muscle and longitudinal sections of cadaveric muscle were washed with 0.1M glycine in PBS for 10 min, washed once with PBS for 5 min, and kept hydrated with PBS. Sections that were not subjected to AR (see below) were directly subjected to immunolabeling. Cross sections of cadaveric muscle and sections of human biopsies from the Wellstone Center (see above) were rehydrated with PBS for 10 min, fixed with 4% paraformaldehyde for 15 min, and washed with 0.1M glycine in PBS, as above.
AR and Immunolabeling
AR was performed on sections by placing them in 10 mM sodium citrate, 0.05% Tween-20, pH = 6.0, and heating them to 90C over a period of 10 min. Samples were then cooled to 30C over a period of 60 min. Sections were removed from citrate buffer, washed twice with PBS for 5 min, and incubated for 10 min with 5 µg/ml avidin (11680, USB Biologicals, Santa Clara, CA) in PBS/0.01%Tx to block endogenous biotin, followed by an additional wash with PBS and incubation for 30 min with PBS/0.01%Tx/BSA to reduce nonspecific labeling. Sections were incubated overnight at 4C with primary antibodies, as described above, washed once with PBS/0.1%Tx for 10 min, washed 3 times with PBS at 5 min per wash, and incubated with suitable secondary antibodies diluted 1:200 in PBS/0.01%Tx for 90 min at room temperature. Antibodies to dysferlin were routinely treated with suitable biotin-conjugated goat anti-mouse or goat anti-rabbit antibodies, for subsequent amplification with fluorescent streptavidin. After sections were washed, as described above, they were incubated for 15 min with streptavidin conjugated to Alexa568 or Alexa488 (S11226 and S11223, respectively; 2.5 µg/ml in PBS/0.01%Tx), washed again twice with PBS, and, where indicated, co-stained with Alexa555-conjugated WGA followed by DAPI, or with DAPI alone. For WGA labeling, an additional fixation step was performed before DAPI staining by applying 4% paraformaldehyde for 10 min. After two additional washes with PBS for 5 min, samples were mounted in Vectashield (H-1000; Vector labs, Burlingame, CA) under a cover slip. Additional details are provided as supplementary material.
Western Blotting of Mouse Muscle Homogenates
Unfixed TA muscles were obtained from mice that were euthanized with an overdose of ketamine and xylazine. Muscles were homogenized with a commercial tissue-grinding kit (sample grinding kit, 80-6483-37; GE Healthcare, Piscataway, NJ). The homogenizing buffer contained 1% neutral detergent (IGEPAL CA-630, I8896; Sigma, St. Louis, MO) in Tris-buffered saline containing protease inhibitors (two tablets per 50 ml, cOmplete, 11697498001; Roche Applied Science, Indianapolis, IN). Ten µl of buffer was added per milligram wet weight of muscle tissue. After homogenization, the samples were subjected to centrifugation at 13,000 RPM (~10,000 × G; 5415 C centrifuge, Eppendorf, Hamburg, Germany). The supernatants were transferred into new tubes, and the pellets were frozen and stored at −80C for later study. The protein content of the supernatant was measured (Protein Assay Kit II, 500-0002; Bio-Rad, Hercules, CA), and samples containing ~2 mg/ml protein were heated to 90C for 5 min with equal volumes of Laemmli sample buffer (Laemmli 1970) containing 5% 2-mercaptoethanol, preparatory to SDS-PAGE. Proteins were separated on 4% to 12% Bis-Tris gels with a NuPage mini-gel electrophoresis system (30 µg of protein per lane, MOPS running buffer, 200 V) and transferred to nitrocellulose membranes overnight at 4C. The blots were incubated for 4 hr with 3% milk in TBS containing 0.1% Tween-20 (blocking solution), incubated overnight with primary antibodies diluted in blocking solution, washed 10 times (5 min per wash) with blocking solution, and incubated for 2 hr with alkaline phosphatase–conjugated secondary antibodies, also in blocking solution. The membranes were washed 5 times with blocking solution (5 min per wash) and then 5 times with TBS containing 0.1% Tween-20 (5 min per wash), and transferred to the appropriate assay buffer for 10 min for chemiluminescent visualization of bound alkaline phosphatase (Tropix CSPD and N-Block; Applied Biosciences, Carlsbad, CA). Chemiluminescence was recorded on X-ray film.
For immunoblotting, we used the three antibodies to dysferlin (see above; Hamlet 1:750, Lifespan 1:1000, and Epitomics 1:500) and a mouse monoclonal antibody to glyceraldehyde phosphate dehydrogenase (GAPDH, G8795; Sigma), diluted 1:20,000.
Results
Labeling of Dysferlin in Frozen Sections of Rat Skeletal Muscle
We first examined the ability of our methods to improve labeling of dysferlin in frozen cross and longitudinal sections of rat TA muscle (Figure 1). In untreated cross sections, labeling for dysferlin (red) appears primarily at or near the sarcolemma, with only dull puncta in the sarcoplasm (Figure 1A), similar to earlier observations (Bansal et al. 2003). By contrast, cross sections subjected to AR are more intensely labeled for dysferlin at or near the sarcolemma and in a clear, reticular pattern in the sarcoplasm (Figure 1A′). Labeling for dystrophin (green), a sarcolemmal protein, does not increase either at the sarcolemma or within myofibers following AR, suggesting that the enhanced internal labeling of dysferlin that we observe after AR is specific.
