
Abstract
Epigenetic modifications, such as acetylation, phosphorylation, methylation, ubiquitination, and ADP ribosylation, of the highly conserved core histones, H2A, H2B, H3, and H4, influence the genetic potential of DNA. The enormous regulatory potential of histone modification is illustrated in the vast array of epigenetic markers found throughout the genome. More than the other types of histone modification, acetylation and methylation of specific lysine residues on N-terminal histone tails are fundamental for the formation of chromatin domains, such as euchromatin, and facultative and constitutive heterochromatin. In addition, the modification of histones can cause a region of chromatin to undergo nuclear compartmentalization and, as such, specific epigenetic markers are non-randomly distributed within interphase nuclei. In this review, we summarize the principles behind epigenetic compartmentalization and the functional consequences of chromatin arrangement within interphase nuclei.
Nuclear Architecture and Gene Expression
I
Osborne et al. (2004) have shown that active genes from different CTs located on decondensed chromatin loops are transcribed by the same RNA polymerase II in nuclear compartments called “transcription factories” (Cook 1995, 1999; Faro-Trindade and Cook 2006). In contrast, Andrulis et al. (1998) have shown that perinuclear localization helps to establish transcriptionally silent chromatins in
The reciprocal impact of higher-order chromatin structure and gene activity, observed at several levels, is intimately related to the specific histone modifications of individual chromatin domains. For example, the acquisition of active chromatin marks, such as H3K9 acetylation and H3K4me2, leads to chromatin decondensation and the formation of chromatin loops, which separate out actively transcribed genes from more compact chromosome territories (Chambeyron and Bickmore 2004). Another important example of the marriage of nuclear architecture and the histone code is seen in X chromosome inactivation (Xi). The X chromosome is highly enriched in heterochromatin marks, such as H3K27me3 and H3K9me2 (Figure 1). This histone pattern is consistent with Xi being localized to the most peripheral region of the interphase nucleus (Barr and Carr 1962; Heard et al. 1997, 2001; Bártová et al. 2001). However, the architecture of the X chromosome is organized further still; all genes of the X chromosome have been found to be positioned at the outer rim of the active X and Xi territories (Clemson et al. 2006). The location of active and inactive genes within specific chromosome territories has been addressed by several authors (summarized by Scheuermann et al. 2004). In many cases, transcriptionally active genes are located on the periphery of related chromosome territories (Kurz et al. 1996; Harničarová et al. 2006), in addition to being oriented to the nuclear center (Zink et al. 2004; Harničarová et al. 2006).
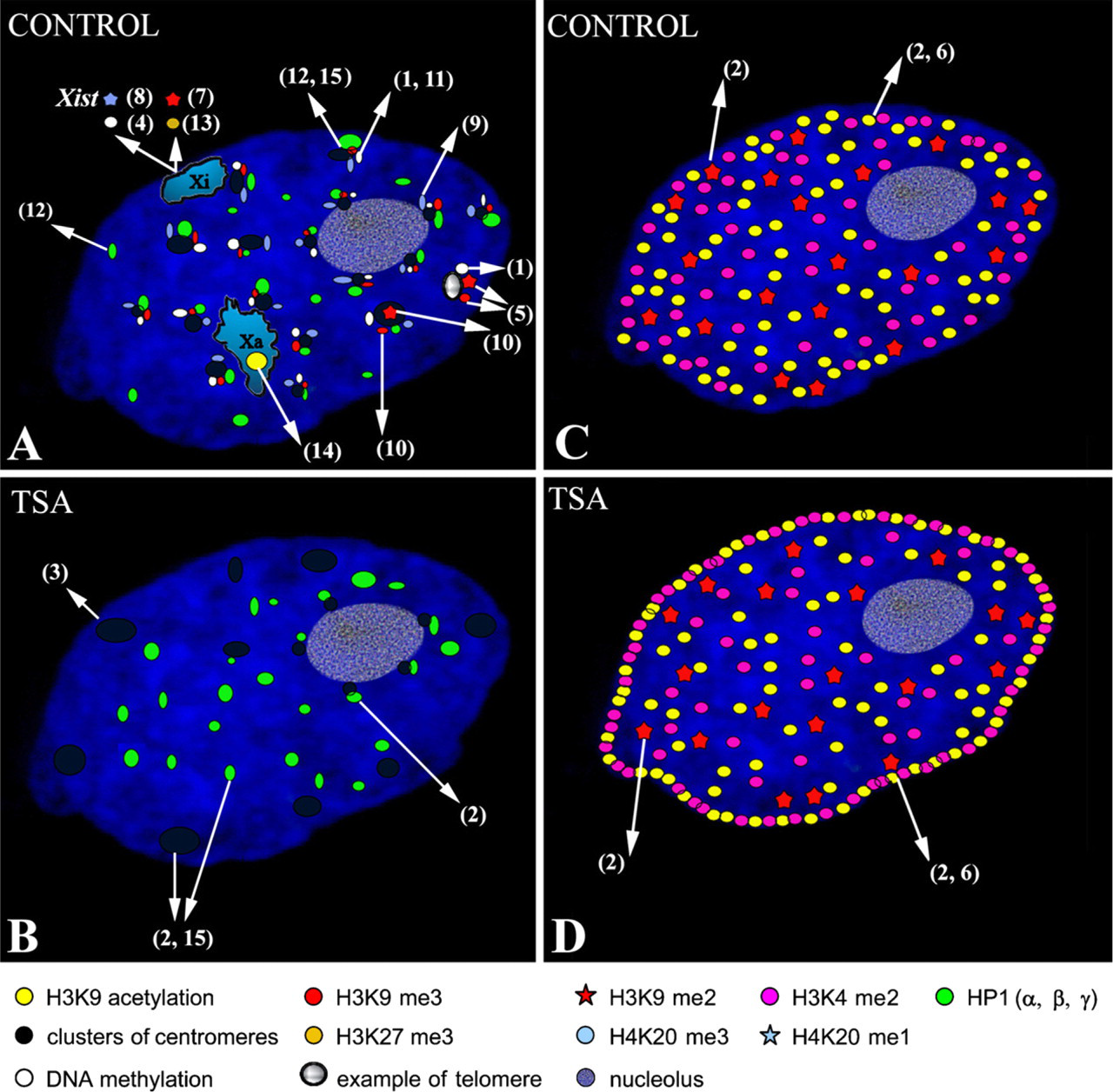
Illustration of the nuclear distribution of histone modifications and their association with heterochromatin protein 1 (HP1) proteins, centromeres, telomeres, and both active and inactive X chromosomes in normal (untreated) cells (
