
Abstract
Versican is an extracellular matrix proteoglycan with key roles in multiple facets of cancer development, ranging from proliferative signaling, evasion of growth-suppressor pathways, regulation of cell death, promotion of neoangiogenesis, and tissue invasion and metastasis. Multiple lines of evidence implicate versican and its bioactive proteolytic fragments (matrikines) in the regulation of cancer inflammation and antitumor immune responses. The understanding of the dynamics of versican deposition/accumulation and its proteolytic turnover holds potential for the development of novel immune biomarkers as well as approaches to reset the immune thermostat of tumors, thus promoting efficacy of modern immunotherapies. This article summarizes work from several laboratories, including ours, on the role of this central matrix proteoglycan in tumor progression as well as tumor-immune cell cross-talk:
Keywords
Structure, Assembly, and Intermolecular Interactions
Versican is a large (>1000 kDa) chondroitin sulfate/dermatan sulfate (CS/DS) matrix proteoglycan. It is classified in the hyalectan family as a proteoglycan with hyaluronan (HA)-binding domains along with aggrecan, brevican, and neurocan. 1 Versican has crucial, non-redundant roles in organ development and disease.2–4
Versican core protein consists of an N-terminal G1 domain, a C-terminal G3 domain, and CS chain-binding regions (Fig. 1). The G1 domain is composed of an immunoglobulin (Ig)-like module, followed by two HA-binding domains (link modules). The G3 domain of versican consists of two epidermal growth factor (EGF)-like repeats, a lectin-like carbohydrate recognition domain (CRD), and a complement binding protein (CBP)-like motif. 5 At least five splice isoforms have been described (Fig. 1).
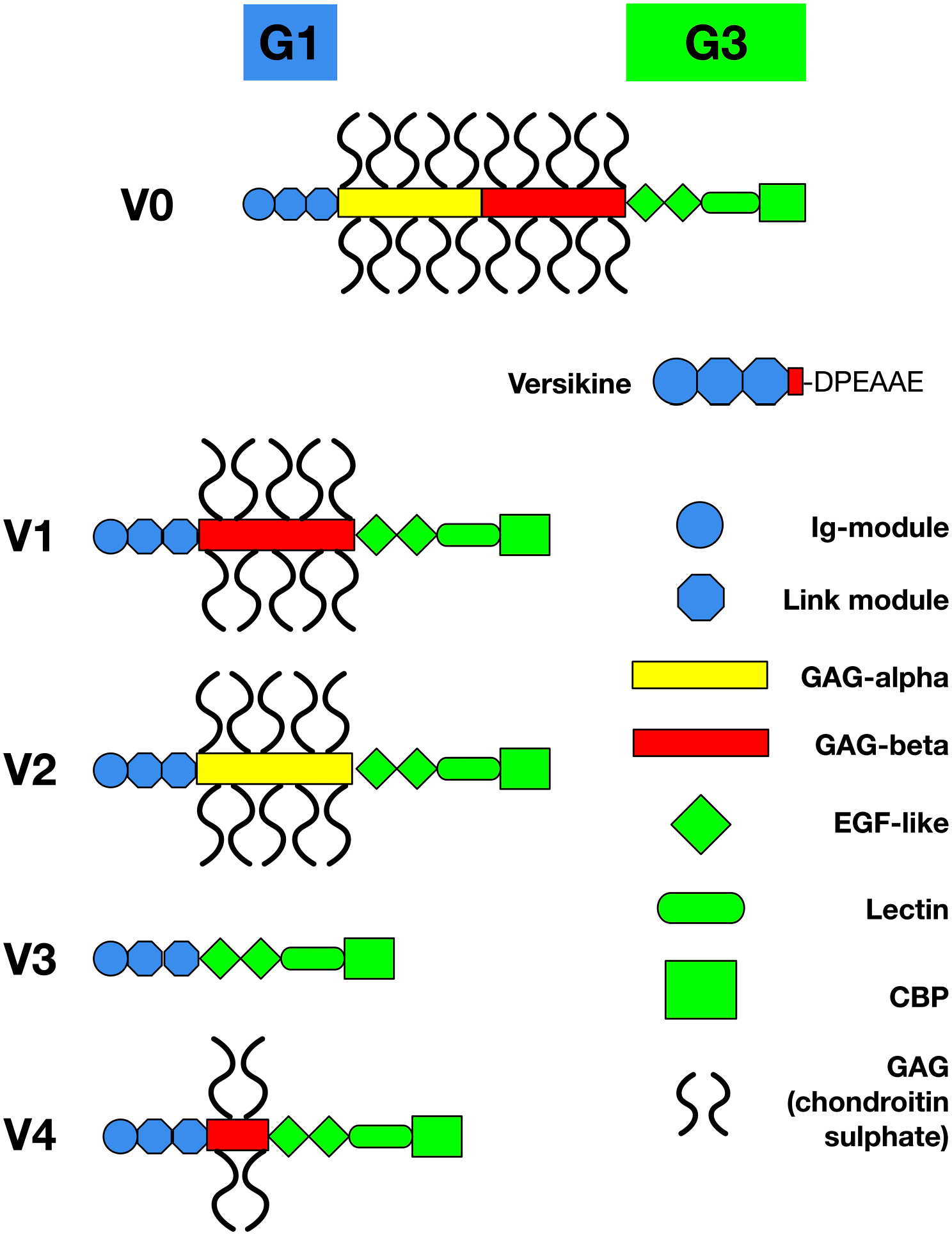
Structure of versican, its splice isoforms, and its proteolytic product, versikine. Alternative splicing of the mRNA encoding the glycosaminoglycan (GAG)-bearing domain of the versican core generates four common splice isoforms (V0, V1, V2 and V3).2,6 An additional isoform, V4, was detected in breast cancers. 60 ADAMTS-mediated proteolysis of the versican V1 isoform at Glu441–Ala442 generates an N-terminal, GAG-bereft fragment (“matrikine”), versikine. The specific cleavage event exposes a unique neoepitope (DPEAAE) that can be detected immunohistochemically. The size of individual domains is not drawn to scale. Abbreviations: ADAMTS, a disintegrin and metalloproteinase with thrombospondin motifs; CBP, complement-binding protein; EGF, epidermal growth factor; GAG, glycosaminoglycan; Ig, immunoglobulin.
