
Abstract
Parkinson's disease (PD) affects >1 million Americans and is marked by the loss of dopaminergic neurons in the substantia nigra. PD has been linked to two causative factors: genetic risks (hereditary PD) and environmental toxins (idiopathic PD). In recent years, considerable effort has been devoted to the development of a Drosophila model of human PD that might be useful for examining the cellular mechanisms of PD pathology by genetic screening. In 2000, Feany and Bender reported a Drosophila model of PD in which transgenic flies expressing human mutant α-synuclein exhibited shortened life spans, dopaminergic losses, Parkinsonian behaviors, and Lewy bodies in surviving dopaminergic neurons. Since then, a number of studies have been published that validate the model or build on it; conversely, a number report an inability to replicate the results and suggest that most protocols for dopaminergic histology underreport the actual numbers of dopaminergic neurons in the insect brain. Here we report the optimization of dopaminergic histology in Drosophila and identification of new dopaminergic neurons, show the remarkable dendritic complexity of these neurons, and provide an updated count of these neurons in adult brains. This manuscript contains online supplemental material at http://www.jhc.org. Please visit this article online to view these materials.
P
Two forms of human PD have been identified: the hereditary form, which accounts for <10% of PD cases, and the idiopathic form, accounting for the remaining 90% (Nussbaum and Christopher 2003). Feany and Bender (2000) reported a Drosophila model of hereditary PD in which A53T and A30P mutant α-synuclein were expressed exclusively in dopaminergic neurons. They observed shortened life spans, an age-related loss of dopaminergic neurons, α-synuclein–positive plaques in surviving dopaminergic neurons that resembled Lewy bodies, and behavioral deficits that mimicked those observed in human PD (Feany and Bender 2000). Subsequently, Pendleton et al. (2002) observed that L-DOPA, a drug used to treat PD patients, could suppress the behavioral defects observed in these flies. Auluck et al. (2002) showed that coexpressing heat-shock/chaperone proteins in this model could correct the misfolding of amyloid α-synuclein molecules. Chaperone proteins observed to suppress dopaminergic toxicity in Drosophila were subsequently identified as colocalizing with mutant α-synuclein in the plaques of PD patients, suggesting that this fly model of PD could be predictive of human pathology.
A number of researchers have attempted to duplicate these data with mixed results (Auluck et al. 2002; Pesah et al. 2005). A recent study showed that misexpressing α-synuclein in Drosophila brains did not actually shorten lifespan and did not produce dopaminergic neuronal losses; these same authors failed to observe Lewy body-like plaques and observed none of the previously reported behavioral deficits (Pesah et al. 2005). Discrepancies may derive from variably effective methods of dopaminergic histology in Drosophila, resulting in underestimates of actual numbers of dopaminergic neurons. A comprehensive analysis of dopaminergic loss in Parkinsonian fly brains—essential for forward genetic screens—requires the ability to accurately detect all dopaminergic neurons in control (wild type) and experimental (Parkinsonian) fly brains. Here we report a systematic comparison of protocols for fixing and staining dopaminergic neurons in insect brains. We describe an optimized protocol for this histology and identify previously unreported dopaminergic neurons. We show the remarkable dendritic complexity of these neurons and provide an updated count of these neurons in adult brains.
Materials and Methods
Animal Dissections
We anesthetized 15-day-old Oregon R flies, initially selecting five males and five females for each group. On analysis, male and female brains showed no differences in staining or anatomy, so the sexes were used interchangeably. Wings, legs, and the lower abdomen of each fly were removed, leaving the thorax and head intact. The proboscis was removed at its attachment to the skull, leaving a hole on the anterior surface of the head. A slit was made in the exoskeleton on the posterior surface of the head, allowing increased fixative penetration.
Drosophila Stocks
Wild-type Oregon R flies and transgenic fly lines expressing Ddc-GAL4 and elav-GAL4 were obtained from the Bloomington Drosophila stock center (Indiana University; Bloomington, IN). Flies expressing UAS-Gap43-GFP were generously provided by Dr. Akira Chiba (University of Miami; Coral Gables, FL). Drosophila were grown on standard cornmeal medium at 25C.
Fixation
Four fixatives were used: (a) Carnoy's fixative (Weinstock and McDonald 1969; Giaccone et al. 2000), (b) Bouin's fixative (Seroogy et al. 1988; Homberg et al. 1991), (c) 8% w/v paraformaldehyde, and (d) Nassel/Elekes fixative (NEF) (Nassel and Elekes 1992) (Supplemental Table 1). All fixations proceeded for a minimum of 24 hr and for a maximum of 48 hr at 4C. We infiltrated tissue using two techniques: (a) microwave infiltration and (b) simple infiltration. For microwave infiltration, Eppendorf tubes containing fixative and dissected brain tissue were placed into the pressure chamber of a histological microwave apparatus (EMS 9000; Electron Microscope Sciences, Hatfield, PA) and vacuum was applied at −20 psi. The microwave was set to a constant temperature setting of 25C; microwaves were applied through three cycles, each cycle consisting of a 2 min microwaving interval followed by a 2 min rest. Subsequently, tissue was stored in fixative overnight at 4C. For simple infiltration, tissues were incubated in solution for identical time periods, without microwaves or vacuum.
Embedding
Paraffin
After fixation, brains were rinsed in PBS five times, each time for 10 min at room temperature. The tissue was dehydrated through an ethanol series to xylenes. Xylenes were replaced with paraffin in increasing concentrations at 56C in a vacuum oven at −20 psi. Brains were placed in TissueTek molds containing molten paraffin, mounted on TissueTek cassettes, and stored at −20C for at least 24 hr.
Agarose
After fixation, samples were rinsed in PBS four times for 15 min per rinse; 8% agarose was micro-waved in a conventional microwave oven for ∼2 min and could be stored in a 56C oven for up to 20 min. Fly tissue, with ventral surfaces down and with anterior head surfaces parallel to the rim of the dish, were positioned in 60 mm disposable Petri dishes. Tissue was covered with 56C agarose, and blocks were chilled at 4C. If blocks were to be stored for several hours, they were immersed in appropriate buffer (see below under Sectioning), and dish covers were sealed with parafilm.
Sectioning
Paraffin
Blocks were mounted and sectioned at 5 μm thickness on either a Leica RM2255 (Wetzlar, Germany) or an American Optical 820 Spencer (GMI; Ramsey, MN) microtome, and slides containing sections were stored upright overnight at 37C. They were transferred to −20C and stored for up to 2 months under desiccation.
Agarose
Trapezoid blocks containing a single fly head were positioned on a vibratome mounting block and attached with Superglue. Mounted blocks were immersed in a vibratome sectioning well (OTS-5000; Electron Microscope Sciences) containing the appropriate buffer [PBS for Carnoy's fix, Bouin's fix, and paraformaldehyde; Nassel/Elekes Buffer (NEB) for NEF], and sections were cut at 50 μm thickness, with oscillations of 8/sec and advance at 5. All sections were placed in embryo filters that were immersed in wells in a 24-well plate containing PBS or NEB. Agarose sections could be stored up to 24 hr at 4C before staining.
IHC
All polyclonal antibodies were preabsorbed against 50 μl of fixed Drosophila embryos for 24 hr at 4C (Mitchison and Sedat 1983). Briefly, preabsorption is achieved by diluting a preliminary volume of antibody (preabsorption concentration should be ∼10X the desired final concentration) in PBT [PBS with 0.1% Triton × (v/v)]. We typically used 50 μl of embryos for every 250 μl volume of preabsorption dilution; embryos are not considered in volume calculations. This stock solution is rocked at 4C overnight. The next day, the entire volume, including embryos, is transferred to a larger conical tube and diluted to the final concentration with PBT. Preabsorbed diluted antibodies may be maintained (with embryos in tube) for up to 4 weeks at 4C. Preabsorption is used to minimize background staining in invertebrates, as an alternative to blocking steps.
Each Drosophila brain section was permeabilized in PBT or NBT [NEB with 0.1% Triton × (v/v)] for 1 hr. Primary antibody incubation was performed at 4C overnight or at room temperature for 2 hr. Rabbit polyclonal antibodies targeting Drosophila tyrosine hydroxylase (dTH) and Drosophila tyrosine phosphatase (dTPh), generated by one of us (WN) (Neckameyer et al. 2000), were used at a final dilution of 1:1000. Tissue was rinsed five times in PBT or NBT, each for 10 min. Preabsorbed Alexa Fluor 488 goat-anti-rabbit secondary antibody (final dilution 1:500; Invitrogen-Molecular Probes, Carlsbad, CA) was applied for 2 hr at room temperature or for 24 hr at 4C. Other secondaries that were used included Alexa Fluor 568 goat-anti-rabbit secondary antibody and Alexa Fluor 488 goat-anti-mouse secondary antibody; these were preabsorbed and diluted identically. Samples were rinsed five times in PBT, 10 min per rinse, mounted in 70% glycerol, and kept at 4C for up to 1 month before imaging. Anti-green fluorescent protein (GFP)/anti-dTH double staining was performed on selected fly brains using an antibody generated against full-length GFP at a 1:1000 dilution (#632381; Clontech, Mountain View, CA).