Together, the data described above portray the nuclear periphery as a dynamic compartment generally associated with transcriptional repression in both yeast and mammals. Therefore, lamins, components of the nuclear envelope (NE), have been suggested to be involved in the regulation of chromatin conformation and nuclear shape. For example, most of the
Functional Consequences of Lamin A/C Deficiency for Higher-order Chromatin Structure
Interaction of chromatin with the NE and nuclear lamina is thought to determine higher-order chromatin organization within interphase nuclei. The nuclear envelope, which isolates the nucleus from the cytoplasm, consists of the outer (ONM) and inner nuclear (INM) membranes separated by the perinuclear lumen. NPCs are inserted into the double membrane and mediate the exchange of components. The nuclear lamina protein meshwork is tightly associated with the INM and provides mechanical stability (summarized by Schirmer and Foisner 2007). A- and B-type lamins are major components of the nuclear lamina, which form a protein meshwork adjacent to the INM. The nuclear lamina is in close contact with the NE, and there are many lamin-binding proteins, which play an important role in chromatin architecture and provide anchorage sites for peripherally localized chromatin (Goldman et al. 2002). A-type lamins (A and C) have been shown to form functional associations with polynucleosomes, histones, and DNA, in a sequence-independent manner (summarized by Dorner et al. 2007 and Lattanzi et al. 2007). In addition, lamin-associated polypeptides (LAPs) influence histone modification, chromatin organization, and gene expression (summarized by Schirmer and Foisner 2007).
Since a functional relationship between A-type lamins and histones was first suggested by Taniura et al. (1995), A-type lamin mutations that are associated with changes in the pattern of histone modifications have been described recently (Filesi et al. 2005; Scaffidi and Misteli 2006; Shumaker et al. 2006, Lattanzi et al. 2007). Scaffidi and Misteli (2006) have shown that heterochromatin markers, such as H3K9 trimethylation and heterochromatin-associated protein HP1γ, are reduced in the cells of Hutchinson-Gilford progeria syndrome (HGPS), a disease associated with mutations in the lamin A gene (LMNA). In addition, HGPS cells display a decreased level of H3K27 trimethylation on the inactive chromosome X. In contrast, H4K20 trimethylation is increased in laminopathy cells (Shumaker et al. 2006). Loss of heterochromatin caused by lamin A/C deficiency is also associated with rearrangement and dispersion of HP1β and H3K9 methylation within the nucleoplasm (Filesi et al. 2005; Galiová et al. 2008). Similarly, inhibition of histone deacetylases, leading to decondensation of heterochromatin domains, is associated with nuclear reorganization of HP1 foci (Taddei et al. 2001; Bártová et al. 2005). All these observations imply that the loss of heterochromatin and its decondensation may be controlled by epigenetic processes.
