
Abstract
Proteoglycans are rapidly emerging as versatile regulators of intracellular catabolic pathways. This is predominantly achieved via the non-canonical induction of autophagy, a fundamentally and evolutionarily conserved eukaryotic pathway necessary for maintaining organismal homeostasis. Autophagy facilitated by either decorin, a small leucine-rich proteoglycan, or perlecan, a basement membrane heparan sulfate proteoglycan, proceeds independently of ambient nutrient conditions. We found that soluble decorin evokes endothelial cell autophagy and breast carcinoma cell mitophagy by directly interacting with vascular endothelial growth factor receptor 2 (VEGFR2) or the Met receptor tyrosine kinase, respectively. Endorepellin, a soluble, proteolytic fragment of perlecan, induces autophagy and endoplasmic reticulum stress within the vasculature, downstream of VEGFR2. These potent matrix-derived cues transduce key biological information via receptor binding to converge upon a newly discovered nexus of core autophagic machinery comprised of Peg3 (paternally expressed gene 3) for autophagy or mitostatin for mitophagy. Here, we give a mechanistic overview of the nutrient-independent, proteoglycan-driven programs utilized for autophagic or mitophagic progression. We propose that catabolic control of cell behavior is an underlying basis for proteoglycan versatility and may provide novel therapeutic targets for the treatment of human disease:
Keywords
Introduction
The extracellular matrix (ECM) is a dynamic meshwork of macromolecules that nourishes and surrounds all cells present in tissues and organs. 1 One major class of molecules that constitute the ECM are proteoglycans (PGs).2,3 PGs are glycosylated extracellular proteins bearing one or more glycosaminoglycan (GAG) chains and/or O- and N-linked oligosaccharides linked to protein cores of various dimensions. The GAG chains are frequently sulfated. Particularly, chondroitin sulfate/dermatan sulfate, keratan sulfate, and heparan sulfate (HS) are the most common. At least 43 different genes encode for PG cores, and these transcribed sequences may undergo additional alternative splicing events. 2
PGs are highly dynamic molecules involved in a wide range of cellular processes including ECM assembly, cellular signaling, tissue repair, migration, inflammation, proliferation, and immune response.4–13 More recently, PGs have been described as active players in cancer biology via their ability to regulate angiogenesis and induce autophagy in the tumor stroma and mitophagy in parenchymal cancer cells.8,14–19 The finely regulated mechanisms of action of PGs in cancer biology depend on the direct interaction of these PGs with receptor tyrosine kinases (RTKs), integrins, and toll-like receptors on the surface of cancer cells, stromal cells, and macrophages.16,20 The intracellular signaling events evoked via PGs binding to cell surface receptors dynamically modulate cell homeostasis by controlling autophagy, an essential homeostatic process where various cytosolic components are degraded and recycled through lysosomes.14,21
The role of autophagy in regulating cancer progression has long been controversial. Initial studies reported on an exclusively protumorigenic and prosurvival function, as the catabolism of intracellular compartments enhances cell survival under nutrient deprivation. However, mounting evidence proposes new roles for autophagy, suggesting that autophagic induction in both cancer cells and surrounding stromal cells can increase tumor cell death, reduce malignant neovascularization, and impede metastases.22–25 PGs can both inhibit and enhance autophagy depending on the cellular context or expression levels.15,26 Importantly, these soluble molecules are composed of several different domains, characterized by the ability to fold on their own. 2 Different domains of the same PG can also inhibit or enhance autophagy depending on the tissue context.14,20 Here, we summarize the major autophagic events evoked by PG signaling in cancer and in the tumor microenvironment. We focused our review on the major players in this field: decorin, perlecan, and its C-terminal fragment endorepellin, with the aim to elucidate their roles in cancer biology and their potential as innovative therapeutic agents.
Decorin Evokes Autophagy in a Nutrient-independent Manner
The inherent proclivity of decorin to bind a multitude of RTKs via hierarchical binding affinities3,16,27 leading to receptor dimerization, downstream signaling, and receptor internalization for lysosomal degradation is only a small piece of the entire portrait that more fully represents the functionality of decorin.28–30 The hypothesis that inducing autophagy results in an oncosuppressive phenotype 31 is supported by seminal genetic observations including the heterozygous deletion of Becn1, in which the partial deletion of Beclin 1, a major component of the autophagic core machinery, 32 promotes tumorigenesis.33,34 An intriguing line of evidence for the antitumorigenic nature of autophagy that explicitly involves an RTK-dependent mechanism akin to that of decorin and endorepellin (see below) originates from the finding that epidermal growth factor receptor (EGFR) phosphorylates and inactivates Beclin 1 35 via Akt. 36 In this manner, EGFR suppresses the function of Beclin 1, leading to increased chemoresistance and tumor progression. 35 The converse also hold trues, wherein augmented autophagy aids to suppress HER2-mediated tumorigenesis. 37
We have serendipitously discovered that nanomolar amounts of soluble, monomeric decorin 38 evoke protracted endothelial cell autophagy 28 and breast cancer cell mitochondrial autophagy (mitophagy) 29 within the tumor microenvironment and parenchyma, respectively (Fig. 1, center). Thereby, decorin contemporaneously targets two distinct histomorphological compartments via cell-type-specific interactions conveyed by the differential expression of cell surface RTKs. Decorin binds vascular endothelial growth factor receptor 2 (VEGFR2) expressed by endothelial cells and the Met receptor that abundantly decorates tumor cells. Decorin triggered the formation of bubble-like structures in endothelial cells reminiscent of autophagosomes. These structures, originally detected by differential interference contrast microscopy, were validated as autophagosomes by co-immunostaining for Beclin 1 and LC3, two key autophagic effectors. 39 This pioneering discovery posited decorin as the first PG to evoke autophagy.
