
Abstract
On the surface of endothelial cells (ECs) lies the glycocalyx, a barrier of polysaccharides that isolates the ECs from the blood. The role of the glycocalyx is dynamic and complex, thanks to not only its structure, but its vast number of components, one being hyaluronan (HA). HA is a critical component of the glycocalyx, having been found to have a wide variety of functions depending on its molecular weight, its modification, and receptor–ligand interactions. As HA and viscous blood are in constant contact, HA can transmit mechanosensory information directly to the cytoskeleton of the ECs. The degradation and synthesis of HA directly alters the permeability of the EC barrier; HA modulation not only alters the physical barrier but also can signal the initiation of other pathways. EC proliferation and angiogenesis are in part regulated by HA fragmentation, HA-dependent receptor binding, and downstream signals. The interaction between the CD44 receptor and HA is a driving force behind leukocyte recruitment, but each class of leukocyte still interacts with HA in unique ways during inflammation. HA regulates a diverse repertoire of EC functions.
Keywords
Introduction
The endothelium is covered in a dynamic, polysaccharide rich layer which lines the apical surface of endothelial cells (ECs) and extends into the lumen of vessels. The EC surface layer is a unique form of extracellular matrix (ECM) distinct in composition and function from the matrix found in other tissues. 1 This ECM is often referred to as the glycocalyx and lies juxtaposed between blood and the endothelial surface. The first ultrastructural visualization of the glycocalyx used the cationic-dye ruthenium red, which binds to acidic and charged polysaccharides producing electron density in the presence of osmium tetroxide. 2 Experimental evidence demonstrates that proteoglycans (PGs), glycoproteins, glycolipids, and glycosaminoglycans (GAGs) together comprise the glycocalyx, and serve as the functional interface between serum, blood cells, and the endothelium.3,4 New imaging techniques combined with fluorescent probes have been used to specifically label major components of the glycocalyx.5,6 These approaches have been combined with enzymatic degradation of the glycocalyx to determine the relative thickness within the lumen vessels, revealing a much thicker glycan layer in large vessels ranging from 2.2 µm to 11 µm and ranging from 0.1 µm to 3.02 µm in microvessels.3,7–10 Due to its location, the glycocalyx plays important roles in homeostatic vascular function which when disrupted contribute to disease, such as in atherosclerosis.11,12 In particular, diabetes and dysregulated glucose metabolism is characterized by significant alterations of the glycocalyx and its components.13–17 These roles are determined by composition, structure, and relative abundance. Components of the glycocalyx regulate many important physiological processes including outside-in signaling, immune cell recruitment, angiogenesis, and coagulation, together playing a key role in vascular integrity.6,18–22
While the specific thickness of the glycocalyx may be dependent on the type of vessel, the major GAG constituents include heparan sulfate, chondroitin sulfate, and hyaluronan (HA). 6 Whether the relative abundance of each GAG varies by vessel or tissue requires further study. HA is distinguished from the other GAGs as it is the only non-sulfated GAG in the glycocalyx, and it is not covalently attached to proteins, but rather bound and tethered to the cell surface by HA-binding proteins. 23 While other GAGs are synthesized in Golgi compartments as a post-translational modification of a PG core protein, HA synthases embedded in the EC plasma membrane assemble HA at the cell surface and extruded the growing HA polymer directly into the extracellular space. At the cell surface, HA can achieve an extremely high degree of polymerization, reaching a molecular weight >1 mDa. HA is presumed to intertwine through elements of the glycocalyx, and can function as a scaffold capable of forming microdomains for binding and clustering of appropriate receptors. 24 At the EC surface, HA is observed as a network covering the luminal surface, intercalated between various PGs forming a “gel-like” layer which acts as a molecular sieve of the luminal capillary. 25 Many functions governed by HA depend upon interaction with binding proteins or HA-receptors reviewed comprehensively by Day and Prestwich. 26 Of these, Stabilin-2, which was discovered as the liver hyaluronan receptor for endocytosis (HARE) and the lymphatic vessel endothelial receptor (LYVE-1) play key roles in HA turnover in the vascular systems.27,28 A growing body of literature implicates HA production and turnover as dynamic events capable of regulating cellular responses in polymer size and binding-partner-dependent context. While many receptors and binding proteins are known to interact with HA, the functions of HA and its receptors as they pertain to EC behaviors are described in detail in this review.