Functional Aspects of Histone Modifications
The term epigenetics refers to a heritable change in phenotype that does not involve a change in the underlying DNA sequence. Therefore, histone modifications are epigenetic regulators of chromatin. The histone code influences higher-order chromatin structure by affecting contacts between different histones and between histones and DNA. Specific histone modifications are responsible for the compartmentalization of the genome into distinct domains, such as transcriptionally silent heterochromatin and transcriptionally active euchromatin (summarized by Martin and Zhang 2005). The ability of the histone code to dictate the chromatin environment allows it to regulate nuclear processes, such as replication, transcription, DNA repair, and chromosome condensation (Kouzarides 2007).
Next to DNA methylation, histone acetylation and histone methylation are the most well-characterized epigenetic marks. Trimethylation at H3K4, H3K36, or H3K79 results in an open chromatin configuration and is, therefore, characteristic of euchromatin. Euchromatin is also characterized by a high level of histone acetylation, which is mediated by histone acetyl transferases (HATs). Conversely, histone deacetylases (HDACs) have the ability to remove this epigenetic mark, which leads to transcriptional repression (summarized in Figure 2). Condensed heterochromatin is enriched in trimethylation of H3K9, K3K27, and H4K20 (Kouzarides 2007) and silencing of euchromatin loci caused by histone deacetylation involves the recruitment of specific K9 histone methyltransferases (HMTs). Methylated H3K9 provides a binding site for the chromodomain-containing heterochromatin protein 1 (HP1), which induces transcriptional repression and heterochromatinization (Figure 2). At euchromatic loci, this process is mediated by co-repressors, such as retinoblastoma protein pRb or KAP1 (Nielsen et al. 2001 and summarized by Kouzarides 2007). Histone demethylases, first described by Shi et al. (2004), have the opposite effect on transcription (Figure 2). The histone demethylase LSD1 is responsible for H3K4 demethylation, which leads to transcriptional inactivation (Figure 2). However, when LSD1 forms a complex with androgen receptors, it demethylates H3K9 and activates transcription (Metzger et al. 2005). Other histone demethylases, such as jumonji (JHDM2A), are responsible for H3K9 demethylation (summarized by Klose et al. 2006 and Kouzarides 2007), whereas JHDM1 has the ability to convert active chromatin marks such as H3K36me2 to an unmodified state (Jenuwein 2006).
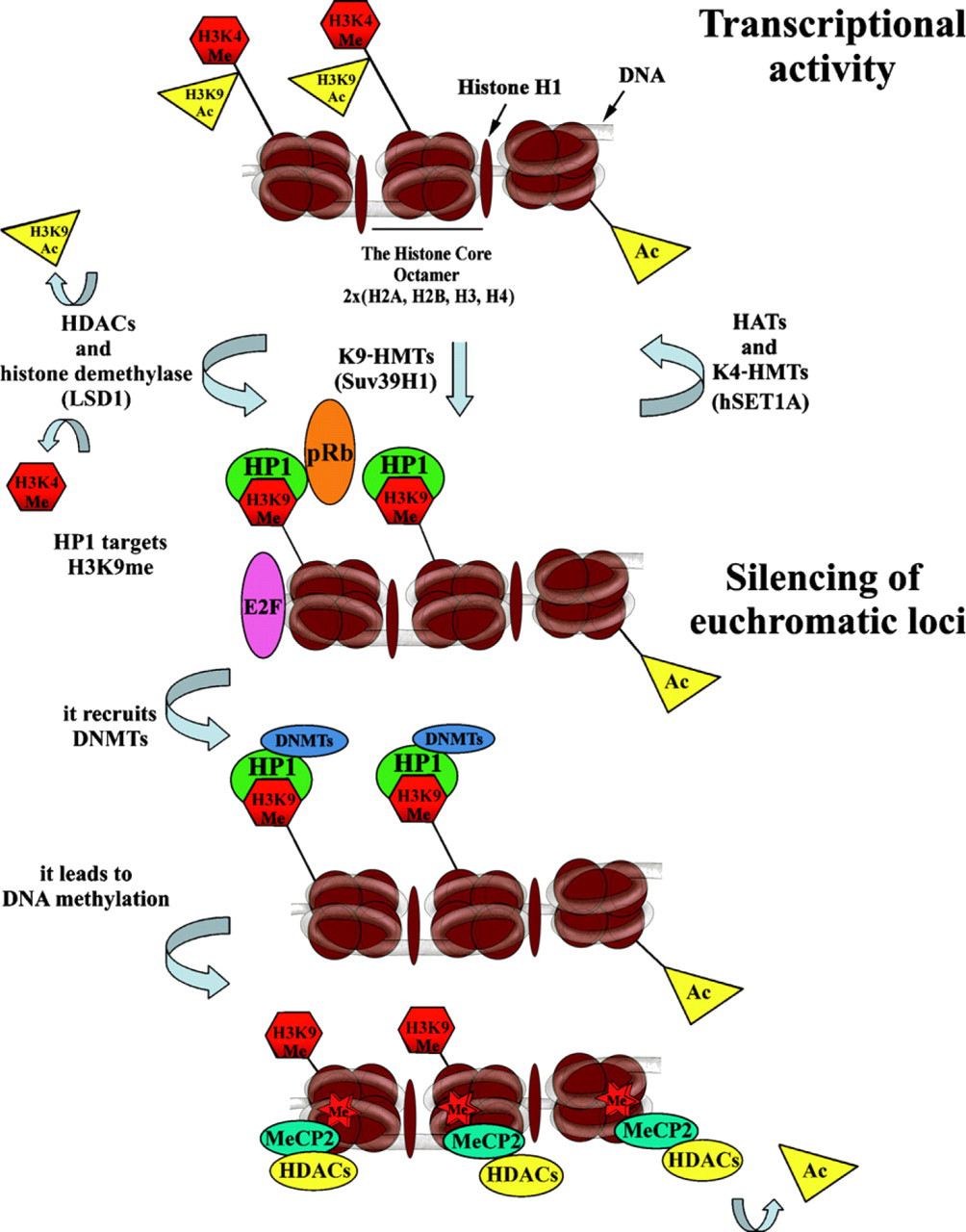
Model of formation of transcriptionally active and inactive chromatin by histone modifications. The scheme of the three core nucleosomes is used to describe chromatin. The acetyl group, especially on H3K9, and the methyl group on H3K4 associate with the transcriptionally active chromatin state of the mammalian genome. Histone acetylation is initiated by histone acetyl transferases (HATs; such as GNAT, MYST, and CBP/p300) and removed by histone deacetylases (HDACs; classes I, II, III). H3K4 methylation is induced by specific histone methylases, such as