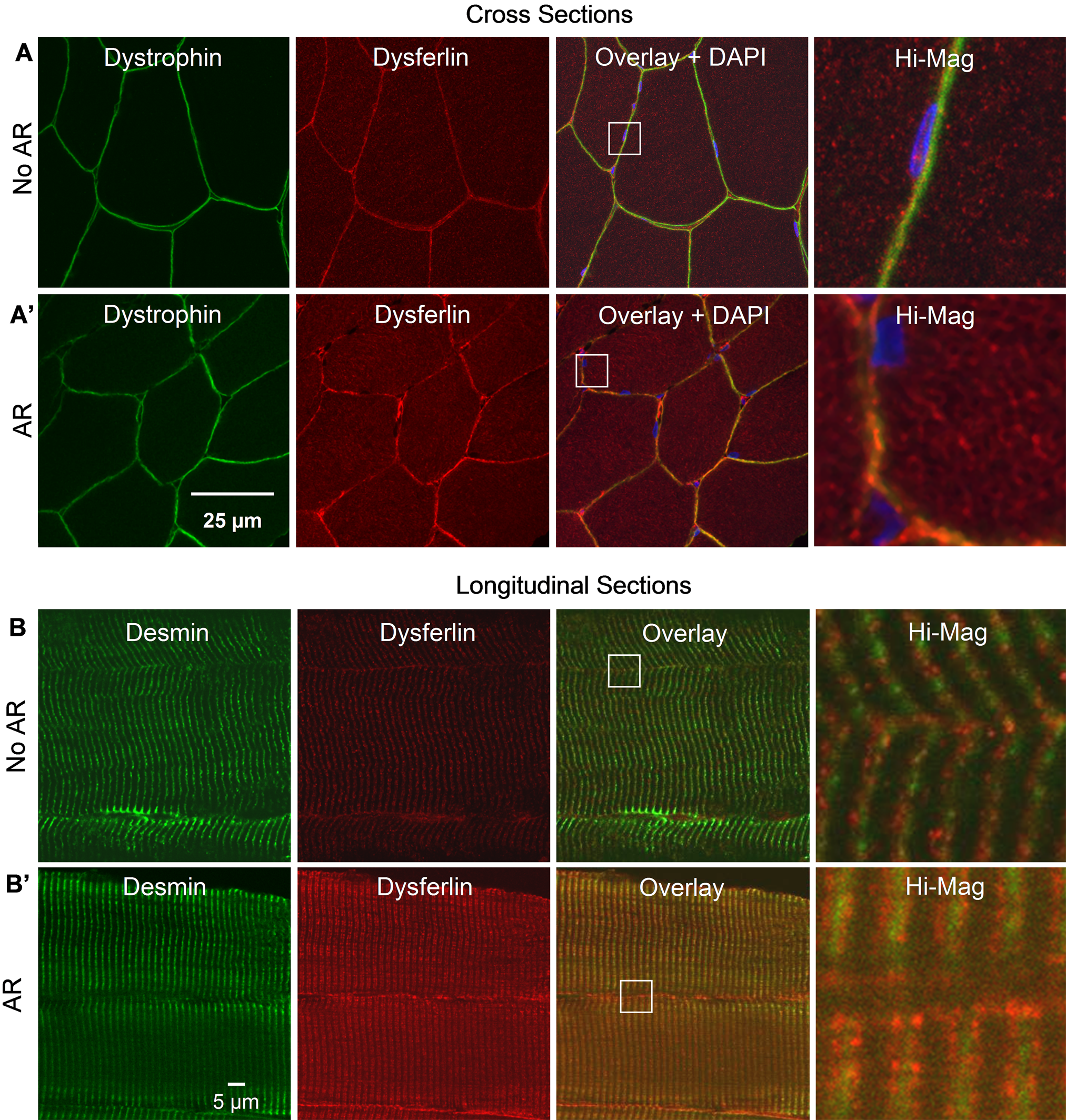
Labeling of dysferlin in rat tibialis anterior (TA) muscle with the Hamlet antibody. (A, A′) Labeling of dysferlin and dystrophin in cross sections of perfusion-fixed rat TA, with and without antigen retrieval, respectively. Antigen retrieval (AR) enhances labeling of dysferlin in an intracellular reticulum but does not alter labeling for dystrophin. (B, B′) Labeling of dysferlin and desmin in longitudinal sections of perfusion-fixed rat TA, with and without AR. Dysferlin appears as puncta adjacent to the Z-disks without AR but appears as a clear doublet flanking the Z-disks following AR. Labeling of desmin is not altered by AR.
Longitudinal sections of rat TA muscles that were labeled without AR show irregular punctate labeling, arrayed more or less transversely adjacent to Z-disks. After AR, dysferlin labeling is brighter and appears as a regular set of puncta arrayed in two lines (a “doublet”) flanking Z-disks (Figure 1B′), marked with antibodies to desmin (green), which surrounds Z-disks. Labeling for dysferlin at or near the sarcolemma is also primarily punctate and aligned with intracellular structures containing dysferlin. As with dystrophin, the improved immunolocalization of dysferlin seen following AR is specific, as labeling for desmin does not change.