Imaging and Analysis
Sectioned brains were imaged on a Bio-Rad (Hercules, CA) Radiance confocal microscope, using X10, X20, and X60 Nikon lenses (Melville, NY). Data were collected at 488- and/or 568-nm excitation. Cell and axon measurements were performed manually, using raw data Z-stacks. Bio-Rad software was used to project each Z-series to form two-dimensional images, which were assembled into figures using Adobe Photoshop (v 7.0) and Macromedia Freehand (v 10.0) software (Adobe Systems Inc.; San Jose, CA). Zeiss LaserSharp 2000 software (Carl Zeiss MicroImaging, Inc.; Thornwood, NY) was used to generate rotational movies from selected Z-series data sets.
Images were scored for presence of dopaminergic neurons in relation to morphological markers, including the esophageal opening, dorsal, ventral, and lateral brain edges, retinal diameter, proboscal musculature, well-characterized central body and core complex structures, and mushroom bodies. Because dendritic complexity was great, it was necessary to distinguish cell bodies from an axon nexus; thus, a structure was determined to be a cell only if an unstained core (nucleus) at the center of the structure could be observed and if the greatest diameter of the structure was 3 μm or larger. Neurons identified by three independent viewers were added to a new map of dopaminergic staining. This map was used as a reference standard for dopaminergic staining, and as a measure of efficacy in fixation and staining.
Results
Because previous reports (which relied on paraformaldehyde fixation of Drosophila brains followed by whole mount imaging or on paraffin sectioning before imaging) generated conflicting data sets, we concluded that a systematic comparison of the protocols developed for dopaminergic staining in Drosophila would resolve an issue that might lie at the source of inconsistency in the field (Davis et al. 2003; Yang et al. 2003; Pesah et al. 2005; Whitworth et al. 2005; Wang et al. 2006). We surveyed the literature describing classical insect neuro-anatomy, focusing on the characterization of aminergic peptides in the adult insect brain (DeGiusti and Ezman 1955; Seroogy et al. 1988; Homberg et al. 1991; Nassel and Elekes 1992). We determined that variability could be found in five major areas: fixative used, method of fixative delivery, embedding medium, sectioning technique, and choice of antibodies. We tested a number of commercially available antibodies directed against vertebrate components of the dopamine synthetic pathway and found none that worked well in Drosophila. One of us (WN) recently published antibodies generated against Drosophila dopaminergic synthetic enzymes (Neckameyer et al. 2000); we found two of these antibodies to work well in Drosophila brains: anti-dTH and anti-dTPh. We used dTH throughout this study. Having chosen an optimal antibody, we varied each of the other parameters independently and determined an optimal method of preserving dopaminergic neurons in fruit flies (Table 1; Figure 1).
Summary of histological protocols for dopaminergic neuronal staining in Drosophila
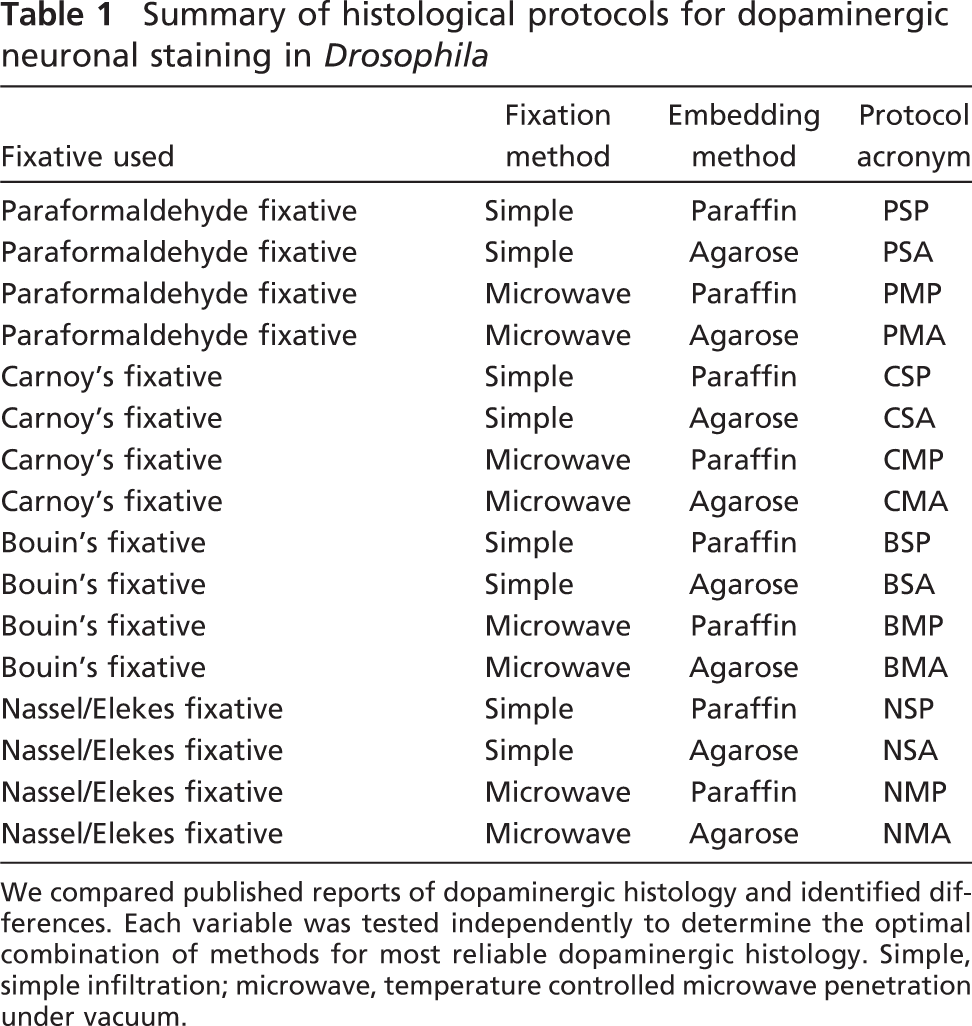
We compared published reports of dopaminergic histology and identified differences. Each variable was tested independently to determine the optimal combination of methods for most reliable dopaminergic histology. Simple, simple infiltration; microwave, temperature controlled microwave penetration under vacuum.
Comparison of Histological Protocols
Optimal dopaminergic staining has never been adequately developed in Drosophila and, as a result, histological inconsistency between groups makes it impossible to compare reported PD models. We observed that the fixative used in these protocols played an integral role in the preservation of dopaminergic neurons as well as in the general preservation of brain tissue. Temperature-controlled microwaving either further improved the efficacy of the fixative or exacerbated its detrimental effects. Paraffin embedding always diminished dopaminergic staining and made it difficult to generate sections of reproducible quality. Thus, we found that a weak fixative delivered with microwave energy under vacuum to tissues that were subsequently embedded in agarose and sectioned on a vibratome produced optimal dopaminergic staining (Figures 1 and 2).
Paraformaldehyde, the fixative most commonly used in previous reports of dopaminergic histology, could partially preserve dopaminergic neurons and brain tissue; however, reported histological methods vary greatly. For example, formaldehyde was used at concentrations of 2–10% v/v (Feany and Bender 2000; Auluck et al. 2002; Davis et al. 2003; Whitworth et al. 2005; Anathbandhu et al. 2007). Some investigators embed in paraffin and section at thicknesses that vary from thin (4 μm) to thick (8 μm) (Feany and Bender 2000; Auluck et al. 2002); others embed unfixed tissue in OCT compound and section on a cryostat before formaldehyde fixation (Pesah et al. 2005). Still others image whole mount brains, performing optical sectioning with a confocal microscope (Anathbandhu et al. 2007). Some investigators use antigen retrieval to improve antigen detection (Feany and Bender 2000); still others add an antibody blocking step that consists of incubating their sectioned tissue in mammalian sera (Feany and Bender 2000; Whitworth et al. 2005; Anathbandhu et al. 2007). In all of these studies and in our own hands, cell bodies and axonal projections could be observed, but immunocytochemical results were highly variable.
We found that paraformaldehyde never produced brains that were thoroughly fixed and optimally stained, and tissue shrinkage was a frequent artifact (Figure 3). Of the 40 brains processed with paraformaldehyde fixation, 19 yielded sections adequate for scoring; 11/19 were embedded in agarose and sectioned on a vibratome, and 8/19 were embedded in paraffin and sectioned on a microtome. When embedded in paraffin, the method of fixative infiltration (i.e., simple vs facilitated by vacuum and microwave) had little effect on the histology. All paraffin sections generated with paraformaldehyde fixation generated shrinkage and sectioning artifacts (chatter). Embedding in agarose and sectioning on a vibratome produced noticeably improved results only when fixative infiltration was not enhanced; three of seven of the brains fixed with paraformaldehyde, infiltrated by simple incubation in fixative, and subsequently embedded in agarose generated sections that did not suffer from shrinkage. When paraformaldehyde was delivered using microwaves and vacuum, 100% of the brains suffered shrinkage artifacts (n = 4), suggesting that shrinkage artifacts are caused by aldehyde fixation as well as dehydration (see Discussion). Note that when dopaminergic staining was successful using aldehyde fixation, it was generally observed in the centers of brains, where rapid infiltration and fixation were unlikely to occur (Figure 3). Paraformaldehyde fixation has been used successfully to generate good maps of dopaminergic cell bodies; it is the ability to do this consistently that is problematic.