hSET1A, and removed by histone demethylase LSD1, which leads to activation of the HMT such as Suv39H1, which mediates H3K9 methylation. The chromodomain of the HP1 protein can recognize methylated H3K9, resulting in the propagation of transcriptionally silent chromatin. In euchromatic loci, this process is additionally mediated by co-repressors, such as retinoblastoma protein pRb, and constitutive heterochromatin is stabilized by HP1 binding to regional H3K9 methylation, which also involves incorporation of RNA (summarized by Lachner and Jenuwein 2002). Subsequently DNA methyltransferases (DNMTs) are activated, leading to DNA methylation. Methylated DNA is recognized by methyl-DNA-binding proteins, such as MeCP2, which, in turn, can associate with HDAC activity to eliminate the rest of the acetylated histones. Therefore, DNA methylation can facilitate additional histone methylation to enhance the repressed state of chromatin.
In contrast to the large amount of available information on the functions of HATs, HDACs, and enzymes that mediate histone methylation (listed in Kouzarides 2007), enzymes responsible for other types of histone modifications, such as phosphorylation and ubiquitination, are not as intensively studied (Grant 2001). This is despite the fact that these modifications have important roles in transcription, DNA repair, the induction of apoptosis, and chromosome condensation (Cheung et al. 2000). For example, the phosphorylation of serine 10 in histone H3 is associated with transcriptional activation in mammalian cells (Thomson et al. 1999), and H2A phosphorylation is responsible for chromosome condensation (Grant 2001). These observations underscore the importance and complexity of histone modifications in the regulation of nuclear and, subsequently cellular, processes.
Studies of the relationships that exist between nuclear architecture and histone marks have begun to show novel principles underlying nuclear compartmentalization (Branco and Pombo 2007; Lanctôt et al. 2007). Several models for the nuclear arrangement of chromosome territories and interchromatin compartments have been published on the basis of distinct experimental and mathematical approaches. The first revelation related to these models was the discovery of peripheral invaginations of chromosome territories, which admit transcriptionally active chromatin into the more interior parts of interphase chromosomes (Verschure et al. 1999). This sponge-like CT architecture is taken into account in the interchromatin compartment model (CT-IC), suggested and summarized by Cremer and Cremer (2001) and revised by Cremer et al. (2006). In contrast, the interchromosome domain (ICD) model (Zirbel et al. 1993) defines CTs as smooth domains that are separated from each other. The concept of mutual untouchability of CTs was later disproved by the discovery of chromosome “intermingling” (Branco and Pombo 2006), which could explain the occurrence of chromosome translocations. As suggested by Kozubek et al. (1997), the physical proximity of chromosome territories caused by radiation increases the probability of specific chromosome translocations frequently observed in tumor cells.