These data suggest that in addition to its presence at low levels at or near the sarcolemma, dysferlin is abundant within the cytoplasm of myofibers. The reticular pattern of dysferlin labeling seen in cross sections, and the doublet flanking the Z disk seen in longitudinal sections, are typical of proteins that are present at the t-tubules or the junctional SR.
Importance of Fixation
Proper fixation of muscle is important for immunolocalization of dysferlin in both longitudinal and cross sections after AR. As shown in Figure 2, longitudinal sections of unfixed rat muscle, labeled with and without AR, fail to show a clear intracellular pattern of dysferlin, although labeling for both dysferlin and dystrophin at or near the sarcolemma is preserved (Figure 2A, A′). Disruption of intracellular structures in these samples is suggested by the fact that labeling for desmin is irregular (Figure 2B, B′). In cross sections (Figure 2C), reticular labeling of dysferlin is difficult to find in unfixed sections of unfixed muscle that have not been treated for AR (unfixed, no AR). In unfixed muscles, internal reticular labeling is improved by briefly fixing sections with paraformaldehyde before AR (postfixed–AR; also see Figure 4). Internal reticular labeling is robust and most defined in sections from perfusion-fixed muscle treated for AR (perfusion fixed–AR), however. These results suggest that earlier reports of the localization of dysferlin predominantly at the sarcolemma may be due not only to partial exposure of the antigen in the intracellular compartment but also to inadequate tissue fixation.
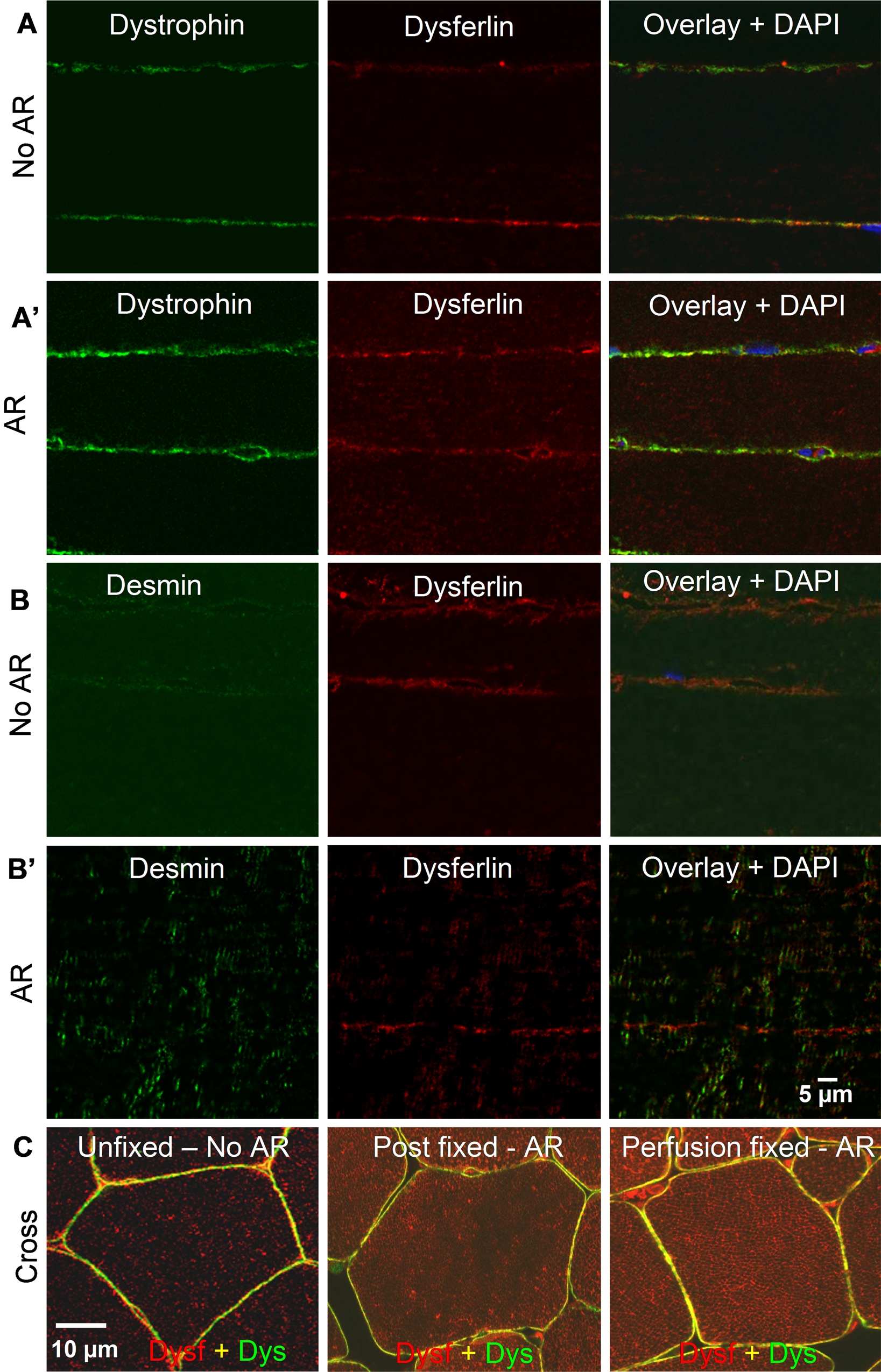
Labeling of dysferlin in unfixed rat tibialis anterior (TA) muscle with the Hamlet antibody. (A, A′) Labeling of unfixed longitudinal sections of rat TA muscles for dysferlin and dystrophin is similar with antigen retrieval (AR) and without AR (no AR). Both proteins are visible at the sarcolemma, but intracellular dysferlin is not labeled. (B, B′) Similarly, for dysferlin and desmin, neither of which are visible in the sarcoplasm of unfixed longitudinal sections of muscle. (C) Labeling of dysferlin in unfixed cross sections not subjected to AR is primarily sarcolemmal and punctate in the cytoplasm. Internal labeling is improved moderately if sections from unfixed muscles are fixed and then subjected to AR. However, the best results are seen in sections from perfusion fixed muscles that are subjected to AR.