Carnoy's fixative proved to be the least effective fixative we tested. Carnoy's fixative is used frequently in studies of olfactory sensillae for its ability to penetrate insect cuticle and more recently for effective fixation of amyloid structures in neurodegenerative diseases (Weinstock and McDonald 1969; Giaccone et al. 2000). We prepared 40 brains using Carnoy's fixative but were able to score only 18 of them. Only 25% of Carnoy's fixed brains designated for paraffin embedding survived sectioning (5/20). Of the 18 scored Carnoy's brains, 17/18 suffered massive cellular destruction with or without the assistance of microwave infiltration techniques, but microwaving protocols produced consistently greater tissue damage than that observed with simple infiltration methods (Supplemental Figure 1). In our hands, Carnoy's destroyed cellular profiles so that neural tissue appeared as uniform tissue slabs (Figures 2 and 4). Occasionally (2/18) dopaminergic neurons could still be observed (Figure 4C, arrow), suggesting that, whereas Carnoy's fixative seemed to have broadly destructive effects on Drosophila brain histology, it did not penetrate tissue evenly and allowed the occasional preservation and detection of dopaminergic epitopes. We never observed dopamine-positive axons or dendrites in Carnoy's fixed tissues. Carnoy's fixative also generated significant shrinkage that was exacerbated by microwaving infiltration (Figure 4B; Supplemental Figure 1) compared with simple infiltration (Figures 2 and 4A).

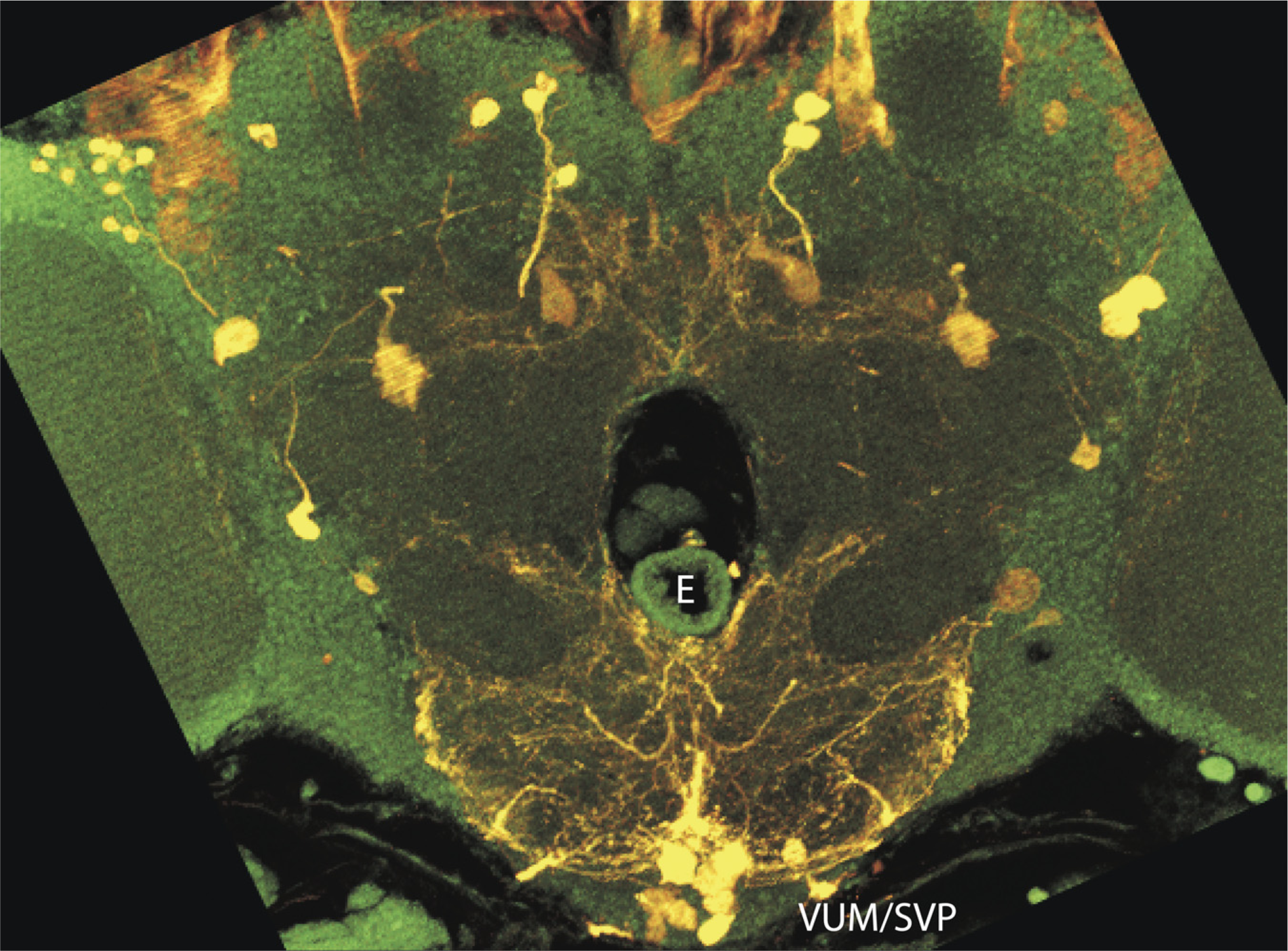
Fixation with the Nassel/Elekes fixative (NEF) protocol, using antibodies specific for Drosophila isoforms of dopaminergic synthetic enzymes generated comprehensive staining. Images were projected from confocal Z-stacks acquired on a Bio-Rad Radiance confocal microscope. Bar = ∼100 μm.
Bouin's fixative was found to preserve neuronal tissue well; however, it failed to preserve dopaminergic epitopes (Figure 2; Supplemental Figure 2). Almost all of the brains fixed in Bouin's fix and embedded in agarose produced good general histology (16/20) but never produced good dopaminergic staining. Surprisingly, Bouin's fixative was the only fixative we tested that absolutely destroyed the ability to detect dopaminergic epitopes, even when fiber tracts were clearly well preserved (Supplemental Figure 2). Of the brains prepared for paraffin embedding and microtome sectioning, 16/20 generated sections. Those infiltrated simply, without microwaves or vacuum, were poorly fixed and suffered a massive disintegration of brain tissue; those fixed with support of microwaves and vacuum generated intact sections but lacked any dopaminergic staining (Supplemental Figure 2).
The NEF with microwave infiltration was found to be the optimal protocol for the preservation of dopaminergic epitopes in Drosophila. Cell bodies and axonal projections were observed more consistently and in greater abundance than with other protocols (Figure 1). Axonal projections were so well preserved and stained that they often masked the position of cell bodies in planar views of the cross-section (Supplemental Movies SM2 and SM3). Of the 10 brains prepared using NEF with microwave/vacuum infiltration and embedded in agarose, 9/10 generated superior (compared with any other fixation method) staining of axonal and dendritic processes, 10/10 showed the additional cell bodies demonstrated in Figure 5, and none suffered from tissue disintegration or shrinkage. Of the 20 brains prepared with NEF but subsequently embedded in paraffin and sectioned using a microtome, only 10 produced sections (the majority of the remainder were not able to be sectioned). Of these 10, 3 were infiltrated using microwave and vacuum techniques, and these produced some dopaminergic staining but suffered from generally poor anatomy, and all 10 produced sectioning artifacts. Of the 10 brains fixed with NEF (but without the benefit of microwave or vacuum during infiltration) and subsequently embedded in agarose, we observed that 3/10 produced partial dopaminergic staining (Supplemental Figure 3) but only 1/10 preserved the additional neurons we report in Figure 5. In the remaining 7/10 brains, there was massive disintegration of tissue but no shrinkage.