Histone Methylation and Acetylation Patterns in Interphase Nuclei
Drawing from the principles of nuclear compartmentalization (summarized by Cremer et al. 2006 and Lanctôt et al. 2007), several structural studies have addressed the distribution of methylated and acetylated histones in interphase nuclei of mammalian cells. The nuclear distribution patterns of methylated histones were studied by Cremer et al. (2004) in three-dimensional-preserved nuclei of normal and malignant cell types. In these studies, interphase histone methylation profiles varied between the nuclei of individual cells, through the cell cycle, and in quiescent cell populations. A homogeneous distribution of multiply methylated lysines was found in normal quiescent lymphocytes. In contrast, distinct clusters of this histone modification were found in leukemia cells, whereas a more peripheral accumulation was observed in a colon carcinoma cell line compared with normal colon epithelium. During the analysis of histone lysine methylation patterns of breast carcinoma cells through the cell cycle, it was observed that histone methylation is concentrated into a few compact clusters when cells exit the cell cycle (G0 phase). The clusters of methylated histones in G0 cells were found predominantly in close proximity to nucleoli, whereas some smaller clusters were found at the nuclear periphery. In cycling cells, sites of methylated histone lysines were found dispersed throughout the nucleus during G1 but were mostly located at the nuclear periphery and around nucleoli in S phase. In S phase, the centromeres (which contain histone-like CENP proteins (Sullivan et al. 1994; Goldberg et al. 1996) were also associated with sites of highly methylated histone lysines, although some clusters of highly methylated lysines were observed outside the centromeric heterochromatin (Cremer et al. 2004).
Zinner et al. (2006) further studied the three-dimensional arrangement of specific histone lysine methylation sites (H3K4me3, H4K20me1, H3K9me1, H3K27me3, H4K20me3, and H3K9me3) in various human cell lines and described the spatial proximity of the methylated regions to centromeres or newly synthesized RNA. Simultaneous visualization of different histone lysine methylation sites showed that methylation patterns are arranged in distinct nuclear layers, with a certain degree of overlap depending on the type of epigenetic modification (Zinner et al. 2006; Skalníková et al. 2007). Moreover, Zinner et al. (2006) reported a co-localization coefficient of 38% for active chromatin markers H4K20me1 and H3K4me3. Meanwhile, a co-localization coefficient of 43% was found for heterochromatin markers, such as H3K9me3 and H3K27me3. This probably reflects the overlapping of heterochromatin-associated histone modifications at the nuclear periphery, where nuclear processes, suchas transcription, rarely occur (Zink et al. 2004; Williams et al. 2006). These results document the existence of distinct nuclear layers with respect to specific histone modifications (Zinner et al. 2006; Skalníková et al. 2007), which correlate well with the model for compartmentalization of interphase chromatin(Cremerand Cremer2001; Lanctôt et al. 2007).
As described above, individual types of histone modifications are thought to occupy distinct nuclear domains. This model has gained support from the finding that chromatin in the nuclear interior is H3K9 monomethylated, whereas H3K9 dimethylation occurs preferentially at the nuclear and nucleolar periphery (Wu et al. 2005). In addition, H3K9me2 is mainly associated with sites that replicate during mid-S phase, whereas H3K9me3 is a characteristic marker of late-replicating pericentric heterochromatin (Figure 1A) (summarized by Lachner et al. 2003), suggesting that specific histone modifications may influence the timing of replication of individual chromatin domains in dividing cells.
Proliferating cells are characterized by a higher level of methylation of H3K4, H3K9, H3K27, and H4K20 than quiescent G0 B lymphocytes. Similarly, heterochromatin protein 1β (HP1β) and the Ikaros factor, normally present at pericentric regions, are absent in G0 B lymphocytes but are restored along with histone methylation after mitotic B-cell stimulation (Baxter et al. 2004). These observations, along with those documented by Gilbert et al. (2003) in the model of cell differentiation, illustrate the plasticity of the histone code. These authors found that, in the nuclei of mouse embryonic erythrocytes, HP1 proteins are abundant, H3K9 trimethylation is increased, and H3K27 trimethylation is lost. In contrast, in maturing chicken erythroid cells, HP1 proteins and trimethylation of both H3K9 and H3K27 are substantially reduced, suggesting that the same cell lineage can use different patterns of histone modification to form heterochromatin. In addition, Lukášová et al. (2005) found that all isoforms of the HP1 protein are absent in terminally differentiated human granulocytes. This suggests that patterns HP1 binding could change significantly during cell differentiation. Interestingly, residual levels of HP1 protein and H3K9 methylation were detectedin in vitro–differentiated myeloid leukemia cells, which may be caused by a disruptionin the maturation process that leads to incomplete chromatin condensation and could be related to disease progression (Lukášová et al. 2005).