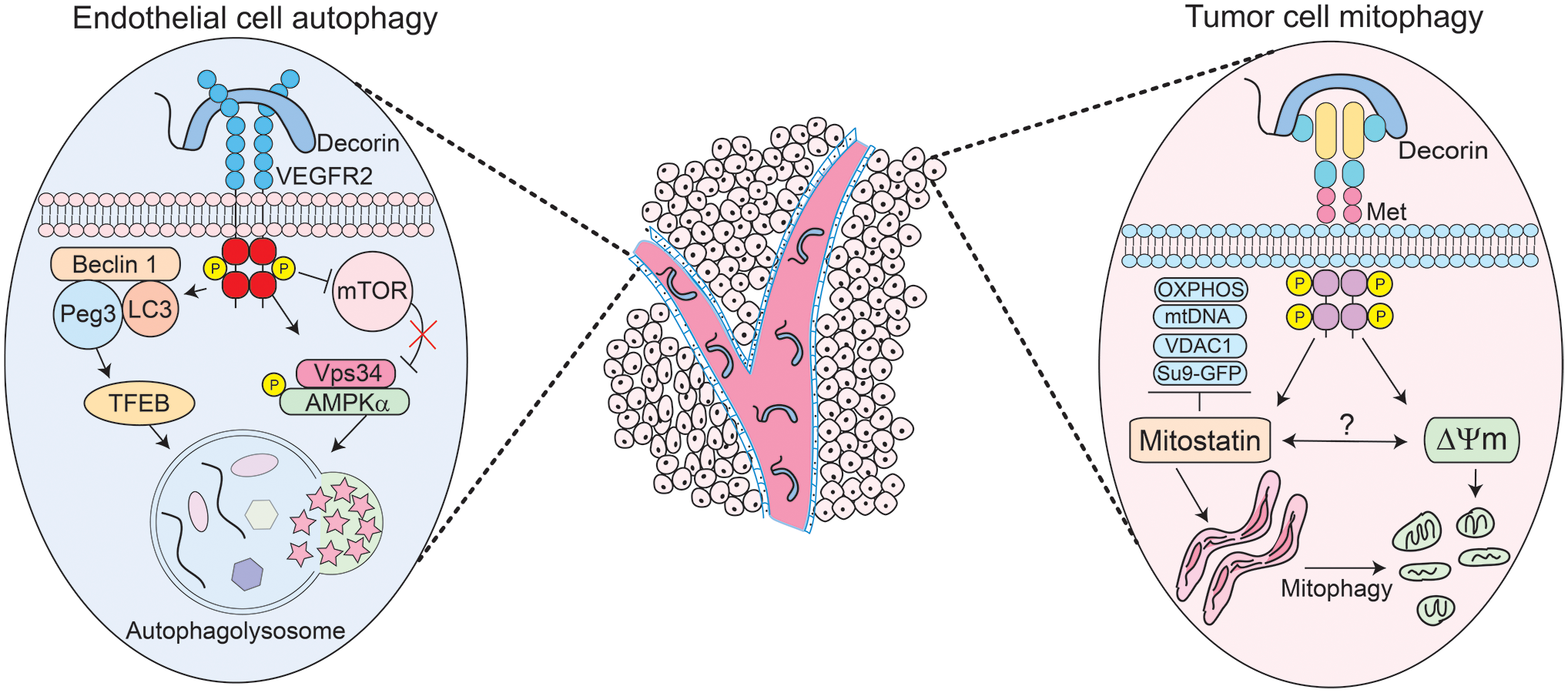
Schematic depiction of the mechanisms of action of decorin-evoked endothelial cell autophagy (left) or decorin-mediated tumor cell mitophagy (right) on distinct compartments within the tumor such as the tumor microenvironment and tumor parenchyma (center). Please consult the manuscript for additional details. Abbreviations: AMPK, activated protein kinase; GFP, green fluorescent protein; mtDNA, mitochondrial DNA; mTOR, mammalian target of rapamycin; OXPHOS, oxidative phosphorylation; Peg3, paternally expressed gene 3; TFEB, transcription factor EB; VDAC1, voltage-dependent anion channel 1; VEGFR2, vascular endothelial growth factor receptor 2.