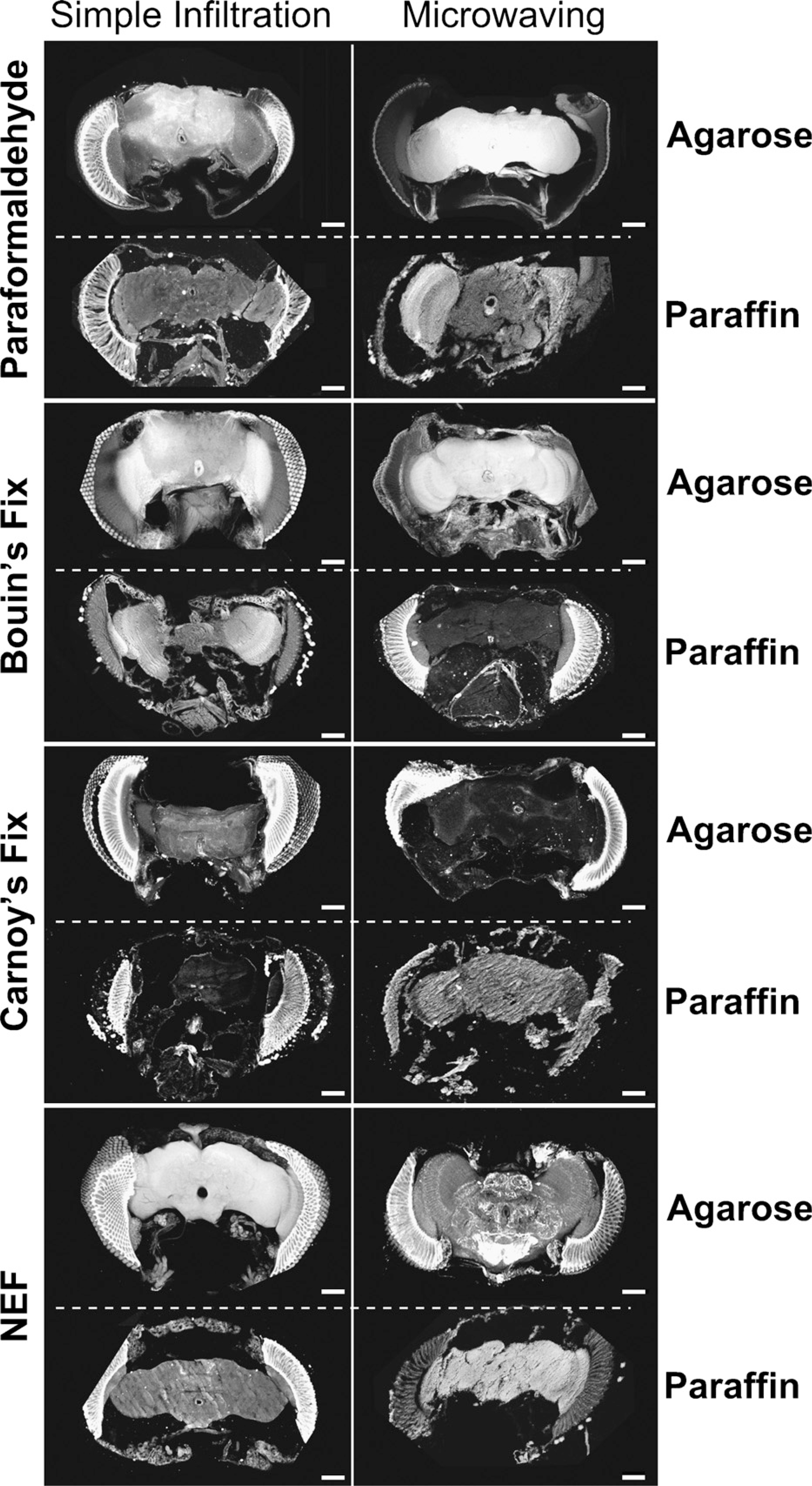
Comparison of histological protocol on efficacy of dopaminergic staining. Dissected brains were fixed with either paraformaldehyde, Carnoy's fix, Bouin's fixative, or NEF (see Materials and Methods and Supplemental Table 1). Fixes were infiltrated simply or by controlled microwaving under vacuum; brains were subsequently embedded in paraffin or agarose, sectioned on a microtome (paraffin) or vibratome (agarose), and stained identically using antibodies directed against dTH. Images are projections of confocal Z-stacks acquired on a Bio-Rad Radiance confocal microscope. Bar = ∼80 μm.
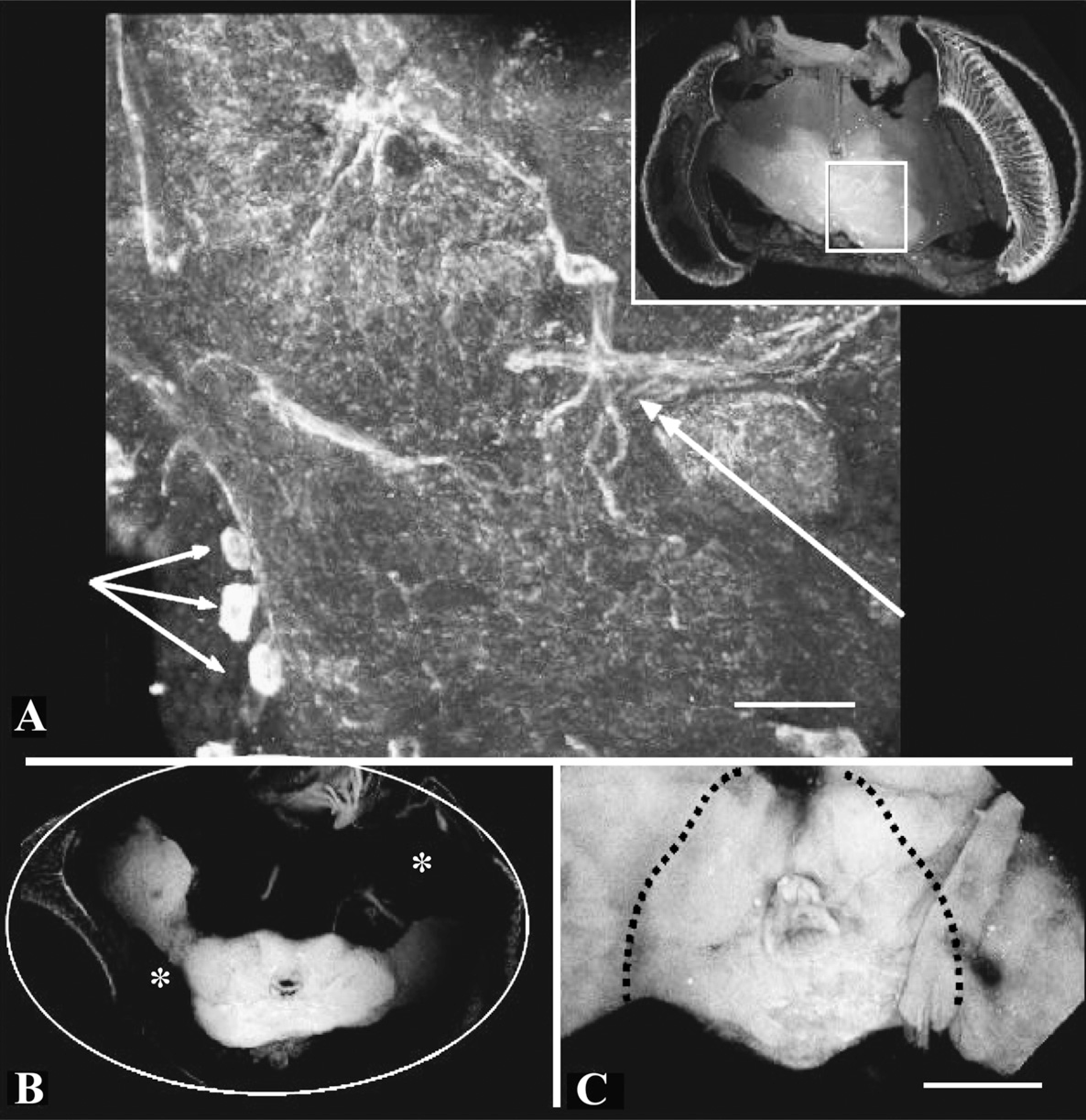
Paraformaldehyde generates artifacts in dopamine histology. Brains were fixed with paraformaldehyde, infiltrated simply or using cool microwaving techniques, embedded in agarose, and stained as described. (
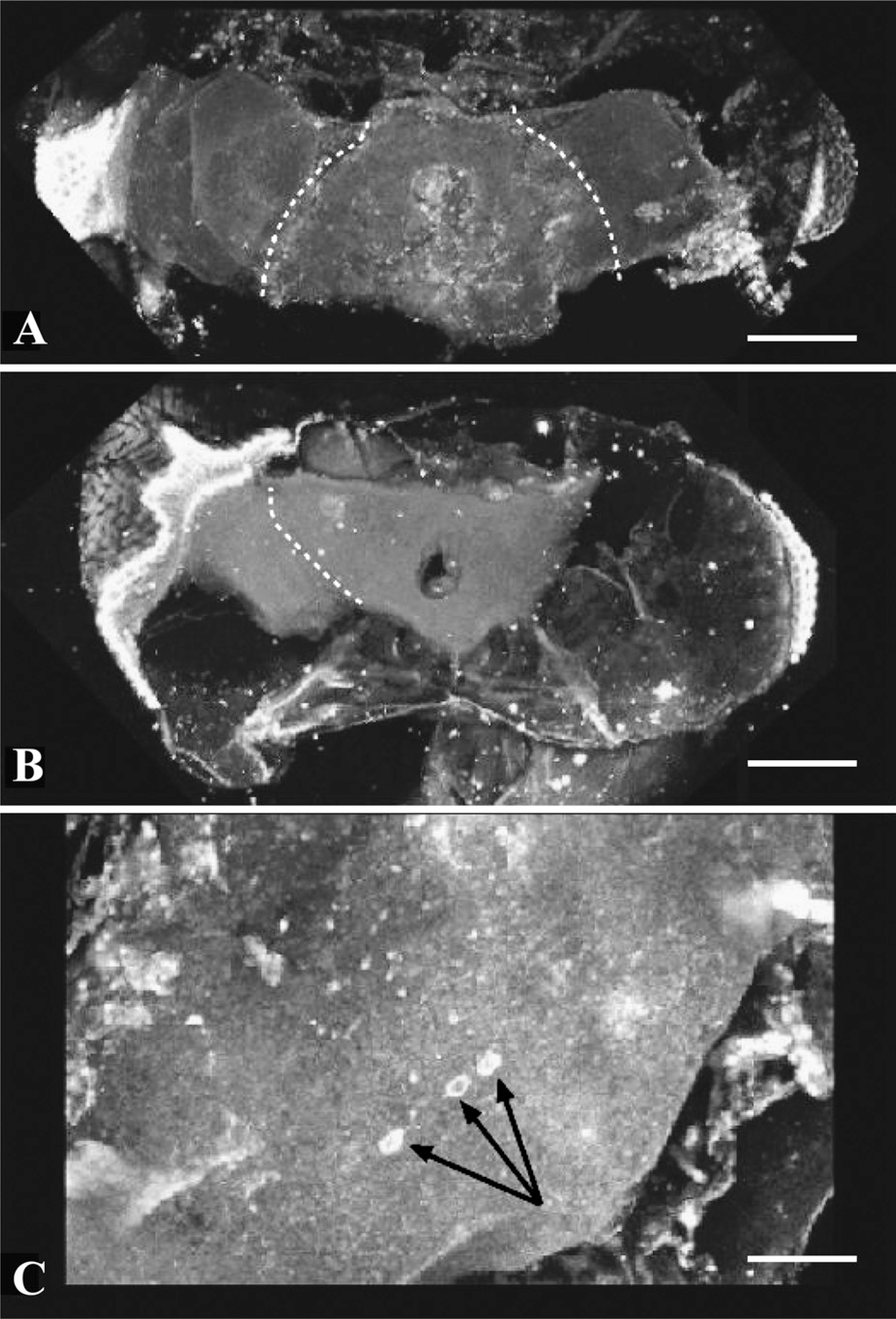
Carnoy's fixative seems to dissolve neural tissue in Drosophila. When brains were fixed in Carnoy's fixative, neural tissue was poorly fixed and lost cellular definition. Shrinkage was exacerbated by microwaving infiltration (
Microwave infiltration for weak fixes, such as NEF, was an essential feature of successful staining. As noted, without it, NEF failed to efficiently penetrate neural tissue and it subsequently disintegrated during tissue processing (NSA method; Table 1; Figures 2 and 6). We found similar results with Bouin's fixative (Figure 2). NEF contains glutaraldehyde in moderate concentrations, as well as paraformaldehyde; glutaraldehyde is known to penetrate tissues slowly and evenly, allowing even prolonged enzymatic activity in fixed tissues (Sabatini 2005). It is also known, however, for