DNA Methylation and Nuclear Organization
DNA methylation is known to affect the nuclear localization of constitutive heterochromatin. For example, DNA methylation has been found to be responsible for the clustering of centromeres during cell differentiation (Brero et al. 2005), and the DNA of centromeric and telomeric regions of many chromosomes is preferentially methylated (Barbin et al. 1994; Montpellier et al. 1994). Mutant mouse embryonic stem cells that completely lack DNA methylation have alterations in the overall levels of histone H3 methylation and acetylation, decreased mobility of linker histones, and increased chromocenter clustering (Gilbert et al. 2007). These observations have uncovered a novel relationship between DNA methylation, linker histones, and chromatin structure.
It is a well-known phenomenon in chromatin structural biology that the average nuclear positioning of gene-poor chromosome 18 (HSA18) is peripheral, whereas gene-rich chromosome 19 (HSA19) occupies more internal regions of interphase nuclei (Croft et al. 1999). Despite the fact that DNA methylation tends to be associated with silent chromatin, nearly all of human chromosomes 19 and 22 are found to contain 5-methylcytosine–rich DNA, but chromosome 18 is relatively deficient in this type of DNA modification (see Barbin et al. 1994). Bearing in mind that in mammalian cells, transcriptionally active sequences and GC-rich regions (irrespective of their activities) are more centrally located (Zink et al. 2004; Harničarová et al. 2006; Williams et al. 2006), centrally positioned gene-rich HSA19 is expected to lack DNA methylation. Importantly, these data originated from the statistical evaluation of nuclear radial distributions of HSA18 and HSA19 from hundreds of cells and do not reflect variability on the individual cell level. However, similar trends in nuclear distribution have also been published by Goetze et al. (2007) for RIDGE (region of increased gene expression) and anti-RIDGE (regions of low transcriptional activity) sequences. This apparent contradiction has recently been clarified by Zinner et al. (2007). These authors reported no differences in the levels of H3K9me3 (often associated with DNA methylation; Figure 2) and H3K27me3 in the territories of human chromosomes 18 and 19, but found that H3K4 trimethylation is more abundant on the gene-rich HSA19 than the gene-poor HSA18 (Zinner et al. 2007). Their data imply that the level of H3K4me3, often associated with promoter regions of active genes (Schübeler et al. 2004), dictates the distinct nuclear locations of chromosomes 18 and 19. In contrast, nuclear distribution of DNA methylation and the occurrence of H3K9me3 or H3K27me3 within these two autosomes do not explain their specific locations in interphase nuclei.
Links between histone methylation and DNA methylation in mammals have been documented by Lehnertz et al. (2003). These authors observed that histone methyltransferases (HMTs), such as Suv39h types, are required for both H3K9 trimethylation and DNA methyltransferase 3b (Dnmt3b)-dependent DNA methylation at pericentric repeats. From these experiments, it is evident that Suv39h HMTs govern H3K9 methylation at constitutive heterochromatin. This is also supported by the finding that, in comparison with wild-type controls, abnormally long telomeres in primary cells of Suv39h1 and Suv39h2 double null mice had less dimethylated and trimethylated H3K9, but increased levels of H3K9 monomethylation were observed (García-Cao et al. 2004) (Figure 1A). In contrast, DNA methylation at centromeric regions has been reported to be independent of Suv39h function (Lehnertz et al. 2003), which is consistent with the observation that centromeres display Suv39h-independent H3K9 dimethylation (summarized by Lachner et al. 2003) and are enriched in H4K20 trimethylation (Kourmouli et al. 2004) (Figure 1A).
Nuclear Arrangement of HP1 Protein
Mammalian heterochromatin protein 1 (HP1α, HP1β, and HP1γ isoforms) is a non-histone protein that can interact with other proteins to regulate transcription and chromatin arrangement. Histone H3K9 methylation, which is characteristic of transcriptionally inactive chromatin, provides the binding site for the HP1 protein. Both Suv39H1 and G9a histone methyltransferases are responsible for the induction of H3K9 methylation, but only Suv39H1 has the ability to recruit HP1 to chromatin (Stewart et al. 2005). Co-localization of HP1α and HP1β has been observed in 3T3 cells, and in HeLa cells, HP1α and a fraction of HP1β and HP1γ have all been found to associate with mitotic chromosomes. In other experiments, HP1α and HP1β have been unambiguously localized to heterochromatin, but HP1γ has been found to be associated with both euchromatin and heterochromatin domains (Minc et al. 1999, 2000, 2001) (Figure 1A). These experiments indicate a role for HP1γ in the silencing of euchromatin loci.