Immunolocalization of Dysferlin in Human Muscle
We next tested our methods on frozen sections of human TA muscle. When co-labeled with antibodies to dysferlin and dystrophin (Figure 3A, A′) or desmin (Figure 3B), sections of human muscle yield results identical to those obtained with rat TA. Consistent with its presence in an intracellular reticulum flanking the Z-disks, dysferlin aligns with, but does not extensively overlap with, labeling for dihydropyridine receptors (DHPR; Figure 3C), present at the triad junctions formed by t-tubules and the junctional SR. Quantitative profiles of the intensities of labeling for dystrophin, desmin, DHPR, and dysferlin, shown in Figure 3D, illustrate the differences in distribution of dysferlin at the sarcolemma and within the sarcoplasm. Scans of dystrophin (Figure 3D.i) at the sarcolemma show several peaks, consistent with its localization at the junctions of the surface membrane with the Z-disks and M-bands where costameres are present, as well as at lower intensities in intercostameric regions (Williams and Bloch 1999). Desmin shows a frequency that corresponds to the Z-disks (Figure 3D.ii). Dysferlin at the level of the sarcolemma and within the sarcoplasm (red and black lines, respectively; Figure 3D.iv) shows a periodicity similar to DHPR (Figure 3D.iii), although its periodicity is not as regular near the sarcolemma as it is internally. This suggests that even near the cell surface, dysferlin associates primarily with t-tubules or nearby structures.
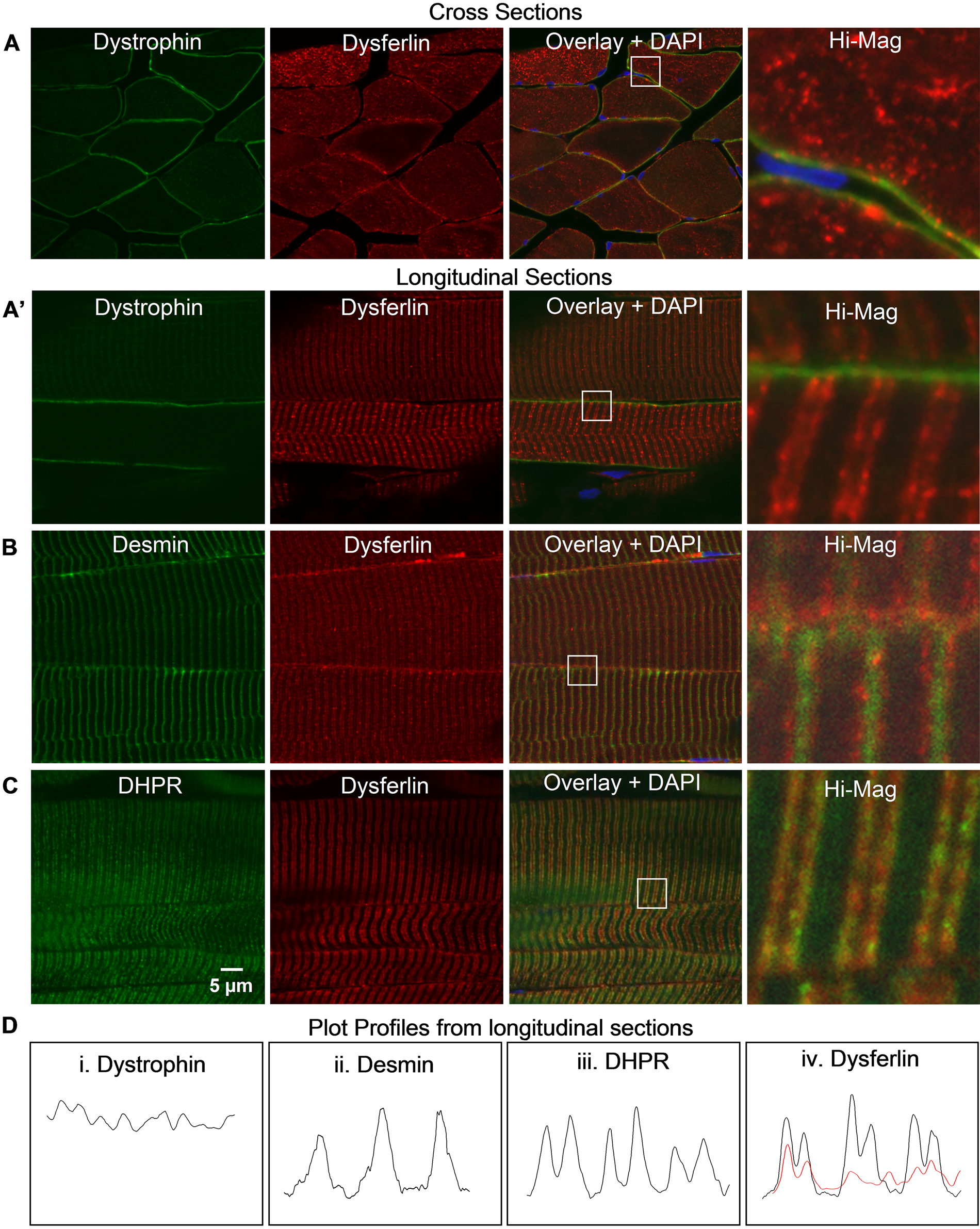