HA Regulates Endothelial Behavior in Response to Shear Forces
ECs are exposed to dynamic extracellular stimuli at the luminal surface by the flow of blood and its components. Within the glycocalyx, HA and its binding proteins can transmit these extracellular signals via “outside-in” signaling, which functions as a mechanosensory transducer regulating several critical EC behaviors. 29 HA synthases are primarily regulated at the level of transcription, and the resulting HA polymer is regulated by the coordinated activity of hyaluronan synthase (HAS), hyaluronidase, substrate availability, and other factors such as reactive oxygen species and ultra-violet light.30–33 The HAS enzymes are present at low levels at the plasma membrane until activated in response to various stimuli. Expression of the HAS enzymes and HA abundance varies based upon vessel origin and size. 34 In macrovessels, both HAS2 and HAS3 mRNAs are present, 35 while only HAS3 is detected in human intestinal microvessels, 36 and all three HAS enzymes are present in corneal ECs. 37 Despite this heterogeneity, HA synthesis in most vascular and lymphatic ECs is responsive to induction by pro-inflammatory mediators such as interleukin (IL)-1β, IL15, TNF-α, and lipopolysaccharide.38,39 Ablation of HA production in mice by administration of 4-methylumbelliferone (an inhibitor of GAG synthesis) results in widespread EC dysfunction, increased macrophage driven vascular plaque inflammation and atherosclerosis. 40
At the EC surface, CD44 tethers HA to the plasma membrane and, through its cytoplasmic tail, interacts with actin-binding ERM proteins. 41 Fluid shear stress may then be transmitted via HA-CD44 in the vessel lumen directly to the EC cytoskeleton. For example, in response to shear forces, CD44 activates Rac1 while inhibiting RhoA and promotes localization of adductin-γ to tight junctions strengthening the EC barrier. 42 Loss of CD44 leads to increased vessel permeability and release of HA from the EC surface. HA itself can be induced by shear forces, though whether this is regulated by CD44 or other binding proteins is not known. In vitro studies of ECs subject to arterial pulsatile flow conditions promotes increased hyaluronan synthase 2 (HAS2) expression and HA production via the Akt pathway. 43
While flow is capable of inducing HA synthesis, HA and CD44 interactions modulate EC spreading and production of the vasodilator, nitric oxide (NO). 44 This is consistent with early implications of the glycocalyx as a regulator of flow-induced vasorelaxation, and with clinical observations indicating that HA destruction at the EC surface in diabetes leads to a decrease in NO production.45–48 A dominant mechanism that defines EC dysfunction is reduced constitutive endothelial nitric oxide synthase (eNOS) expression. In the context of vascular lesions, continuous exposure of endothelia to high shear can impair NO production, which in turn correlates with the loss of pericellular HA. Femoral arteries perfused with hyaluronidase decreased endothelial NO synthesis, consistent with the aforementioned direct correlation of HA and NO. 49 Hyaluronidase treatment of cultured, primary bovine aortic ECs also results in a dramatic loss of shear-induced NO, but does not affect prostacyclin synthesis. 50 As hyaluronidase treatment had no effect upon prostacyclin production, these data suggest that the HA-dependent mechanism exists independent of the cellular compartment containing NO production.