PG-mediated autophagy wholly manifests from PG/RTK interactions. 16 In genetically stable endothelial cells, decorin physically binds the VEGFR2 ectodomain, mapping at IgG folds 3–5, which partially overlaps with the binding region for vascular endothelial growth factor A (VEGFA) at IgG domains 1–3. 28 Decorin competes with VEGFA and can inhibit its proangiogenic signaling cascades. This high-affinity decorin/VEGFR2 interaction results in protracted downstream activation of the α-catalytic subunit of 5′-AMP-activated protein kinase (AMPK), the master energy sensor kinase previously implicated in cancer inhibition. 40 AMPK coordinates a multitude of fundamental catabolic processes, including autophagy. Canonically, AMPK activates autophagy under situations of nutrient scarcity and bioenergetic stress where the AMP:ATP ratio is severely elevated. 41 This is in stark contrast to PG-mediated autophagy which occurs in an RTK-dependent fashion and in nutrient-rich conditions. 28 Furthermore, using RNA interference (RNAi) to silence VEGFR2, or using small molecule inhibitors to pharmacologically impair the VEGFR2 kinase domain, abrogates the ability of decorin to evoke endothelial autophagy. 28
Mechanistically, autophagy initiates with the formation of the phagophore assembly site (PAS) which contains the non-oncogenic p110 class III PI3K Vps34, ULK1/2, Atg13, and FIP200.42,43 Decorin requires Vps34 and significantly induces AMPK phosphorylation at Thr172 44 (Fig. 1, left). Pharmacologically inhibiting Vps34 with 3-methyladenine or AMPK with Compound C abrogates the autophagic response 28 and precludes PAS formation. AMPK diametrically opposes mTORC1, which is responsible for cell growth, cell size, and numerous anabolic processes, 45 thereby making mammalian target of rapamycin (mTOR) antiautophagic. Decorin severely attenuates the mTOR axis (Fig. 1, left) by dephosphorylating mTOR at Ser2448, Akt at Ser476, and its predominant downstream effector p70S6K at Thr389. 44 Autophagy is a fluidly dynamic process 46 that proceeds via specific phase separations to form cytosolic multiprotein condensates.47,48 Decorin embodies this basic principle by combinatorially favoring the formation of key proautophagic condensates while disfavoring the formation of inhibitory, antiautophagic ones. 44 This “dynamic reciprocity” is best represented by the active coalescence of LC3/Peg3 (paternally expressed gene 3) (see below) with the Beclin 1 scaffold protein (Fig. 1, left) and the concomitant dissociation of Bcl-2, an autophagic inhibitor,49–51 from Beclin 1. This increases Beclin 1 bioavailability for sustained autophagy. 44 It remains to be elucidated whether decorin actively promotes specific phase separations to drive protein condensate formation downstream of RTK signaling. Interestingly, autophagy has been implicated in promoting organismal longevity via the disruption of the Beclin 1/Bcl2 complex in mice. 52 Therefore, decorin may have a role in life span extension via autophagy induction.
These signaling cascades result in a specific proautophagic signature, converging in the synthesis and accumulation of Peg3. 28 The identification of Peg3 originated from a small subset of differentially expressed genes specifically within the Mus musculus tumor stroma of triple-negative orthotopic tumor xenografts.28,53,54 Peg3 encodes for a genomically imprinted, Krüpple-like zinc finger–containing transcription factor. Initially characterized as a tumor suppressor, we discovered that Peg3 is a nexus for decorin-mediated autophagy28,29 (Fig. 1, left). Peg3 is silenced in different tumors, including breast cancer, via promoter methylation and/or loss of heterozygosity,55,56 substantiating its role as a tumor suppressor. As Peg3 non-canonically disrupts the Wnt/β-catenin pathway 57 in a mechanism akin to how decorin suppresses β-catenin in HeLa cells, 58 we pursued Peg3. We found Peg3 localized to autophagosomes in human and murine microvascular and macrovascular endothelial cells. This was confirmed empirically by a series of experiments that conclusively demonstrated colocalization of Peg3 with Beclin 1 and/or LC3. 28 Mechanistically, Peg3 is indispensable for decorin-evoked endothelial cell autophagy and for activating BECN1 and MAP1LC3A expression. 29 Importantly, Peg3 is necessary and sufficient28,59 to maintain basal levels of BECN1 and to ensure appropriate levels of BECN1 and MAP1LC3A are available when the cell encounters an autophagic stimulus.
One of the major factors involved in driving autophagic flux is TFEB (transcription factor EB). TFEB recognizes and binds CLEAR-box sequences present in the proximal promoters of many lysosomal genes necessary for long-term autophagy.60–63 Protracted autophagy is a key characteristic of decorin. TFEB is normally kept inactive via mTOR phosphorylation that enables 14-3-3 scaffolding proteins to bind and sequester it within the cytosol.63–65 However, TFEB is rapidly dephosphorylated by calcineurin phosphatase under autophagic stimuli and undergoes nuclear translocation where it promotes target gene expression 60 for sustained autophagy. We discovered that decorin evokes TFEB expression in a VEGFR2- and Peg3-dependent manner. 66 Moreover, decorin inactivates mTOR and promotes TFEB nuclear translocation in endothelial cells. 66 Genetic depletion of Peg3 is sufficient to inhibit decorin-mediated TFEB expression and translocation. Increasing amounts of Peg3 proportionately and saturably boost TFEB expression. Therefore, Peg3 functions as an upstream regulator of TFEB. 67 Pharmacologically targeting the VEGFR2/AMPK axis with SU5416 or Compound C, respectively, abolishes TFEB induction. This places TFEB as a major downstream effector of the decorin/VEGFR2/Peg3 autophagic cascade 66 and provides the first work that links decorin to lysosomal homeostasis. The formation of LC3-positive autophagosomes by decorin is abolished when TFEB is silenced, underlining the key role of TFEB in the induction of crucial autophagic genes and in the structural formation of autophagosomes (Fig. 1, left).