Labeling of dysferlin labeling in human tibialis anterior (TA) muscle with the Hamlet antibody. (A, A′) Labeling of dysferlin and dystrophin in sections of fixed, cadaveric human TA muscles after antigen retrieval. Dysferlin appears in an intracellular reticulum in cross sections and as a transverse, striated doublet in longitudinal sections. Dystrophin is limited to the sarcolemma. DAPI, in blue, marks nuclei. (B) Labeling, as above, for dysferlin and desmin shows that the doublets labeled for dysferlin flank desmin at Z-disks. (C) Labeling, as above, for dysferlin and dihydropyridine receptors (DHPR) is similar but only partially overlapping. (D) Scanned profiles of dystrophin, desmin, and dysferlin in longitudinal sections were obtained across approximately three sarcomere lengths in the magnified region. Dysferlin at the surface (light dotted line) and within the myofiber (dark solid line) appears as twin peaks on either side of desmin (D. ii) and much more closely resembles that of DHPR (D. iii) than dystrophin (D. i).
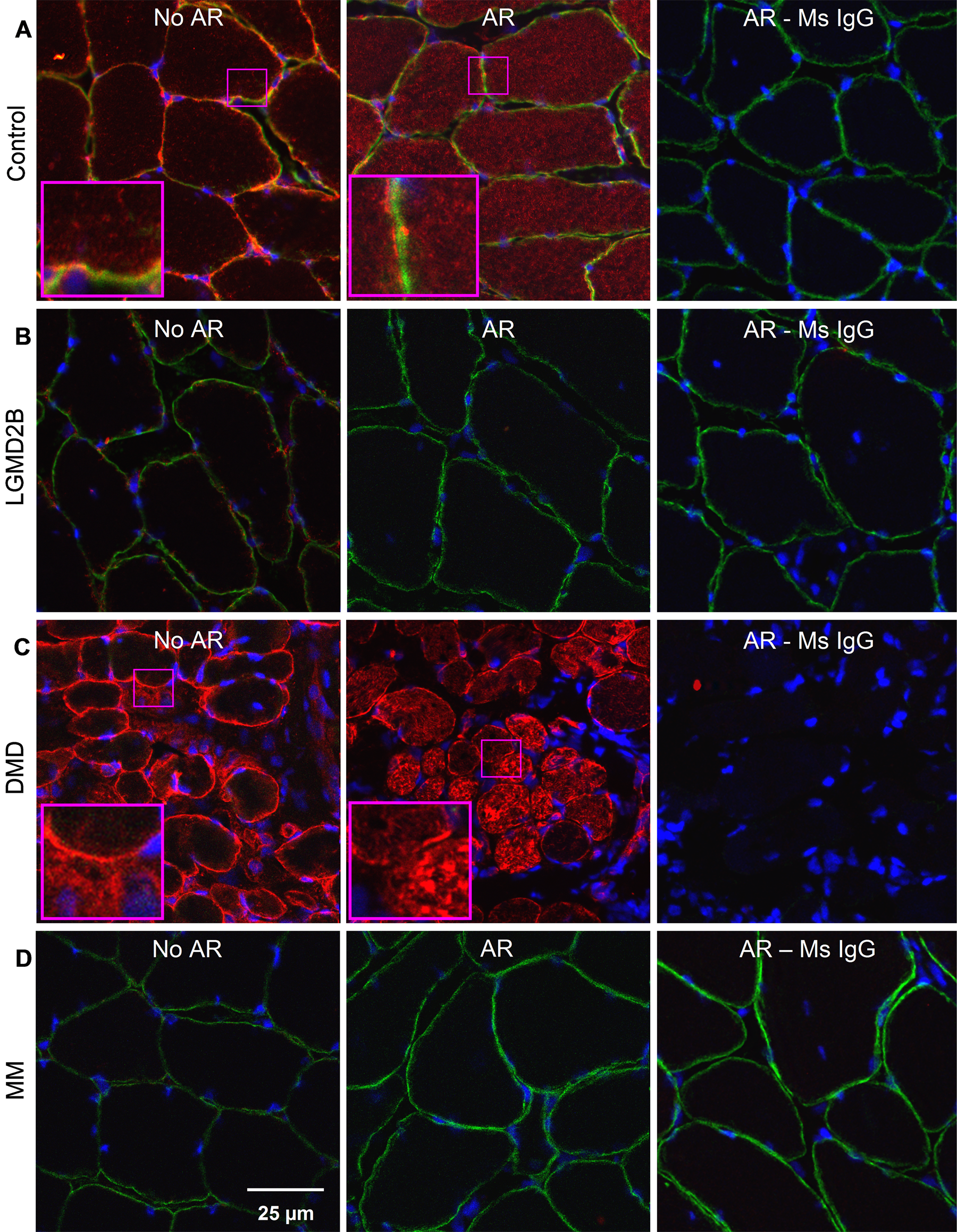
Labeling of dysferlin in cross sections of control and dystrophic human muscle. (A) In sections labeled with the Hamlet antibody after fixation with antibodies to dysferlin (red) and dystrophin (green), control muscle shows an internal reticulum rich in dysferlin when antigen retrieval (AR) is used (middle), but not when it is omitted (left), or when a non-immune mouse IgG is used (right). Dysferlin labeling at the sarcolemma, co-labeled for dystrophin, requires specific antibody but not AR. (B, D) Labeling for dysferlin at the sarcolemma and in the intracellular reticulum is absent in biopsies from patients diagnosed with dysferlinopathies (LGMD2B: B; MM: D). (C) Internal labeling of dysferlin is brighter in muscle from a patient diagnosed with Duchenne muscular dystrophy, and this labeling also appears reticular after AR.