introducing elevated levels of autofluorescence and so must be used with caution in immunocytochemical protocols (N. Strausfeld, unpublished data). NEF also contains sodium metabisulfite, a commonly used anti-oxidant, believed to act in preserving the dopamine epitope, although its mechanism of action in this context is unknown.
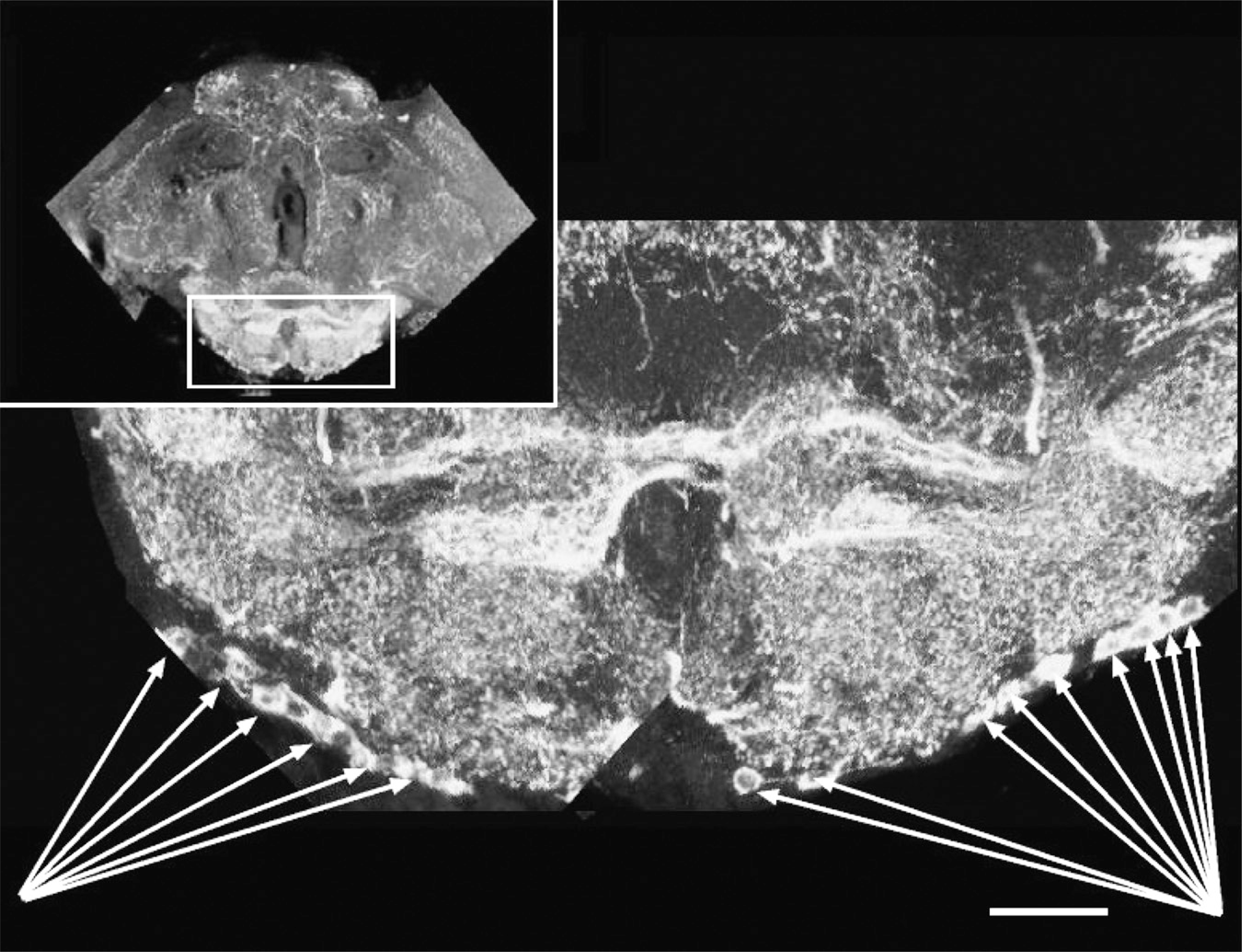
Newly identified dopaminergic neurons at ventral surface of the brain. Five to eight neurons were observed in a chain-like configuration extending from the midline laterally. Arrows demarcate cell bodies. Rectangle in inset orients subesophageal ganglion below. Images are projections of Z-stacks collected on a Bio-Rad Radiance confocal microscope. Bar = ∼55 μm.
Embedding
Our paraffin-embedded tissues produced dopaminergic histology that was significantly inferior to that generated by agarose preparations. We prepared 80 brains for agarose embedding and vibratome sectioning and an additional 80 for paraffin embedding and microtome sectioning. In the paraformaldehyde fixation group, 8/20 produced stained sections. With Carnoy's fixation, that number dropped to 5/20; using Bouin's fixative or NEF increased that number slightly to 10/20. Thus, fewer than one half of the brains we handled for paraffin embedding generated data; we believe this was primarily because of the varying levels of fixation produced by these differing protocols. This may also be caused, in part, by the increased manipulation associated with paraffin processing: paraffin processing requires more solution changes, harsher solvents, and longer exposure to elevated temperatures. Agarose embedding and sectioning were far simpler, less harsh, and produced consistently superior results.