Regulation of the Endothelial Barrier
Maintenance of endothelial barrier function is a critical process in which HA plays numerous roles. On the luminal surface of the capillary, HA maintains and controls microvascular permeability by acting as a molecular sieve, restricting access of inflammatory molecules and cells while also regulating endothelial cell–cell junctions. Several studies have demonstrated that treatment with hyaluronidase leads to a permissive capillary barrier allowing plasma proteins to enter tissue resulting in edema and proteinuria.3,10,51 Enzymatic degradation of HA at the EC surface can result in a compromised EC barrier, as small HA fragments can induce blood–brain barrier permeability in a CD44-dependent manner. 52 While CD44-null mice do not exhibit elevated vascular permeability at baseline compared with controls, upon histamine challenge, barrier function is significantly compromised in CD44-deficient mice. 53 Loss of CD44 leads to a corresponding decrease in platelet-endothelial cell adhesion molecule-1 (PECAM-1) and VE-cadherin in cultured ECs and inactivation of the Hippo pathway. 54 Restoration of CD44 expression leads to a recovery of both VE-cadherin and CD31 expression as well as barrier function. Loss of CD44 appears to lead to widespread downstream EC dysfunction with impaired formation and maintenance of both adherens and tight junctions. While HA likely plays a role in these observations, its specific role remains to be defined.
Other aspects of barrier function by CD44 have been demonstrated to depend upon its interactions with HA at the EC surface. For instance, high molecular weight (HMW)-HA bound to CD44 recruits the S1P1 receptor into lipid rafts leading to AKT-mediated phosphorylation of S1P1 and enhancement of EC barrier function. 55 HA fragments may compete for HMW-HA binding to CD44 leading to downstream activation of the S1P3 receptor via ROCK1/2 and subsequent loss of barrier function. 55 Administration of HMW-HA in a rat model of ventilator-induced acute lung injury exhibits protective effects by enhancing EC barrier function and reducing fluid accumulation. 56 HA also has been shown to bind angiopoietin-1 and regulate Tie2 receptor signaling in the control of glomerular endothelial barrier function. 57
Thrombin is a potent inducer of EC permeability and disrupts the EC barrier by activation of protease activated receptors (PARs). In a similar fashion, the activity of factor VII activating protease is also known as HA binding protein 2 (HABP2), and promotes EC barrier disruption in a mechanism similar to thrombin. Treatment of ECs with HABP2 leads to barrier disruption by activation of PAR-1 and 3. Co-stimulation of ECs with HABP2 and either HMW-HA or HA fragments demonstrates that HMW-HA inhibits HABP2 activity while HA fragments promotes protease activity. 58 Thrombin has also been shown to bind to HA and its proteolytic activity can regulate monocyte adhesion to HA in inflammatory settings. 59 Whether HA may play a general role in the regulation of PAR activation by thrombin and other proteases requires further study.
Alteration of HA at the EC surface at either the level of synthesis or degradation can control permeability of both transmigrating cells and cytokines and when dysregulated may promote disease.
Control of Angiogenesis and Vascularization
The regulation of EC proliferation, migration, and capillary formation is regulated not only by the activities of well-studied growth factors (e.g., VEGF, fibroblast growth factor [FGF]) but also by components of the ECM, including HA. 60 Angiogenesis is initiated by localized release of pro- and anti-angiogenic factors from ECs in response to disease, injury-induced inflammation, hypoxia, and other conditions. Inhibiting HA synthesis through treatment of ECs with 4-MU suppressed proliferation in cultured cells, as well as angiogenesis in a rodent model of endometriosis without significant changes in the number of proliferating or apoptotic cells. 61 Large polymers of HA in the range of 106-107 Da support EC homeostasis by inhibiting proliferation, migration, angiogenesis, and inflammation, while HA fragments generated by hyaluronidases in turn drive EC proliferation,62,63 migration, 64 and secretion of angiogenic factors. 65
Control of angiogenic and proliferative responses in ECs by HA depends upon polymer size and differential involvement of at least two HA receptors: CD44 and receptor for HA-mediated motility (RHAMM). Both receptors are present at the EC surface and interact with HA, yet govern different EC functions. Neutralization of CD44 with blocking antibodies suppresses the proliferative effect of HA, while blockade of RHAMM has no effect on proliferation under the same conditions. 66 Under steady-state conditions, CD44-HA interactions can suppress EC proliferation and expression of early response genes c-fos and c-jun in an HA-size dependent fashion. 67 In this context, small HA oligosaccharides promote proliferation while high-molecular weight polymers demonstrate a suppressive effect. 52 Neutralization of HA via a soluble recombinant HA-binding domain of CD44 inhibits EC proliferation, similar to antibody-mediated inhibition, and also led to an inhibition of tumor-associated angiogenesis in vivo in both chick and murine models. 68 It is possible that HA may exert these opposing effects based on whether cellular receptors are engaged in monovalent clusters or in a distributed, polyvalent manner. Furthermore, treatment with either HA oligosaccharides, high-molecular weight HA, or soluble receptor decoys may alter receptor organization at the cell surface.