Changes in HP1 protein localization through the cell cycle have been observed. During interphase, HP1β associates preferentially with centromeric heterochromatin, whereas HP1α and HP1γ are mostly found in the promyelocytic leukemia (PML) nuclear compartment (Hayakawa et al. 2003). During metaphase, only HP1α co-localizes with the centromeric heterochromatin and association of HP1α and HP1β proteins with metaphase and interphase centromeres has been ascribed to certain motifs of these protein variants (Hayakawa et al. 2003).
The specific ability of HP1 to bind to centromeric sequences has also been studied for endodermal cell differentiation (Cammas et al. 2002, 2004, 2007; Bártová et al. 2007), during which there is a dynamic nuclear rearrangement of HP1 and the transcriptional intermediary factor 1β (TIF1β), whose activity is dependent on HP1. In undifferentiated cultures, TIF1β is diffusely distributed throughout the nucleoplasm, whereas in differentiated cells, TIF1β and HP1 co-localize at centromeric regions (Cammas et al. 2002). Further analyses has shown that the TIF1β–HP1 interaction is not important for the differentiation of mouse embryonal carcinoma cells into primitive endoderm, although it is essential for further differentiation into parietal endoderm-like cells (Cammas et al. 2004). These data imply that interaction between TIF1β and HP1 is probably important for induction of the endodermal pathway and possibly regulates the expression of endoderm-like markers. The pattern of nuclear distribution of HP1 subtypes and TIF1β has been found to be differentiation-specific and more fundamental to the differentiation of embryonal carcinoma cells into primitive endoderm than are changes in protein levels, which are relatively stable during this process (Bártová et al. 2007). In undifferentiated cells, TIF1β/HP1 interaction occurs only within euchromatin and involves HP1β and HP1γ, but not the HP1α subtype. In cells undergoing differentiation, TIF1β selectively associates with HP1β within heterochromatin, and TIF1β/HP1γ is present exclusively within euchromatin. TIF1β does not interact with HP1α in either undifferentiated or differentiated cellular systems (Cammas et al. 2007), consistent with data published by Bártová et al. (2007) showing clustering of TIF1β only at those centromeric regions that contain both HP1α and HP1β subtypes. Results published by Bártová et al. (2007) and Cammas et al. (2007) imply that HP1β plays an important role in the transition of TIF1β from euchromatin to heterochromatin during cell differentiation.
Inhibition of HDACs Induces Reorganization of Chromatin and Histone Modifications
Histone acetylation, which can be modified by histone deacetylase inhibitors (HDACIs), such as trichostatin A (TSA) and sodium butyrate (NaBt), influences many nuclear and downstream cellular processes. Global hyperacetylation induced by HDACI causes reversible decondensation of chromatin (Tóth et al. 2004). Moreover, histone acetylation plays a central role in the dynamic regulation of chromatin accessibility during interphase (Gorisch et al. 2005). Several experiments have used HDACIs to show the extent to which certain biological functions are associated with changes in chromatin structure. Prolonged treatment with a low concentration of TSA caused centromeric regions to be repositioned at the nuclear periphery and to lose the ability to bind HP1 protein (Taddei et al. 2001). These changes in nuclear organization persisted after TSA was removed from the cell culture (Taddei et al. 2001). Bártová et al. (2005, 2007) observed a similar peripheral repositioning of centromeres after TSA treatment. However, the dissociation of HP1α was only found to occur at peripherally localized centromeric sequences (Bártová et al. 2005), which formed large clusters in TSA-treated mouse embryonal carcinoma cells (Bártová et al. 2007) (Figure 1B). Gilchrist et al. (2004) have also showed that heterochromatin is highly sensitive to HDACIs. In their experiments, TSA stimulated an increase in the level of H4K5 and H3K9 acetylation at the nuclear periphery (compare Figures 1C with 1D). In contrast, H3K9 methylation and nuclear positioning of centromeres were relatively conserved after a short treatment with a high dose of TSA (Gilchrist et al. 2004). Like Gilchrist et al. (2004), Bártová et al. (2005) reported a dispersed pattern of H3K9 dimethylation, which did not change at the nuclear periphery of HDACI-treated cells (Figures 1C and 1D). In control cells, H3K9 acetylation and H3K4 dimethylation were absent from the most peripheral regions of interphase nuclei, whereas TSA and NaBt induced a high level of acetylation at H3K9 and dimethylation at H3K4 at the nuclear periphery (Bártová et al. 2005) (compare Figures 1C and 1D). Although all of these experiments gave similar results, certain differences in cell sensitivity to HDACI stimulation were reported. Taddei et al. (2005) have summarized the biological effects of HDACIs and highlighted the importance of the dose and exposure time. Further experiments have shown that HDACIs can block mitotic progression at prometaphase. In addition, HDACIs cause decreased HP1β binding to pericentromeric heterochromatin in the S and G2 phases of the cell cycle. This may be related to increased acetylation of H3K9 in pericentromeric heterochromatin, which in turn leads to mitotic defects resulting from the inhibition of histone deacetylation (Robbins et al. 2005).