Labeling of Dysferlin in Cross Sections of Human Muscle Biopsies
We also tested our labeling methods on frozen cross sections of control and dystrophic human muscle biopsies (Figure 4). AR is necessary to visualize a reticular pattern of dysferlin in control human sarcoplasm with the Hamlet antibody (Figure 4A). The reticulum is absent in biopsies from patients diagnosed with dysferlinopathies (LGMD2B and MM; Figure 5B, D, respectively), indicating that labeling for dysferlin is specific. Consistent with this, non-immune mouse IgG fails to label the sarcoplasm or the sarcolemma (AR–Ms IgG, panels on right). Our data confirm earlier reports that cytoplasmic labeling for dysferlin is increased in Duchenne muscular dystrophy (Piccolo et al. 2000), which after AR appears brighter and also appears reticular (Figure 4C). Images of sections processed without AR resemble those in earlier reports ( Lovering et al. 2007; Piccolo et al. 2000; Guo et al. 2010), in which dysferlin appears predominantly at or near the sarcolemma, with faint, ill-defined puncta in the sarcoplasm of control muscle and brighter punctate labeling in muscular dystrophies not linked to dysferlin. These data emphasize the value of our improved methods to immunolabel dysferlin in human muscle for diagnostic as well as research purposes.
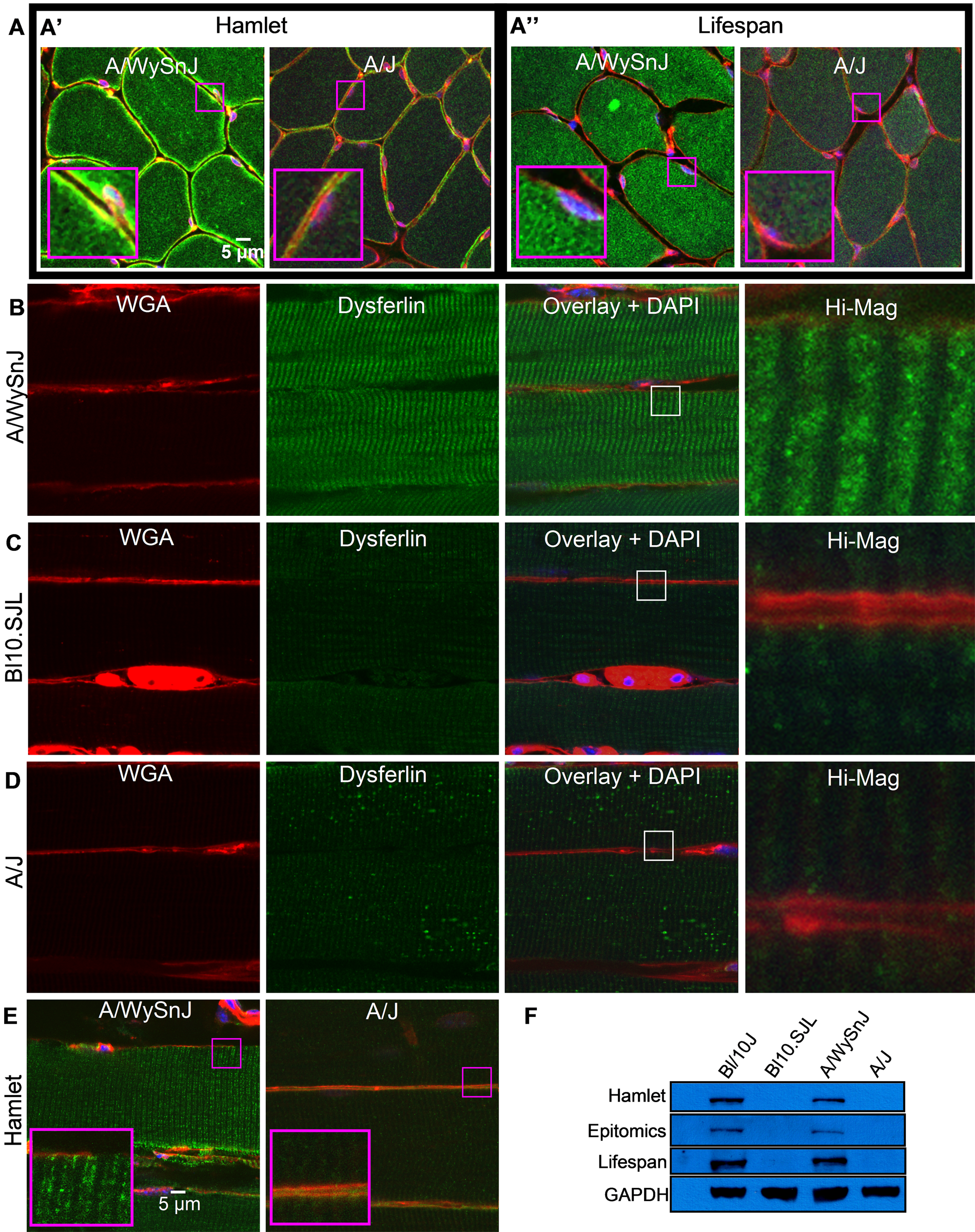
Labeling of dysferlin in mouse tibialis anterior (TA) muscle. (A′, A″) Cross sections of control A/WySnJ muscle labeled with Hamlet and Lifespan antibodies show reticular labeling of dysferlin, which is very faint in dysferlin-null A/J muscle. Hamlet does, however, show enhanced sarcolemmal labeling in A/WySnJ muscle, which is likely to be nonspecific as it is also seen in A/J muscle. (B–D) In longitudinal sections, rather than a clear doublet, Lifespan specifically labels a thick transverse band along sarcomeres in A/WySnJ muscle, which is very faint in Bl10.SJL and A/J, suggesting that the labeling is dysferlin specific. (E) Hamlet labels a clear doublet in A/WySnJ muscle, which is very faint in A/J muscle. (F) Hamlet, Epitomics, and Lifespan antibodies label a band at ~230 kDa in control A/WySnJ and Bl10/J muscle homogenates but not dysferlin-deficient Bl10.SJL and A/J muscle homogenates, suggesting that these antibodies specifically bind to dysferlin.
Labeling of Dysferlin in Mouse Skeletal Muscle
Our methods are also applicable to mouse muscle labeled with the Hamlet monoclonal antibody. As with human and rat muscle, Hamlet shows bright clear reticular labeling of the sarcoplasm (Figure 5A′), but labeling at or near the sarcolemma is brighter than in human and rat samples. This is likely to be artifactual, as it occurs in both control (A/WySnJ) and dysferlin-null (A/J) muscle, probably because of residual mouse immunoglobulins (Figure 5A′; Supplementary Figure 1). To test this, we examined two rabbit anti-dysferlin