CD44 and RHAMM also differ in their ability to support EC adhesion to HA. Whereas CD44 promotes HUVEC adhesion to immobilized HA, 69 RHAMM does not promote adhesion, but can retain soluble HA at the EC surface. 66 This seems in agreement with data suggesting CD44-HA interactions with HA fragments generated during angiogenesis can promote proliferation, as adhesion and spreading are essential in EC proliferation. Both CD44 and RHAMM can support EC migration, and neutralization of either receptor inhibits EC tube formation in vitro. However, examination of a murine neovascularization model, in which vessels form within subcutaneous Matrigel plugs containing bFGF, antibody-mediated neutralization of CD44 shows little effect on vessel formation, while anti-RHAMM inhibits the angiogenic response entirely. 66 Modification of HA with the heavy chains of inter-alpha trypsin inhibitor (IαI) promotes CD44 binding 70 and mice deficient for IαI also have complete loss of angiogenesis in the matrigel model, but only impaired angiogenesis in a model of bleomycin-induced lung injury. 71
CD44 and RHAMM likely act in a synergistic fashion in some contexts but have differential requirements in HA-mediated angiogenesis. In support of this, studies have indeed demonstrated that CD44 and RHAMM work in concert. HMW-HA (~1 mDa) induces activation of PKCδ via CD44, leading to co-localization of RHAMM with ERK, and activation of TGFBR1. Activation of TGFBR1 is independent of smad2, and promotes vessel remodeling by induction of plasminogen activator inhibitor-1 and MMP2. 72 Single knock down studies of CD44 or RHAMM both inhibit responses to low molecular weight HA induced EC tube formation in matrigel through regulation of differential pathways but share responses via ERK1/2 and cdk1/2. Knock down of CD44 abolishes PKC-α, actin stress fiber formation, and induction of MMP9, whereas knock down of RHAMM appears to lead to activation of glycogen synthase serine kinase 3a. 73