In comparing vibratome sectioning to microtome sectioning, we observed that considerable expertise was needed to generate good microtome sections and that this was virtually impossible to do with inadequately prepared tissues. For example, we experienced significant problems with chatter (typically an artifact of sectioning related to inadequate fixation followed by rapid dehydration; see Discussion) in paraffin sections (Klatt 1994). However, we found that of all of the fixes we tested—when combined with paraffin embedding—only paraformaldehyde under microwave infiltration produced levels of dopaminergic staining that were marginally acceptable (Figure 2). Paraffin sectioning techniques were effective in producing excellent results when testing for other neuronal markers (data not shown) but seemed to fail in preserving dopaminergic epitopes consistently.
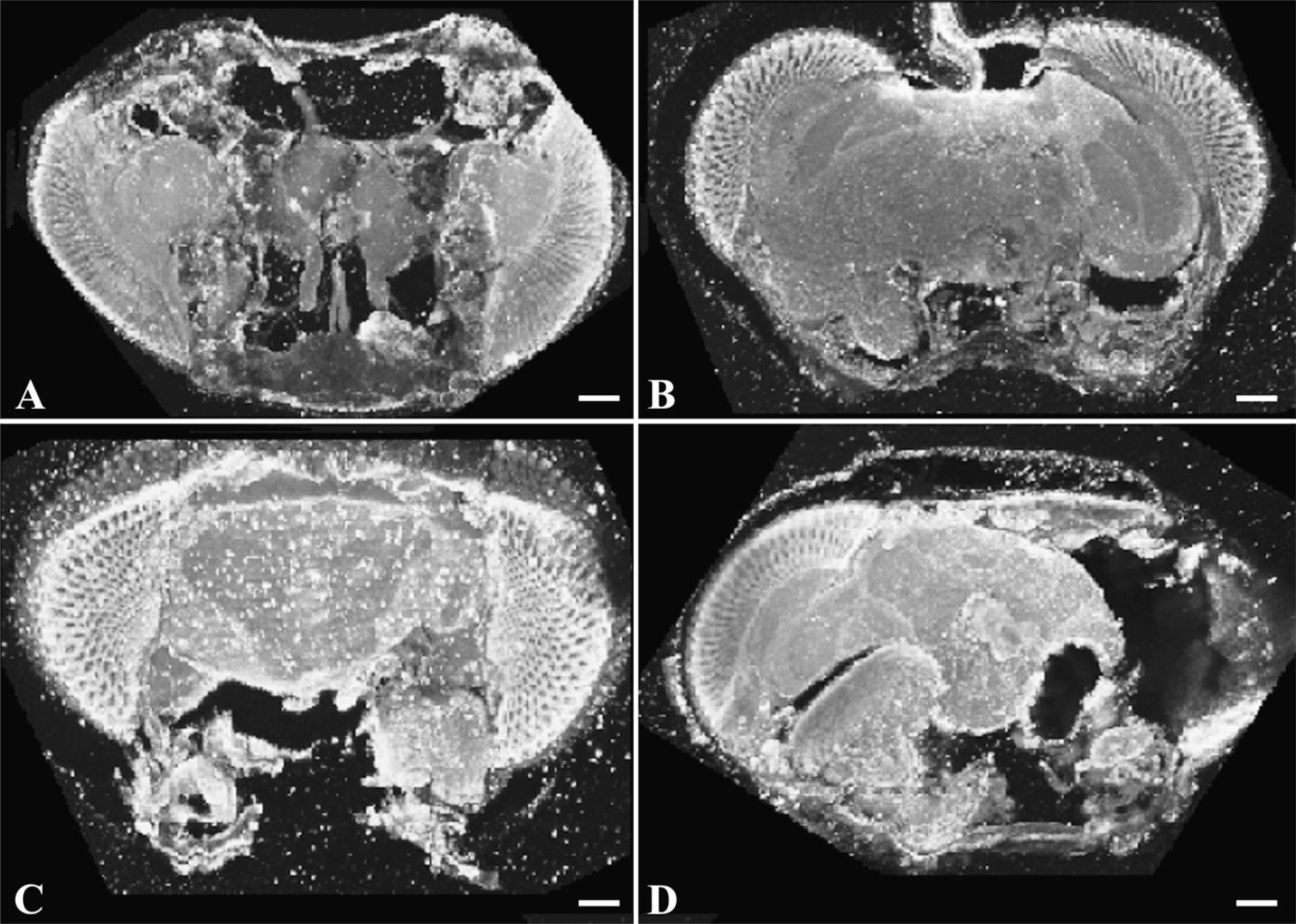
Weak fixes require microwave infiltration for good preservation of tissue. Brains were fixed in NEF, infiltrated by simple infiltration (consisting of overnight immersion at 4C), and processed in agarose as described. In every case, tissue was poorly preserved, with holes evident throughout (
The Drosophila Dopaminergic Neuronal Map
The supraesophageal insect brain consists of three major parts: the protocerebrum, the deutocerebrum, and the tritocerebrum (Figure 7, inset). The protocerebrum is contiguous with the optic lobes and comprises the majority of the brain structures, including the mushroom bodies and the core complex. The deutocerebrum consists primarily of the antennal lobes, and the trito-cerebrum consists of the brain matter on either side of the gut tube but not ventral to it (Chapman 2004). Nassel and Elekes (1992) analyzed aminergic neurons in the brains of blowflies (Calliphora) and, to a lesser extent, in Drosophila and identified the clusters of neurons listed in Table 2 and Figure 7; this nomenclature has become standard in referring to all dopaminergic staining in the adult insect brain. For our purposes, we will consider only the main portion of the brain, excluding the optic lobes. Using the margins connecting this centered protocerebrum to the optic lobes, we can divide the brain along the midline by three planes. Each of the previously defined groups of neurons can therefore be assigned to a coordinate position in the brain by the relative position to the optic lobe margins and the midline. The measurements listed in Table 2 are defined as approximate x-, y-, and z-positions, where the x-position is defined as the linear displacement from the midline (0) to the junction with the optic lobe (the lobula) (100); where the y-position is defined as the distance from the dorsal-most point of the brain (0) to the ventral most surface of the brain (100); and where the z-position refers to the anterior-posterior position, (necessarily the most difficult to quantify), with the proboscal opening (0) and the posterior surface of the head (100) as the margin extremes (Figures 1 and 2; Table 2). There was variability in the assignment of anteroposterior coordinate position, possibly because the proboscis was pulled from the anterior surface, generating some variability in the margins determining z-scaling. Because the clusters are largely symmetrical, each cluster can be considered in terms of hemispheric positioning. Note that the dorsomedial posterior protocerebral cluster 3 (PPM3) has never been reported as existing in the right half of the adult insect brain by us or any of the authors we surveyed. Although we were able to identify each of the neuronal clusters identified by Nassel and Elekes (1992) in Calliphora and Phornia, we found additional dopamine-positive neurons scattered throughout the brain that could not be unequivocally assigned to a previously identified cluster. We found ∼20 such neurons and, in addition, identified a new cluster that extends along either side of the ventral surface of the brain, originating close to the midline and extending as a chain of five to eight neurons laterally (Figures 1 and 5). Most strikingly, we observed complex neurite arborizations in the protocerebrum, long projections of the main neurite from the cell body, frequently bifurcating neurons, and highly complex staining in the subesophageal ganglia (Figures 1, 6, and 8; Supplemental Movies SM1, SM2, and SM3 and data presented elsewhere).