It is well established that decorin is a potent antiangiogenic factor68–73 that also possesses proautophagic functions 74 and can counteract disease progression of severe blistering diseases. 75 A picture of the functional connections between PG-mediated angiostasis and proautophagic induction is coming into focus. 20 A key paradigm from this image suggests that soluble ECM components that are proautophagic in nature are also antiangiogenic, whereas components that are antiautophagic are staunchly proangiogenic. Recently, the exact molecular details of how decorin reconciles its antiangiogenic nature with strong proautophagic propensities have started to emerge by evaluating the autophagic degradation of intracellular VEGFA in endothelial cells. 76 Intriguingly, decorin-evoked catabolism of VEGFA is independent of mTOR, but requires Peg3. Peg3 is necessary and sufficient for VEGFA mobilization into LC3-positive autophagosomes in a variety of endothelial cell types. Furthermore, we found that VEGFA is a basal autophagic substrate as pharmacological inhibition via chloroquine (CQ) or Bafilomycin A1 (BafA1) or genetic inhibition via transient ATG5 silencing results in the rapid accumulation of intracellular VEGFA. Mechanistically, decorin facilitates intracellular VEGFA clearance by augmenting autophagic flux. This process required RAB24, a small GTPase that facilitates the disposal of basal autophagic components.77–79 Importantly, starved mice show a sharp decrease in overall cardiac and aortic VEGFA levels, which is rescued by systemic administration of CQ. This study described, in earnest, the process of mechanistic unification for the metabolic control of endothelial VEGFA via autophagic clearance in response to decorin and other canonical, proautophagic stimuli. Overall, decorin signals through the VEGFR2/AMPK/Peg3 axis to curb tumorigenesis and impede rampant angiogenesis.
In the same vein of PG and metabolism, it is known that autophagic and metabolic irregularities are implicated in cardiovascular disorders. We found that decorin itself is induced in cardiac tissue after starvation in vivo. 26 Thus, decorin is the first PG whose expression is controlled by canonical autophagic stimuli. Dcn−/− mice exhibit impaired cardiac autophagy as measured for LC3 and via transgenic GFP-LC3 mice. Following fasting, there is a significant increase in decorin immunostaining as well as GFP-LC3 puncta when compared with ad libitum–fed littermates. However, double transgenic GFP-LC3;Dcn−/− mice show no significant increase in GFP-LC3 puncta. Notably, a cohort of important genes are upregulated following an in vivo fast in a decorin-dependent manner. Among them, Cdkn1a, which encodes the cell cycle inhibitor p21Waf1, is significantly increased in the cardiac tissue from wild-type (WT) mice but not in the Dcn−/− animals. This suggests that decorin is necessary for p21Waf1 induction in vivo upon fasting. In addition, in a recently published metabolomics study 80 that was editorially highlighted, 81 cardiomyocytes undergo extensive metabolic reprogramming following genetic ablation of decorin. Thus, decorin is a matrix-centric nutrient sensor responsible for proper cardiac physiology.
Tumor Cell Mitophagy Is Evoked by Soluble Decorin and Requires Mitostatin
Decorin elicits specific outcomes that are dictated by the differential expression of cell surface RTKs. As discussed above, decorin utilizes VEGFR2 to evoke protracted endothelial cell autophagy in a non-canonical manner. Soluble decorin also directly evokes mitochondrial turnover within the tumor proper via mitophagy through Met receptor signaling. 82 Upon decorin treatment, we discovered that subunits of the mitochondrial respiratory chain complexes were suppressed in tandem with several established mitophagy markers, including depletion of mitochondrial DNA (mtDNA) 82 (Fig. 1, right). Thus, the tandem induction of tumor cell mitophagy and endothelial cell autophagy may be the molecular basis for inhibiting tumorigenesis and angiogenesis by decorin. Interestingly, decorin evokes non-canonical mitophagy as decorin-evoked mitophagy occurs irrespective of the prevailing nutrient conditions and depends on the Met receptor and mitostatin.