antibodies for their ability to label dysferlin in mouse muscle: a monoclonal antibody from Epitomics and a polyclonal antibody from Lifespan. Like Hamlet, these rabbit antibodies recognize epitopes close to the C-terminus of dysferlin. In Western blots, both antibodies specifically label a band at ~230 kDa in extracts of control C57Bl/10J and A/WySnJ muscle but not of muscles from the dysferlin-deficient Bl10.SJL and A/J strains (Figure 5F). Although the Epitomics antibody labels frozen sections treated for AR only faintly, even at high concentrations (not shown), the Lifespan antibody robustly labels an intracellular reticulum as well as structures at or near the sarcolemma in control (A/WySnJ) mouse muscle (Figure 5A″, B). Labeling is faint or absent in myofibers of Bl10.SJL (Figure 5C) and A/J muscle (Figure 5D), with very little labeling of either a reticulum or the sarcolemma. This is consistent with a nonspecific contribution of Hamlet’s labeling of the sarcolemma (Figure 5A′, E). Nevertheless, rabbit–anti-dysferlin also very faintly labels transverse structures in the sarcoplasm of dysferlin-deficient muscle (Figure 5C, D), as does Hamlet (Figure 5E). Our data suggest that, like the Hamlet antibody, the Lifespan rabbit antibody clearly distinguishes dysferlin-deficient from control mouse muscle by immunolabeling. However, unlike Hamlet (Figure 5E), the Lifespan antibody does not yield a sharp intracellular doublet in longitudinal sections of control mouse muscles (Figure 5B), for reasons that are not yet clear.
Discussion and Conclusion
Understanding how the absence of functional dysferlin leads to muscular dystrophy is essential for developing reliable therapies for dysferlinopathies. Perhaps because most research so far has focused on the role of dysferlin at the plasma membrane, the presence of apparently small amounts of dysferlin within the cytoplasm in control muscles and the elevated amount of this population of dysferlin in several muscular dystrophies was initially proposed to be due to mislocalization (Piccolo et al. 2000). Dysferlin has been reported to be associated with t-tubules during development but to be predominantly sarcolemmal in mature muscle (Klinge et al. 2010). Here, we report that intracellular dysferlin is in fact the preponderant population of the protein in skeletal muscle but that its visualization requires careful fixation, followed by unmasking by heat-induced AR in citrate buffer. Using this procedure, we find abundant amounts of dysferlin in the sarcoplasm of healthy muscle and even more sarcoplasmic dysferlin in dystrophin-deficient muscle. Our results demonstrate that most of the dysferlin that can be visualized in skeletal muscles is in fact present in an intracellular reticulum and suggest that this reticulum, rather than (or in addition to) the sarcolemma, is the primary site of dysferlin’s activity in muscle.