HA as a Novel Immune Cell Recruitment Molecule
The presence of a “cable-like” monocyte-adhesive form of HA at the endothelial surface in response to inflammatory stimuli was first reported by de la Motte and colleagues and is implicated as an early event in the progression of inflammatory diseases (Fig. 1). 21 Earlier studies from Siegelman and colleagues demonstrated that induction of HA by both TNF-α and IL-1 stimulated ECs from small vessels (but not from large vessels) is capable of mediating leukocyte adhesion 23 and sufficient to support rolling adhesions under physiological laminar flow similar to the selectins. 74
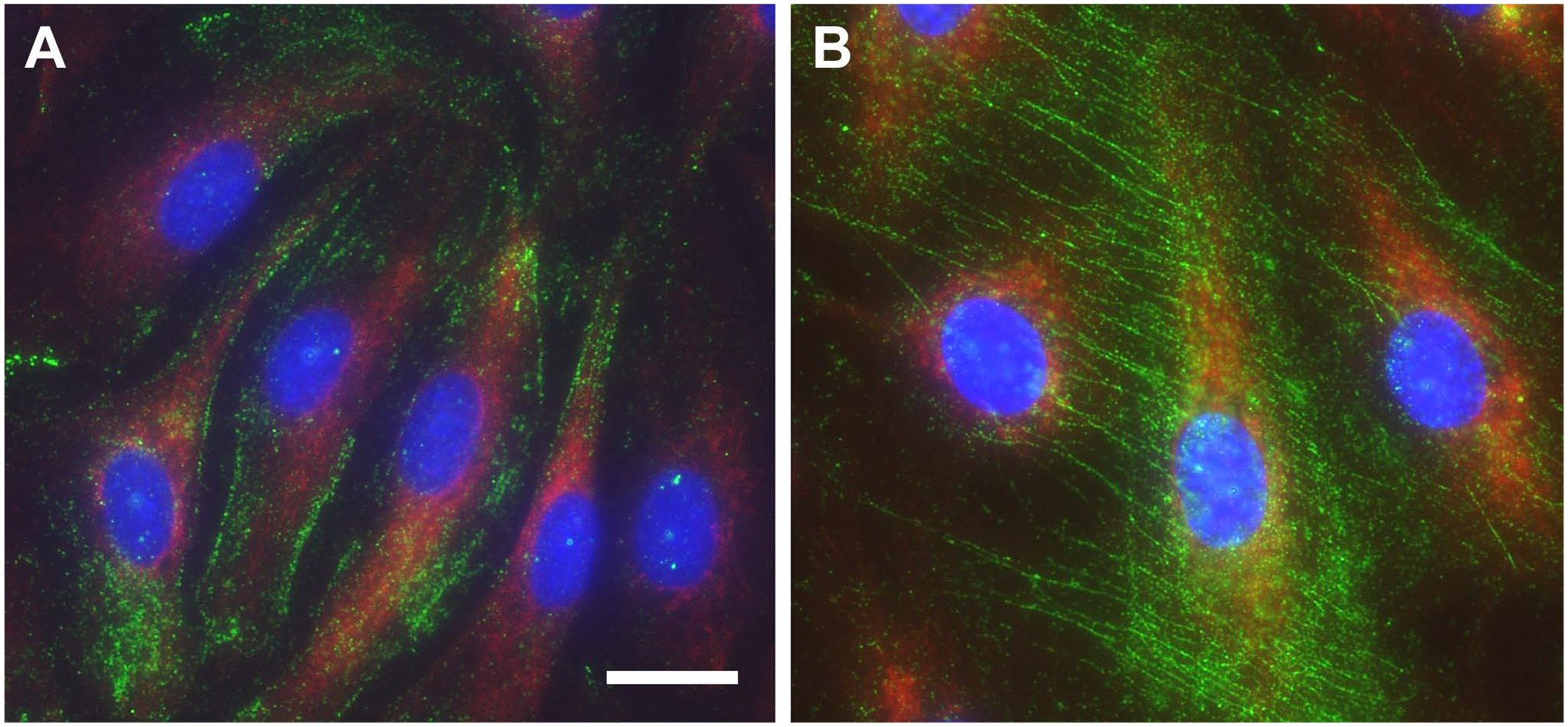
Endothelial cells produce adhesive “cable like” hyaluronan in response to inflammatory stimuli. Human lung microvascular endothelial cells were stained for HA (green), CD31(red), and nuclei (blue). (A) Under normal conditions HA is present as a pericellular coat that is non-adhesive. (B) In response to an inciting stimulus (e.g., poly (I:C)), HA synthesis is increased, and HA adopts a “cable-like” adhesive structure. Scale bar indicates 20 μm. Abbreviation: HA, hyaluronan.
CD44, the best studied receptor for HA, is constitutively expressed on leukocytes in an inactive form under resting conditions. On the EC surface, HA acts as a ligand for this receptor, allowing leukocytes to roll on pericellular HA. The interplay between receptor and ligand is not only limited to leukocyte-endothelial cell binding, as it also performs differential roles in leukocyte recruitment and activation.