A tumor suppressor gene also referred to as trichoplein (TCHP), mitostatin is vital for decorin-evoked mitophagy. The genetic locus encoding TCHP is physically located at 12q24.1. Thus, initially identified as Ts12q, for Tumor suppressor at 12q, the resulting protein product of TCHP was renamed mitostatin, for mitochondrial protein with oncostatic activity. 83 Differential (or subtractive) hybridization of cDNA libraries was utilized to discover TCHP as a decorin-inducible gene. 83 Histologically, mitostatin is found in many normal tissues in differing amounts and is evolutionarily conserved across multiple species. 83 In breast and bladder cancer, mitostatin is frequently lost and/or mutated,84,85 suggesting that it might function as a putative tumor suppressor. Indeed, restoration of mitostatin in prostate cancer cells significantly prevents invasion. 85
Mitostatin immunostaining reveals a punctate pattern that is predominantly localized to the mitochondria. 85 Fractionation experiments demonstrated that mitostatin is enriched at ultra-specialized, interorganelle contacts known as mitochondrial-associated membranes (MAMS). 86 MAMs are specialized juxtapositions of endoplasmic reticulum (ER) with the outer mitochondrial membrane (OMM) that permits bidirectional communication at dedicated, but transient, microdomains. 87 Mitostatin physically interacts with mitofusin 2 (MFN2) 86 and could represent an important tethering component that regulates ER and mitochondrial physiology. Overexpression of mitostatin results in aberrant changes to mitochondrial ultrastructure, including a disrupted mitochondrial matrix, disordered cristae architecture, and the coincident emergence of a swollen, stouter appearance. 84 Coincident with changes in mitochondrial morphology, mitostatin overexpression leads to a decreased phosphorylation of heat shock protein 27 (Hsp27) at Ser82 without overt modulations in total Hsp27 levels. 84 Whether modulating Hsp27 phosphorylation underlies the overt morphological changes elicited by decorin remains to be investigated.
Downstream of both decorin–Met interactions and the initiation of the promitophagic signaling cascade is peroxisome-proliferator activated receptor-γ coactivator 1α (PGC-1α), a nuclear receptor coactivator and master regulator of mitochondrial biogenesis.88,89 PGC-1α is dynamically regulated over time, undergoes nuclear translocation, and directly binds TCHP mRNA for rapid stabilization, leading to increasing amounts of mitostatin 82 (Fig. 1, right). Mechanistically, the interaction between PGC-1α and TCHP mRNA occurs via the C-terminal RNA recognition motif (RRM). 82 Truncating the RRM compromises the overall ability of PGC-1α to stabilize TCHP mRNA, leading to reduced levels of mitostatin. PGC-1α is an RNA-binding protein that is subjected to arginine methylation, a key posttranslational modification (PTM) that regulates its activity. 89 Functionally akin to truncating the RRM, RNAi-mediated silencing of protein arginine methyltranferase 1 (PRMT1), responsible for attaching methyl groups to the C-terminal regions of PGC-1α, 89 phenocopied the loss of TCHP stability. 82 Deciphering the molecular determinants of this cascade uncovered an unlikely alliance between mitostatin, a putative tumor suppressor, and PGC-1α, a known oncogene. Increased oxidative metabolism, mediated by PGC-1α, MITF, and B-Raf, 90 is responsible for driving a subset of aggressive and metastatic melanomas characterized by improved mitochondrial capacity and resistance to oxidative stress. 91 The dynamic regulation of PGC-1α by decorin may alleviate these protumorigenic phenotypes and ameliorate these aggressive properties.
Silencing mitostatin impedes depletion of established mammalian mitophagy markers,92,93 including subunits of the electron transport chain, mitochondrial transcription factor A (TFAM), voltage-dependent anion channel 1 (VDAC1), mtDNA, and fragmentation of the mitochondrial network. 82 Similarly, mitochondrial fragmentation is consistent with mitostatin overexpression 84 and represents a critical mitophagic step 82 (Fig. 1, right).
Immediately preceding lysosomal engulfment, decorin triggers mitochondrial depolarization (ΔΨm) 82 with a magnitude comparable to FCCP (2-[2-[4-(trifluoromethoxy)phenyl]hydrazinylidene]-propanedinitrile), an established ionophore for the chemical uncoupling of mitochondria for depolarization. 94 As the molecular impetus for ΔΨm induction, decorin may elevate cytosolic [Ca2+].95,96 Therefore, mitochondria that have lost membrane polarization may represent the terminal effect of increased cytosolic [Ca2+] following decorin/RTK interactions. Moreover, as mitostatin is localized to the MAM that physically interacts with MFN2, it may permit an efflux of Ca2+ from the ER into the mitochondria to trigger mitophagy. The loss of membrane potential across the OMM is an early harbinger of impaired and/or damaged mitochondria and represents an effective signal for PINK1/Parkin-mediated mitophagy. 97 Parkin is an RBR-domain containing E3-ubiquiting ligase that localizes to the OMM following mitochondrial damage. PINK1 is a kinase that accumulates on the OMM as it is no longer degraded by the N-end rule 98 following ΔΨm. 99 Stabilized PINK1 begins phosphorylating several substrates, including ubiquitin (Ub) 100 that activates the inherent E3 activity of Parkin.101,102 Binding to a multitude of OMM receptors (Bnip3/Nix, FUNDC1, and NDP52), 99 Parkin utilizes phospho-Ub to ubiquitinate several OMM components, such as MFN2 and VDAC1. 103 There is some evidence that even the translocase of the OMM (TOMM) complex is a Parkin receptor and acts as a molecular hub for PINK1/Parkin mitophagy. 104 Recognition of the phospho-Ub substrates by various Ub-binding autophagy receptors (p62, optineurin, or NBR1) results in engulfment by LC3-positive autophagosomes and clearance. 99 Indeed, PINK1/Parkin requires p62/VDAC1 engagement for mitochondrial incorporation into autophagosomes. 105 As Parkin is required to maintain mitochondrial homeostasis 106 and is vital for respiratory chain and mitochondrial turnover in vivo, 107 it is plausible that decorin may recruit Parkin to the OMM following ΔΨm in a mitostatin-dependent manner (Fig. 1, right). Conducting a bioinformatics analyses, it appears that mitostatin harbors a series of five coiled-coil domains that may mediate recruitment and/or binding of Parkin to cognate receptors by acting as a protein–protein interface, such as MFN2, for accumulation on the OMM.