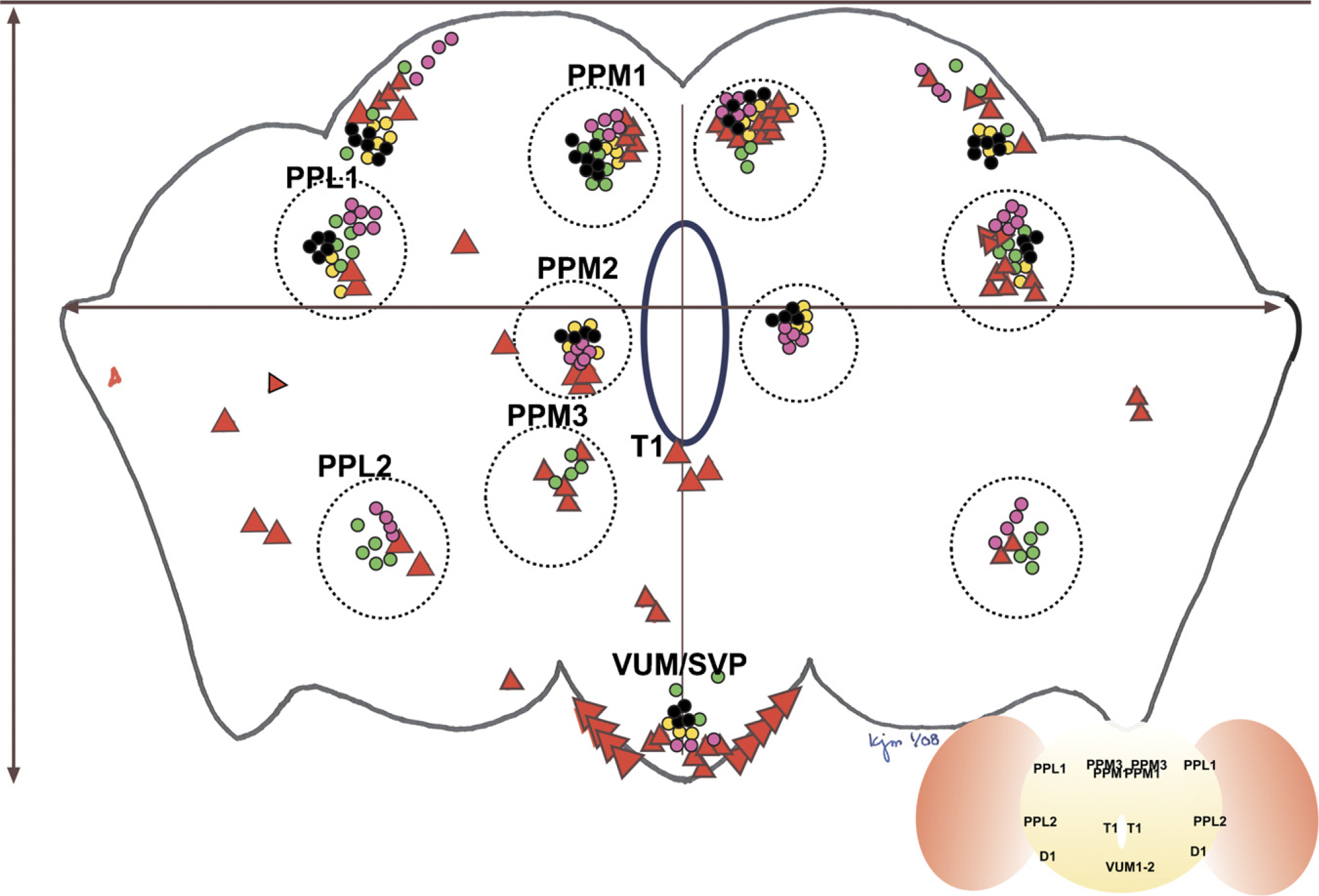
Map of dopaminergic neurons derived from representative studies reporting dopaminergic neuronal anatomy and from this report. Dotted circles surround clusters originally identified by Nassel and Elekes. Each report's neurons are marked by color-coding. Wang 2006, green circles; Friggi-Grelin 2002, pink circles; Auluck 2002, yellow circles; Pesah 2005, black circles; this report, large red triangles. A red triangle was placed when three viewers independently identified neurons in the marked location. Numbers of circles correspond to numbers or neurons reported in the reference and are not meant to identify unequivocally the number of neurons detectable by the report's staining method. Clusters that were anterior (PAL, PAM) are not included in this map; the VUMs and subesophageal ganglia have been grouped together here for the sake of simplicity. The esophageal opening is demarcated in blue. Measurement coordinates are visible as gray lines, intersecting at the esophageal opening.
It should be noted that the map of dopaminergic neuron distribution provided in Figure 7 is not meant to strictly identify the number of neurons/cluster that can be identified with that report's histology protocol. In making up the map, neurons from reported results were counted and reported; larger red triangles are derived from our own studies and are representative of the number of neurons detectable with the histological method reported here. Each of the red triangles represents the scoring of cell position by at least three independent viewers. A more comprehensive analysis of dopaminergic anatomy in the Drosophila brain is in preparation.
To determine the comprehensiveness of our staining protocol, we crossed flies homozygous for a Dopa-decarboxylase GAL4 driver (Ddc-GAL4) to flies homozygous for a membrane-bound GFP reporter gene (UAS-memGFP). Progeny expressed GFP in all subcellular compartments of dopaminergic neurons. Processing of these flies by our optimal method of fixation was followed by immunocytochemistry using two antibodies: a monoclonal antibody generated against the entirety of fluorescent proteins and a rabbit polyclonal antibody directed against dTH (Figure 8). We found these antigens to be perfectly overlapping in their cellular localization. There were no cells that stained exclusively for GFP without colocalizing dTH staining, suggesting that our results are internally consistent and validated by defining molecular expression.
Nomenclature of dopaminergic neurons as designated originally by Nassel and Elekes (1992)
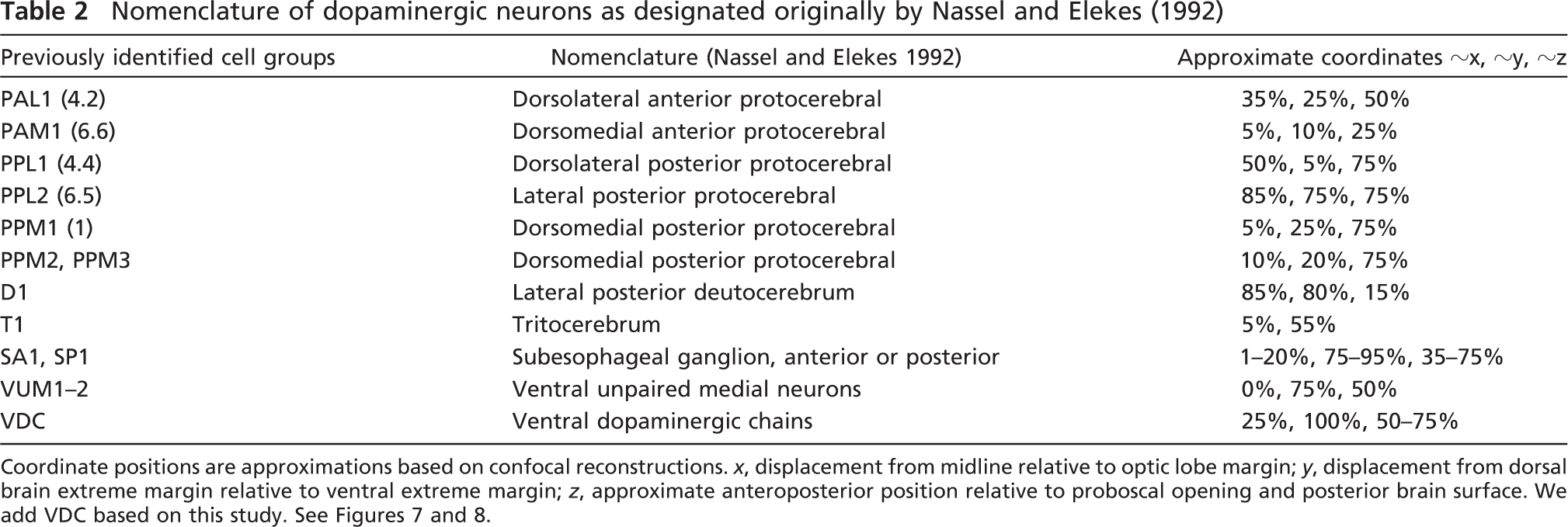
Coordinate positions are approximations based on confocal reconstructions. x, displacement from midline relative to optic lobe margin; y, displacement from dorsal brain extreme margin relative to ventral extreme margin; z, approximate anteroposterior position relative to proboscal opening and posterior brain surface. We add VDC based on this study. See Figures 7 and 8.
When we began these studies, we tested a number of antibodies for their ability to stain Drosophila dopaminergic neurons and found that all antibodies specific for vertebrate epitopes failed to stain fly brains. To determine whether these antibodies (generated against vertebrate epitopes of dopaminergic synthetic enzymes) could stain dopaminergic neurons reliably when brains were prepared by our optimized histological protocol, we repeated our initial staining experiments using four previously tested antibodies (Supplemental Figure 4). In no case did we observe staining comparable to that obtained with either the monoclonal antibody directed against GFP used in DDC-GAL4 × UAS-GFP brains or with the dTH or dTPh antibodies in wild-type brains (Supplemental Figure 4).
Discussion
Achieving excellent histological results requires balancing adequate fixation (for optimal preservation of cellular structures) against hardening techniques, for effective tissue embedding and sectioning. When used to perform modern IHC protocols, these traditional techniques can generate artifacts that are difficult to interpret.
Components of fixatives can include alcohols, acids, aldehydes, various salts, and heavy metals; these are believed to improve tissue permeability by extracting lipids, to dehydrate and harden tissue, to coagulate proteins and form rigid covalent bonds between protein side chains, to inhibit oxidation states, and to change pH (Kiernan 1981). However, a fundamental understanding of the effects of these chemicals on biological tissues remains elusive. Kiernan (1981) describes 12 ways in which fixative components might differentially affect biological specimens; these include rapidity of tissue penetration by chemicals, change in volume of tissue, rate and extent of tissue hardening, oxidation of carbohydrates, inhibition of enzyme activity, destruction of organelles, subsequent ability to stain with charged dyes, and the ability to function as a fixative alone. We considered these characteristics in relation to the four fixatives used in this study.