Histone Methylation and Acetylation Patterns of Human X Chromosome
Dosage compensation of the mammalian female X chromosome is a well-studied phenomenon (Heard et al. 1997, 2001; Park and Kuroda 2001). The inactive X chromosome (Xi) is distinguished from its active homolog (Xa) only by the expression of the
While studying the differentiation of ES cells, Keohane et al. (1996) discovered that histone deacetylation is essential for the maintenance of X chromosome silencing but is not needed for its initiation. Apart from H4 hypoacetylation, H3K9 dimethylation (Khalil and Driscoll 2006) is also present in the Xi of female mammalian cells (Figure 1A). H3K9 methylation occurs at the same time as the induction of H3K9 hypoacetylation and H3K4 hypomethylation, suggesting that histone H3 modifications are particularly crucial for X chromosome inactivation (Heard et al. 1997, 2001). Knockdown of Suv39h HMTases abolished H3K9 methylation in constitutive heterochromatin but not in the Xi. This is explained by the observation that HP1 proteins do not accumulate on the Xi and indicates the existence of a Suv39h-HP1–independent pathway regulating H3K9 methylation of the facultative heterochromatin of chromosome X (Peters et al. 2002).
It is known that increased H3K9 methylation is a distinguishing mark of facultative heterochromatin, associated with promoter of inactive genes. Conversely, H3K4 methylation is absent at Xi, except for three specific sites representing transcriptionally active loci (Boggs et al. 2002). Khalil and Driscoll (2006) have reported that, although H3K4 dimethylation is largely absent from the inactive X in female somatic cells, the inactive X in male meiotic cells is rich in H3K4me2. In contrast, the Xi chromosome in female somatic cells and both the X and Y chromosomes in male meiotic cells were found to be devoid of H3K4me3, which was mainly present at discrete regions along most of the autosomes. H3K4me2 was homogenously distributed throughout the genome, whereas the Y chromosome lacked H3K4me2 and H3K4me3 in somatic cells. However, during spermatogenesis, the Y chromosome was rich in H3K4me2 but not H3K4me3 (Khalil and Driscoll 2006). Taken together, these observations show that distinct epigenetic patterns for X chromosomes of female somatic cells and male germ line cells exist. In addition to being H3K9 methylated (Mermoud et al. 2002), the mammalian female chromosome Xi also has intense H3K27 trimethylation (Rougeulle et al. 2004) (Figure 1A). Dynamic changes in H3K9 and H3K27 dimethylation on the Xi suggest that these epigenetic modifications compliment each other during the X inactivation process (Rougeulle et al. 2004). Moreover, the appearance of H3K27 methylation and ubiquitinated H2A is associated with the transient recruitment of Polycomb group protein complexes (PcG) to Xi during the initial stages of both imprinted and random X inactivation (Martin and Zhang 2005).
Future Directions
Epigenetic modifications are dynamic, and their nuclear distributions undergo specific changes during various cellular processes, such as cell cycle progression and differentiation. The identification of enzymes responsible for the initiation of epigenetic processes represents a very important milestone in chromatin biology (summarized by Klose and Zhang 2007), which has enabled the study of the dynamics of epigenetic regulation. It is likely that the next several years will be dedicated to translating the histone code to discover its role in both normal biological processes and disease states. Similarly, knowledge of how epigenetics influences genomic reprogramming during embryonic development will lead to improved therapeutic approaches for regenerative medicine (Surani et al. 2007). Moreover, inhibitors of HDACs, DNMTs, and probably HMTs are promising tools for anticancer therapy (Yoo and Jones 2006).
Footnotes
Acknowledgements
This work was supported by Grant Agency of the Czech Republic Grant 204/06/0978 and by Ministry of Education of the Czech Republic Grants MSM0021622419 and LC535. The research projects of the Institute of Biophysics AVOZ50040702 and AVOZ50040507 also supported this work.
The authors thank Dr. Adrian Sumner for text revision.