Collectively, these findings culminate in a novel signaling pathway that transduces information via decorin/Met interactions for excessive tumor mitophagy. This occurs in a mitostatin-dependent manner in breast and prostate carcinomas.108,109 These catabolic processes (mitophagy and autophagy) may represent a molecular nexus for integrating and functionally manifesting the decorin-mediated RTK regulation into the well-known matrix phenotypes ascribed to decorin, such as the suppression of angiogenesis, tumorigenesis, and metastasis. The fact that decorin exclusively evokes autophagy in endothelial cells and not in tumor cells, via differential RTK interactions, presents an advantage for decorin as a future protein therapy to combat cancer.
Perlecan and Endorepellin, Two Opposite Regulators of Intracellular Catabolism
Perlecan, encoded by the HSPG2 gene, is a heparan sulfate proteoglycan (HSPG) with a ubiquitous distribution in basement membranes, cell surfaces, and pericellular zones. 110 Perlecan is a very large molecule composed of a multidomain protein core of ~500 kDa that acts in presenting growth factors to membrane receptors 2 via its three HS chains at the N-terminus. 111 Perlecan is uniquely localized in both vascular and avascular tissues,112,113 and abnormal expression of perlecan has been implicated in the metastatic process of melanomas 114 and prostate carcinomas.115–117 A wide cohort of studies have shown the numerous biological functions of perlecan both as a whole molecule and as its individual domains.2,113,118–122 Most of the cellular signaling events evoked by perlecan are mediated by its interaction with different integrins, RTKs, and ligands in different tissues and organs. 123 The multivariable ability of perlecan depends on the release of various pro- and antiangiogenic factors upon cleavage of the HS chains by different proteases.115,117,124 Perlecan is a master regulator of physiological vascularization and tumor angiogenesis 112 via binding VEGFA and fibroblast growth factors through its N-terminal HS chains and presenting these growth factors to their respective receptors evoking proangiogenic signaling events.125–127 In support of these findings, in vivo animal models lacking or preventing Hspg2 expression have shown severe cardiovascular deficiencies.128,129 In other studies, the HS chains of perlecan have been shown to be critical determinants of angiogenesis in mouse hind-limb ischemia. 130 The importance of perlecan in regulating vascular biology via receptor-mediated intracellular signaling pathways has been validated in a lethality-rescued Hspg2−/− where perlecan was reintroduced into the cartilage. In this model, the absence of perlecan caused a profound cell dysfunction due to inhibition of the endothelial expression of nitric oxide synthase. 131
In stark contrast to the proangiogenic N-terminal domain, the C-terminal domain V of perlecan, named endorepellin, exerts an opposite function by inhibiting endothelial cell migration, in vivo angiogenesis, and capillary morphogenesis.132–138 The antithetical roles of perlecan and endorepellin in controlling angiogenesis are matched with their contrasting effects in regulating autophagy. In vivo studies utilizing Hspg2−/− mice showed that the absence of the entire PG, in the muscle, significantly enhances intracellular catabolism by inducing the formation of autophagosomes and by inhibiting the mTOR signaling pathway, suggesting that perlecan acts in an antiautophagic manner. 139 On the contrary, endorepellin is a potent proautophagic agent that, through transient phosphorylation of VEGFR2 at Tyr1175 in endothelial cells, evokes AMPK, Beclin 1, LC3, and p62; inhibits mTOR; and induces the formation of autophagosomes (Fig. 2).20,140 The mechanism of action of endorepellin has been finely elucidated. Endorepellin engages in a dual receptor antagonism by simultaneously binding VEGFR2 and the α2β1 integrin. 141 However, the endorepellin-mediated induction of autophagy relies entirely on the direct binding of its two laminin globular domains LG1/2 to VEGFR2. Interestingly, the third domain of endorepellin, LG3, binds to the α2β1 integrin and inhibits autophagic gene expression. 140 Thus, perlecan and its different domains by inhibiting or evoking autophagy finely regulate intracellular catabolism that maintains physiological cellular homeostasis.