Carnoy's Fixative
We first tested Carnoy's fixative because a previous study had shown it was a superior fixative for neural tissues embedded in insect cuticle, such as olfactory sensillae (Richards 1952; Chapman 2004; Haider et al. 2007 for representative reports). Carnoy's fixative consists of chloroform, ethanol, and glacial acetic acid (Supplemental Table 1). Chloroform increases its ability to permeabilize tissue quickly, enabling fixative to penetrate membranes quickly (Kiernan 1981). This high concentration of chloroform, however, typically destroys the lipid-rich cellular integrity characteristic of brain structures. In addition, the high concentration of ethanol in this fixative (50% v/v) initiates a coagulation effect that denatures proteins and their epitopes. Ethanol fixation is not considered a good fixative alone and will produce tissues that are brittle, shrunken, and prone to chatter during sectioning (Klatt 1994). Concentrated glacial acetic acid is corrosive and oxidative and destroys organelles. Carnoy's fixative rarely produces satisfactory staining with charged dyes. Thus, the ability of Carnoy's fixative to penetrate dense tissues quickly does not adequately compensate for the widespread and extreme destruction observed in neural tissue; microwave infiltration, which accelerates speed of fixative delivery, enhanced this damage.
Ddc-GAL4 × UAS-memGFP transheterozygous fly brains show that our optimized method of dopaminergic histological detection is comprehensive for all dopamine-positive neurons in the adult Drosophila brain. Sections were stained with an MAb generated against full-length green fluorescent protein (GFP) and with a polyclonal antibody directed against Drosophila tyrosine hydroxylase (dTH). Alexa Fluor-conjugated goat secondary antibodies were used to identify GFP (green) and dTH (red). Images were projected from confocal Z-stacks collected on a Bio-Rad Radiance confocal microscope.
Bouin's Fixative
We tested Bouin's fixative because it had been commonly used in studies of neuronal peptides (Seroogy et al. 1988; Nassel and Elekes 1992 for representative reports). It contains picric acid, formaldehyde, and glacial acetic acid. Bouin's fixative was surprisingly effective in fixing insect neural tissues for general axonal morphology and for relatively imprecise staining protocols. This surprised us because we expected picric acid, a highly acidic phenol, to be corrosive; this may have been an unfair bias deriving from its use in manufacturing explosives (Cooper 1996). The urban mythology associated with exploding picric acid may also unfairly predispose investigators against its use, when in fact we found it to be a gentle yet effective fixative and quite worthwhile for preserving axonal and dendritic processes (Supplemental Figure 2). However, although Bouin's fixative was an effective method for preserving neuronal histology generally, it was completely ineffective in preserving insect dopaminergic epitopes specifically. Picric acid's coagulation effect may have contributed to the destruction of dopaminergic epitopes, or alternatively, tyrosine hydroxylase may be susceptible to oxidation, and the strongly acidic picric acid may alter these epitopes. Bouin's fixative penetrates tissues slowly, preserves organelles, and generates satisfactory staining with charged dyes, but is unable to act as an effective fixative by itself (Kiernan 1981). In our hands, it was observed to have improved fixative properties with the assistance of microwaving infiltration techniques.
8% Paraformaldehyde
Paraformaldehyde is the most commonly used fixative in Drosophila histology; in almost all of the studies developing a genetic model of human PD in Drosophila, it was the fixative of choice for the quantification of dopaminergic neuronal loss (Feany and Bender 2000; Auluck et al. 2002; Pendleton et al. 2002; Yang et al. 2003; Pesah et al. 2005). It has been described as penetrating tissue fairly rapidly, extracting lipids slightly, and is sufficiently strong to be used to fix tissue independent of other fixes (Kiernan 1981). We found it preserved dopaminergic epitopes but was ineffective for complete preservation of neural tissue. Paraformaldehyde fixes allowed detection of some dopaminergic neurons, but never all of them, and never identical subsets or clusters of neurons. In addition, we frequently observed brain shrinkage when using paraformaldehyde, suggesting inadequate fixation before dehydration (Klatt 1994). Shrinkage is problematic in studies of invertebrate brain histology because the brain's avascularity means that fixatives must diffuse across hundreds of cell diameters to reach their targets; when shrinkage occurs, tissues become more compact and presumably this diffusion of fixative—and subsequently, antibodies—across cell layers is additionally impeded.
Paraformaldehyde produces fixation by generating cross-linking disulfide bonds; it does not fix by coagulation. Its mechanism of action did not seem to consistently alter the dopaminergic epitopes recognized by our antibodies.
NEF
NEF is weak, containing very low concentrations of paraformaldehyde and moderate concentrations of glutaraldehyde; it also contains reducing agents (sodium metabisulfite salts) thought to preserve dopaminergic epitopes. However, we found that without the supportive effects of microwaving under vacuum for improved fixative delivery, tissue was so inadequately fixed that it disintegrated under the rigors of subsequent tissue processing (Figures 2 and 6). Thus, combining this comparatively weak fixative with rapid penetration enabled by the microwave permeabilization techniques provided an ideal combination of gentle fixation with dopaminergic epitope preservation.
In an effort to determine whether this method of fixation was indeed optimal for Drosophila dopaminergic histology, we fixed brains derived from transheterozygotes expressing Ddc-GAL4, driving membrane-bound GFP. These brains were stained with a monoclonal antibody directed against GFP and with the polyclonal antibody directed against the Drosophila ortholog of tyrosine hydroxylase used throughout this study (Figure 8). We observed three important findings. First, because the stains overlapped exactly, it is possible to use antibodies against GFP in these types of studies as long as dopaminergic epitopes are properly preserved with the protocol we report here. Although the antibodies generated against Drosophila tyrosine hydroxylase and tyrosine phosphatase are specific and work well, they are limited in availability; however, this study showed that alternatives can be used. Second, all of the dopaminergic neurons within the protocerebrum colocalized these antibodies (i.e., there were no cells that were exclusively dTH- or GFP-positive within the protocerebrum), suggesting that we are detecting all of the dopaminergic neurons present in these brains. Finally, we did detect autofluorescence in the cell body cortex that entirely encapsulates the protocerebrum and the inner margins of the two lobulas, a common artifact derived from moderate to high levels of glutaraldehyde fixation (N. Strausfeld, unpublished data). We take this autofluorescence to be an acceptable artifact of fixation, because it does not confound observations related to dopaminergic staining.
Embedding and Sectioning
Artifacts that appear in stained sections are usually the result of poor fixation, poor embedding medium infiltration, or poor sectioning techniques. Clearly the efficacy of fixation varied between our treatment groups and was the major source of variability in histological results. However, when we compared embedding techniques across treatment groups, we observed poor infiltration only when shrinkage seemed to be problematic (see 8% Paraformaldehyde and Carnoy's Fixation). Infiltrating brains with paraffin while under vacuum (supplying −20 psi vacuum pressure for 24 hr at 56C) pulled paraffin into tissue. We used Paraplast Plus, containing a substantial concentration of DMSO, which also facilitated tissue infiltration. Altogether, infiltration was not likely a source of error in tissues that produced unsatisfactory staining results.
It is more likely that choice of fixative, method of fixative delivery, and denaturing techniques affected our results most profoundly. Some tissues were so poorly fixed that infiltration and subsequent sectioning were extremely difficult (Figures 2 and 6). Extended exposure to heat may denature poorly fixed proteins, further affecting epitope conformation and the ability of antibodies to recognize their molecular targets. Ethanol and xylenes dehydration may further harden and fix tissues, providing an additional source of difficulty in achieving effective immunocytochemical staining.
Agarose embedding circumvents most of these problems. Tissue is fixed, rinsed, blotted, and embedded, without dehydration or extended heating. One disadvantage of agarose embedding is the lack of a vacuum step: as a result, one might expect embedding medium to pull away from brain tissue, causing brain sections to be lost during solution changes. Agarose did appear to occasionally pull away from brain specimens that had been inadequately blotted. However, careful drying of the exoskeleton before agarose embedding solved this problem; it appeared that agarose adhered to exoskeleton adequately in the absence of a water interface.
We believe that we have found the optimal protocol for dopaminergic neuronal staining in Drosophila. It combines a weak fixative with cooled microwave infiltration under vacuum, buffers that may provide anti-oxidant protection of dopaminergic epitopes, and agarose embedding combined with thick sectioning on vibratomes. Specific, comprehensive, and reliable staining can be achieved using antibodies specific for the insect orthologs of enzymes needed for the synthesis of dopamine or using antibodies specific for GFP when GFP has been expressed in dopaminergic neurons, thus relieving dependency on an antibody that is not commercially available. We are in the process of applying this technique and newly developed maps of dopaminergic neurons in Drosophila brains to studies of dopaminergic toxins in models of idiopathic PD in Drosophila.
Footnotes
Acknowledgements
We thank Dr. Kathryn Swoboda for helpful advice and financial support; Dr. Ken Metzger and his colleagues at the University of Utah pathology laboratories for expertise and guidance; Dr. Nicholas Strausfeld and members of the Strausfeld laboratory for continued advice and help; Drs. Jacinda Sampson, Anthea Letsou, Nina Tang-Sherwood, Anna Salazar, and Rachel Kraut for helpful comments on the manuscript; and the members of the Schmid laboratory past and present for insights and scientific discussion.