For T-cells, rolling is not the only CD44-HA interaction during inflammation. Anti-inflammatory HMW-HA has been shown in vitro to upregulate FoxP3 in human regulatory T-cells, as well as upregulate the suppressive ability of these Treg cells. 75 While cytotoxic T-cells express the necessary CD44 receptor to bind to and roll along HA surfaces, these cells cannot effectively travel to sites of inflammation without a mechanism that halts their circulation. In vitro studies in liver sinusoids show that CD8+ T-cells require platelets, aggregating to the tissue by HA-CD44 interactions, as docking sites before continuing to roll along the ECM toward the inflamed site.76,77 However, the mechanism for T-cell-platelet docking has not been resolved. While this T cell-platelet interaction has only been scrutinized within liver cells, this platelet CD44-dependent recruitment mechanism likely occurs in many other inflammatory disease states.
While platelets do not bind to HA on the surface of ECs under normal conditions, platelets interact with HA produced by the inflamed vasculature during disease. 78 Once bound, platelets translocate hyaluronidase-2 (HYAL2) from within alpha-granules to the platelet surface, and subsequently degrade HA polymers from the cell surface.79,80 HA fragments generated by HYAL2 can stimulate activation of monocytes, macrophages, and ECs presumably through toll-like receptors (TLR) 2 and 4.36,81 This initiates activation of these cells, as well as the release of pro-inflammatory cytokines including IL-12, IL-1b, and TNF-a. 80 Degradation of HA by platelets has been demonstrated to regulate leukocyte transmigration, 78 and may function to control leukocyte recruitment to sites of inflammation in various disease states. In the case of lung inflammation, both TLRs 2 and 4 play crucial roles in the inflammatory response to HA fragments, but the specific contributions of platelets and ECs in this process is not well studied. 82
In the context of eosinophils, Ohkawara et al. 83 have shown that the interaction of soluble HA and CD44 expressed on eosinophils lead to aggregation of the granulocytes and the release of TGF-B. Eosinophil-derived TGF-B has been implicated in airway remodeling during allergic asthma, and TGF-B at low concentrations can induce eosinophil recruitment; together these suggest that HA-CD44 interactions could be a main driver for inflammatory reactions such as asthma.84,85 This interaction has not yet been studied in neutrophils, but as both cells are granulocytes, there is a high likelihood that neutrophils utilize this same mechanism as well.
Like other leukocytes, neutrophils bind to the endothelium in response to inflammation in part due to CD44-HA interaction, as seen in liver injury. However, the interactions between HA and neutrophils are hardly limited to HA at the EC surface. 86 Soluble HA acts as a priming agent of neutrophils, activating GSK-3 pathways, but does not mediate neutrophil aggregation and movement, unlike HMW-HA. 87 Recently, Leng et al. proposed that the peptide ECRG4 is released by healthy liver tissue and downregulates the expression of CD44 in neutrophils. During tissue injury or inflammation; however, the concentration of produced ECRG4 decreases, resulting in the increased expression of neutrophil CD44. This regulation, coupled with the HA fragments released during inflammation that activate CD44, possibly recruits active neutrophils to inflamed areas. 88
Dendritic cells (DC) and macrophages are unique among the leukocytes as they present extracellular HA on their surface.89–91 This HA “coat” allows DCs and macrophages to bind to LYVE-1), a receptor structurally related to CD44, expressed in lymphatic endothelial tissues. 89 While CD44 is the predominant HA receptor on vascular ECs, LYVE-1 exists as the principle receptor for pericellular HA in the lymphatic endothelium, where it is located at distinct overlapping junctions of the lymphatic capillaries. This unique expression pattern regulates the entry and migration of antigen-presenting DCs within the lymph node. DCs migrate toward the CCL21 gradient at the surface of the lymphatic vessels where LYVE-1 engages HA present on the DC surface, allowing the DC to dock with lymphatic trans-migratory cup structures which mediate DC adhesion and subsequent entry into the lymphatic system.92,93 In the case of macrophages, LYVE-1 interactions with HA is required for macrophage clearance by the lymphatics and resolution of myocardial infarction. 91 Recent studies demonstrate that cortical actin networks regulate LYVE-1 lateral diffusion and receptor clustering in the lymphatic endothelium, supporting the observation that selective engagement of LYVE-1 with HA within the glycocalyx facilitates leukocyte adhesion and transmigration. 94 In contrast to LYVE-1 which does not interact with the cytoskeleton but rather appears constrained by it, CD44 binds to the actin cytoskeleton via actin adapter proteins to promote CD44 clustering.41,95 Understanding cues that promote in vivo dynamic clustering which allows LYVE-1 to discriminate HA present on the surface of leukocytes from soluble HA requires further study. Unlike other endothelial beds, LYVE-1 is present in liver and spleen vascular sinuses, but its role in these locations is not established.