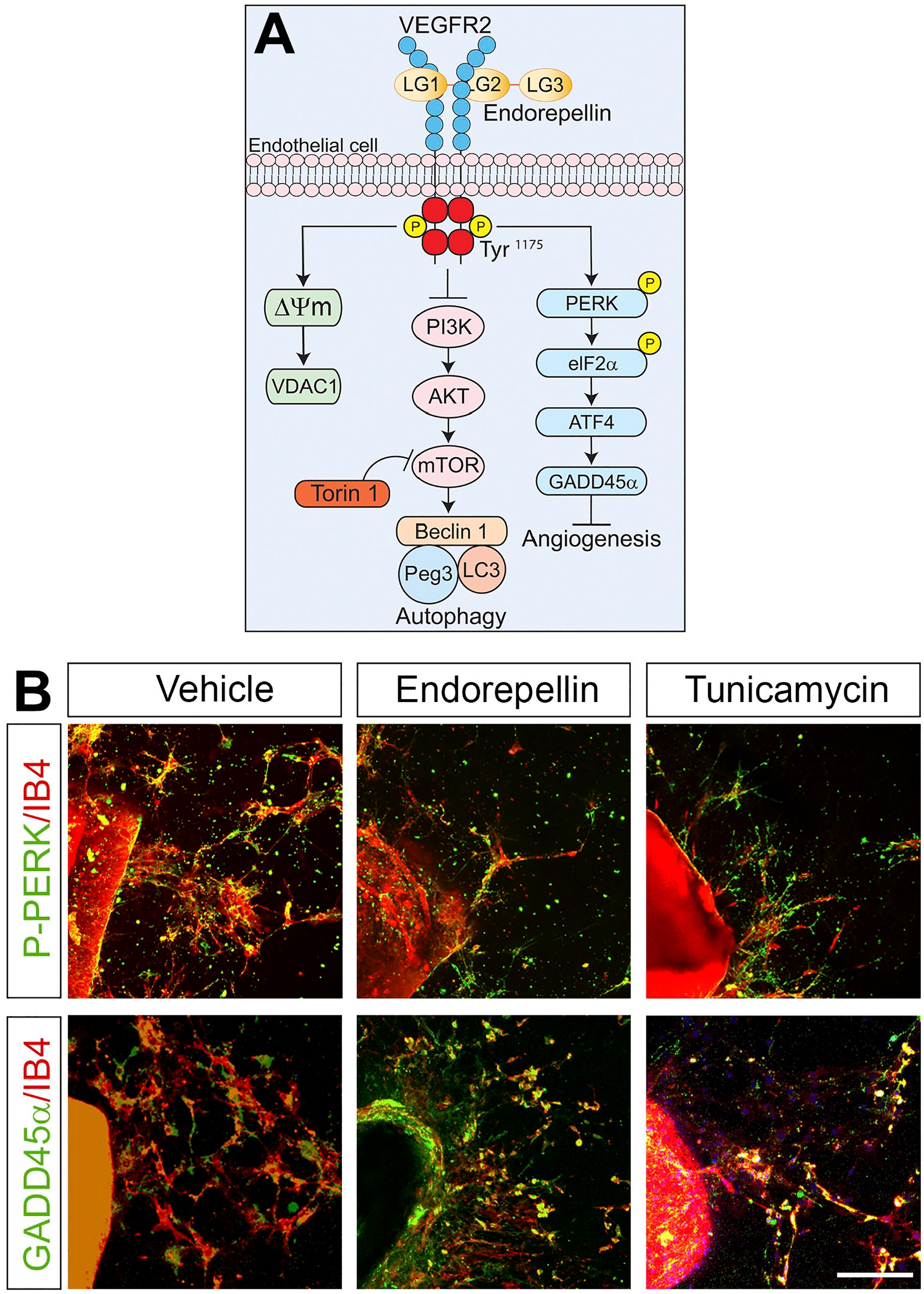
Endorepellin stimulates proautophagic and antiangiogenic pathways. (A) Representative scheme elucidating the major mechanism of endorepellin-based signaling in endothelial cells via VEGFR2. From left to right: induction of mitophagy, induction of autophagy, and inhibition of angiogenesis. (B) Confocal images representing thoracic aortic rings treated with vehicle, endorepellin (200 nM), or iunicamycin (10 µg/ml) for 2 hr and subsequently stained with the endothelial cell marker, isolectin IB4 (red), and phospho-PERK or GADD45α antibodies (green). Scale bar, 250 µm. Abbreviations: ATF4, activating transcription factor 4; eIF2α, eukaryotic initiation factor 2α; GADD45α, growth arrest- and DNA-damage-inducible; IB, isolectin B4; LG, laminin-G-like domain; Peg3, paternally expressed gene 3; PERK, PKR-like endoplasmic reticulum kinase; PI3K, phosphatidylinositol-3-kinase; VDAC1, voltage-dependent anion channel 1; VEGFR2, vascular endothelial growth factor receptor 2.