Although our results show that antigen unmasking is required for clear labeling of dysferlin in the intracellular compartment, it is unclear why labeling internal dysferlin is so challenging when many other sarcoplasmic proteins can be labeled effectively without our AR methods. Studies in progress at our laboratory show that internal labeling of dysferlin goes up in muscles injured by lengthening contractions, suggesting that stressing sarcomeres and their associated internal membrane systems could unmask dysferlin (also see Waddell et al. 2011). The antibodies we use here recognize epitopes close to dysferlin’s C-terminal transmembrane domain. It seems likely that these epitopes are masked by proteins that closely interact with dysferlin. We speculate that dysferlin’s internal masking and the ability of lengthening contractions or hot citrate buffer to unmask it may be related to its function. We are currently testing antibodies targeted to additional regions of dysferlin to ascertain if certain epitopes along the molecule are more or less exposed than others.
As dysferlin in the sarcoplasm appears reticular in cross sections and at the level of the A-I junctions in longitudinal sections, it is a component of either the t-tubules or the junctional SR. The improved labeling procedures we describe here strengthen the evidence for the presence of high levels of dysferlin in the sarcoplasm, consistent with earlier reports of dysferlin associated with t-tubules (Ampong et al. 2005; Lostal et al. 2010); unfortunately, the limited resolution of the light microscope, even with the highest resolution afforded by confocal optics, does not allow us to distinguish between the junctional SR and t-tubules in mature muscle. In initial experiments using double immunofluorescence labeling with markers of both compartments, such as the DHPR and triadin, we did not find significant levels of co-localization with dysferlin, raising the possibility that, if it is present in t-tubules or the junctional SR, dysferlin may occupy a unique membrane compartment or microdomain. Experiments are in progress in our laboratory to address this question.
We are also investigating the reasons behind the faint labeling that we see in A/J and Bl10.SJL muscle with the Hamlet and Lifespan antibodies to dysferlin. Although low levels of expression of dysferlin are expected in muscles of SJL mice, it is unclear why we also see faint labeling in A/J muscle, which has no detectable levels of dysferlin (Ho et al. 2004). Others have also reported images showing faint sarcolemmal and cytoplasmic labeling in A/J muscle with the Hamlet antibody (Guo et al. 2010). Intriguingly, we do not see faint dysferlin labeling in the LGMD2B and MM biopsies that we studied, nor do we see detectable levels of dysferlin in immunoblots of homogenates prepared from Bl10.SJL or A/J muscle. We speculate that mouse muscle may contain other proteins with homology to the epitopes recognized by the Hamlet and Lisfespan antibodies. Alternatively, as in some patients with dysferlinopathies, small amounts of truncated forms of dysferlin may be expressed in A/J mouse muscle (Wenzel et al. 2006; Krahn et al. 2010). This possibility, if true, may help explain phenotypic differences between various dysferlin-deficient mouse strains (Ho et al. 2004). Interestingly, impaired endoplasmic reticulum (ER)–associated degradation of truncated dysferlin by the ubiquitin/proteasome pathways (ERAD-I) has been reported, suggesting a possible link between ER stress and the pathology of dysferlinopathies (Fujita et al. 2007). The link between ER stress and dysferlin is also suggested by the enrichment of dysferlin in tubular aggregates, which develop in response to ER stress in various muscle pathologies (Ikezoe et al. 2003).
To our knowledge, ours is the first evidence that most of the dysferlin detectable in normal adult human and rodent skeletal muscle is present in an intracellular reticulum, rather than at the sarcolemma. Our previous results indicate that dysferlin is not necessary for the sarcolemma to reseal after a physiological injury in vivo (Roche et al. 2008, 2010). The presence of large of amounts of dysferlin in the either the t-tubules or the junctional SR suggests that its primary function is to maintain the integrity of one or both of these internal membrane systems under physiological as well as pathophysiological conditions. This new focus on dysferlin’s function within muscle cells, as well as the methods we describe here, have implications both for the diagnosis of muscular dystrophies and for designing therapies for dysferlinopathies.
Footnotes
Acknowledgements
We thank Dr. Amy J. Wagers, assistant professor of pathology, Harvard Medical School, Cambridge, Massachusetts, for her generous gift of breeding pairs of Bl10.SJL mice. We also thank Dr. Steven A. Moore and Ms. Terese Nelson of the Wellstone Project at the University of Iowa, Iowa City, for generously contributing cross sections of control and dystrophic muscle biopsies.
This work is dedicated to the memory of Dr. Louise V. Anderson, who developed the Hamlet antibody to dysferlin.
The authors declared no potential conflicts of interest with respect to the authorship and publication of this article.
This work was funded by a fellowship to J.A.R. and grants to R.J.B. and J.A.R. from the Jain Foundation Inc.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.