In conclusion, the study of HA and its interacting partners with respect to the endothelium in both health and disease has revealed that HA is capable of regulating a diverse repertoire of functions summarized in this review (Fig. 2). The elucidation of HA-dependent mechanisms that regulate EC function is crucial as it has significant roles in health and clear impact upon virtually all inflammatory disease states. Importantly, work discussed in this article highlights the functions of HA beyond that of a structural component of the ECM. While there has been significant emphasis on particular HA receptors such as CD44, more extensive studies are needed to delineate the roles of other HA-binding proteins such as LYVE-1 and HABP2, both of which may play significant roles in vascular disease. A number of elegant studies discussed within this review demonstrate that HA and LYVE-1 interactions are critical regulators of leukocyte egress from tissue during disease. Understanding how LYVE-1 and HA are regulated in the context of chronic inflammatory diseases may provide a unique opportunity to tip the balance in favor of resolution by developing therapeutic strategies to promote LYVE-1 clustering. The endothelium is intimately associated with inflammation, progression, and resolution of disease in essentially all tissues and organs. Although HA has emerged as a novel immune recruitment molecule, it is still not well understood how HA modified with the heavy chains of IαI contributes to disease progression or resolution. Circulating HA levels associate with several inflammatory conditions, as does circulating levels of heavy chain modified HA, suggesting HA is released by the endothelium. The regulatory networks which control HAS expression and HA synthesis have received considerably more study than the corresponding hyaluronidases and represent an important area of future study. Understanding the primary mechanisms by which HA is released from the endothelium either by the activity of hyaluronidases, proteolytic “sheddases” releasing the HA-binding CD44 ectodomain, or direct secretion of HA by HAS enzymes would allow for a more nuanced understanding of