Endorepellin induction of the autophagic program results in increased expression of the master regulator of autophagy Peg328,140 which recruits Beclin 1 and LC3, two known autophagy markers (Fig. 2A). Moreover, like decorin (see above; cf. Fig. 1), a recent study suggests that endorepellin induces not only canonical autophagy but also mitophagy. NanoString, which is a next-generation digital PCR platform, revealed a promitophagic transcriptome in endothelial cells stimulated with endorepellin. This was confirmed by endorepellin-mediated mitochondrial depolarization concurrent with protracted expression of mitostatin and Parkin. 142 Importantly, the autophagic and mitophagic programs initiated by endorepellin are, like decorin, strictly dependent on its binding to VEGFR2 and take place independently of nutrient conditions. The powerful angiostatic activity of endorepellin can, therefore, be explained by its fine control of these critical catabolic events that ultimately lead to a marked decrease in vessel growth. In aortic ring assays, a widely known and excellent ex vivo model utilized to study angiogenesis,143,144 it has been shown that the natural sprouting of endothelial cells in collagen is significantly suppressed by endorepellin. 145 The angiostatic mechanism of action of endorepellin is strictly connected to its proautophagic nature as blocking auto-phagy with Compound C, a potent AMPK inhibitor, prevents endorepellin-mediated blood vessel growth reduction. 145 Moreover, the mTOR inhibitor, Torin 1, equally suppresses endothelial cell sprouting, similarly to endorepellin, strengthening the link between angiogenesis and autophagy. Recently, we discovered that endorepellin modulates the stress signaling pathway in endothelial cells where binding of endorepellin to VEGFR2 via its proximal LG1/2 domains transcriptionally induces the expression of growth arrest- and DNA-damage-inducible (GADD45α) protein (Fig. 2A). 146 This protein belongs to the canonical PERK (PKR-like endoplasmic reticulum kinase)/eIF2α (eukaryotic initiation factor 2α)/ATF4 (activating transcription factor 4))/GADD45α pathway which is activated in response to stress factors such as nutrient deprivation, oxidative stress, or ultraviolet light. 147 Bridging this new discovery with previous finding is the antiangiogenic and proautophagic role of GADD45α. By interacting with mTOR, GADD45α prevents VEGFA expression, thereby acting as an inhibitor of angiogenesis. 148 At the same time, GADD45α induces autophagy by evoking LC3-II through inhibition of mTOR. 149 As shown in an aortic ring assay, endorepellin evokes GADD45α and the phosphorylation of PERK, its upstream regulator, in a similar fashion as the ER stress inducer, tunicamycin, and inhibits sprouting of endothelial cells vis-à-vis vehicle-treated rings (Fig. 2B).144,146 We have recently discovered that endorepellin evokes autophagic downregulation of Hyaluronan Synthase 2, 150 the major enzyme involved in the synthesis of hyaluronan. 151 Taken together, these studies postulate a translational role for endorepellin as a natural, circulating antiangiogenic agent that exerts its role by stimulating critical intracellular catabolic programs.
A major advancement in the field of matrix biology that is vital for understanding PG biology came with the pioneering discovery that select constituents directly regulate intracellular, catabolic pathways.135,140,152,153 These soluble members, primarily represented by decorin and endorepellin, evoke autophagy in a non-canonical, RTK-dependent manner in a variety of diverse cell types. These functions add to the versatility, interactome, 73 and utilitarian functions3,154 that enhance our knowledge for how PGs regulate homeostatic processes and attractive therapeutic targets for when these pathways go awry, especially in cancer.155–157 Of note, other PGs, which we have not covered here, such as biglycan 158 and lumican, 159 are directly involved in autophagy by either stimulating or inhibiting this intracellular catabolic process.
Fully decoding the multitude of signaling pathways and soluble cues involved in such an intricate system, immortalized as “dynamic reciprocity,” is currently being expedited by comprehensive, innovative, and high-resolution “-omics” techniques.160–162 A prime example is provided by these approaches aided in the identification of a master autophagic regulator, Peg3, 53 and the discovery of RTK-dependent autophagy operating under nutrient-rich conditions evoked by soluble PGs. This viewpoint resulted in a paradigmatic shift where soluble matrix components override the plethora of controls (e.g., the balance between mTOR and AMPK) to favor activation of catabolic pathways, even in the presence of rich nutrient bioavailability. This concept is further underscored that PGs other than decorin or perlecan also modulate autophagy, despite signaling via a different subset of cell surface receptors. It is possible that a common core of base autophagic machinery acts as a conversed signaling node that integrates signals emanating from this disparate array of receptors. The master regulator, Peg3, may serve as the common link that is permissive for PG-mediated autophagy. Circumventing these evolutionary controls results in excessive autophagy that impedes rampant tumorigenesis and aberrant angiogenesis. The multidimensional bioactivities of decorin and endorepellin offer exciting clinical relevance as advanced, next-generation protein therapies to combat cancer.
Footnotes
Acknowledgements
We thank C.G. Chen for critical reading of the manuscript and all past and present members of the Iozzo’s laboratory for their contribution to this area of research. We apologize for not referencing many valuable contributions due to space limitations.
Competing Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported, in part, by National Institutes of Health Grants RO1 CA39481 and RO1 CA47282 (R.V.I.).