how HA regulates immune cell recruitment and EC functions. While glycocalyx degradation can promote immune cell and platelet adhesion to the endothelium, it may also be possible that HA shed into circulation might act as a decoy ligand or act as a signaling molecule for circulating immune cells. From this point of view, further defining the mechanisms by which HA and its binding proteins participate in endothelial regulation may present innovative strategies for vascular pathologies.
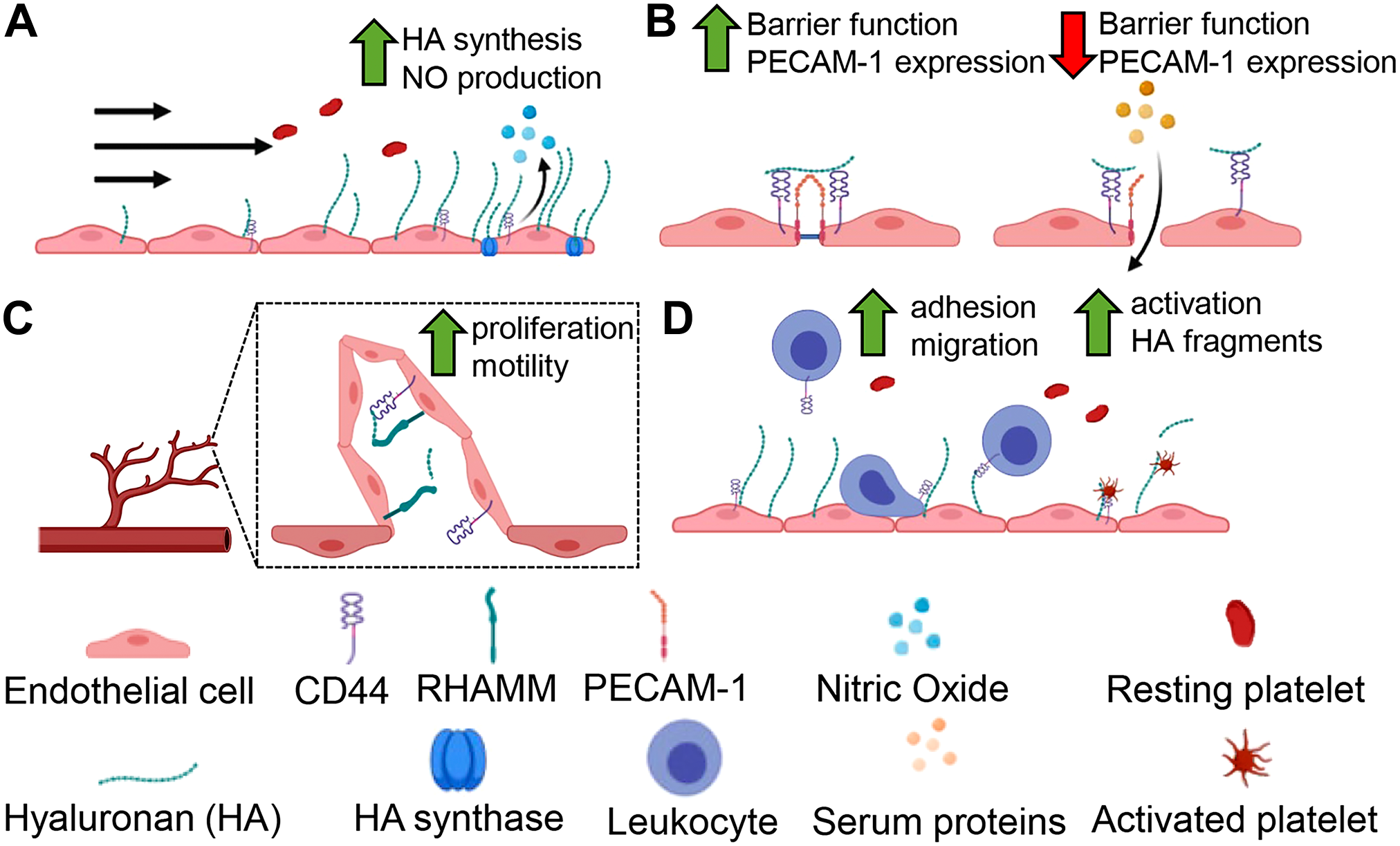
Critical endothelial cell functions are regulated by hyaluronan (HA). HA plays a role regulating diverse but important endothelial cell behaviors. In response to shear forces (A) HA synthesis is increased and promotes NO production. (B) At endothelial cell junctions, HA participates in signaling responses in barrier integrity mediated by CD44. Upon loss of CD44 and tonic HA signals, PECAM-1 is disrupted leading to barrier dysfunction. (C) HA supports vessel integrity, regulating angiogenesis through overlapping activities with RHAMM and CD44. (D) Under normal conditions, HA is non-adhesive. But in response to inflammation, HA supports leukocyte recruitment to the EC surface. Platelets also interact with HA, and can degrade it generating bioactive HA fragments capable of regulating inflammation. Abbreviations: PECAM-1, platelet-endothelial cell adhesion molecule-1; RHAMM, receptor for HA-mediated motility; EC, endothelial cells.
Footnotes
Acknowledgements
We thank Dr. Carol de la Motte, Dr. Vincent Hascall, and our colleagues in the International Society for Hyaluronan Sciences for their helpful discussions, generosity, and support.
Competing Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
KAQ drafted the manuscript and arranged figures; RAM drafted the manuscript and reviewed the manuscript and figures for form; and ACP drafted the manuscript, critically reviewed manuscript and figures for intellectual content and form. All authors read and approved the final version of the manuscript.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was financially supported by the National Institutes of Health [HL135265] to A.C.P.