Versican interacts with several other extracellular matrix (ECM) components. One of the most studied interactions is between the amino-terminal domain of versican (G1 domain) and HA, mediated through link modules. 6 Versican interacts with diverse ECM components that are important in inflammation, such as tumor necrosis factor–stimulated gene-6 (TSG-6), fibulins and fibrillin, inter-alpha-trypsin inhibitor (IαI), fibronectin, tenascin-R and tenascin-C, and CD44. Interactions of versican with TSG-6 and IαI are indirect and mediated by thrombospondin-1.7,8 Versican may interact directly with CD44 through its glycosaminoglycan chains, independent of HA. 9 Tenascin-R binds to versican at its C-terminal lectin-like domain (CRD) through protein–protein interactions. 10 Versican binds to fibulin-2 and fibrillin-1 through its C-terminal lectin-like domain in a calcium-dependent manner.11,12 Fibulin also may serve as a bridge between versican and fibrillin, forming highly ordered multimolecular structures important in the assembly of elastic fibers. 2 Versican also interacts with fibronectin and collagen type I.13,14
Roles of Versican in Inflammation
Versican is a major contributor to tissue inflammation caused by infection and tissue injury. Its production is highly regulated by cytokine networks and in turn regulates downstream mediators to amplify inflammatory cascades. 15 Versican regulates leukocyte traffic across and beyond vascular structures. 16 Upon extravasation in the subendothelium, leukocytes encounter cable-like ECM structures enriched in versican and HA that act as scaffold for leukocytes, having an impact on their cell adhesion and subsequent retention and activation. 17 Blocking versican accumulation in the ECM generated by cultured human lung fibroblasts impedes monocyte adhesion in vitro. 18 Versican interacts with receptors on the surface of leukocytes such as P and L selectins and provides intrinsic signals that influence immune and inflammatory phenotypes.5,9,19,20 Once bound to the versican-containing ECM, leukocytes degrade the ECM to generate proinflammatory fragments, mostly derived from laminin, elastin, and type IV collagen, which further drive the inflammatory response by increasing monocyte/macrophage-dependent secretion of proteases and proinflammatory cytokines.21–24
An effective immune response requires T-cell adhesion and migration through the ECM. 25 Versican binding to HA may interfere with the binding of HA to CD44 on immune cells, such as T-lymphocytes, thus attenuating the immune response. Consistently, versican hindered human T-cell invasion of collagen gels, having a negative effect on T-cell migration and generating immunosuppression. 26 Cleavage of versican by ADAMTS (a disintegrin and metalloproteinase with thrombospondin motifs) proteases is crucial for T-cell adaptive responses in mouse models of influenza virus infection. Accumulation of versican in Adamts5-knockout mice, which lack ADAMTS-5 versican-degrading enzyme, causes impaired influenza virus clearance and prevents CD8+ T-cell egress, leading to compromised antiviral immunity. However, when Adamts5−/−Vcan+/hdf (versican-haploinsufficient) mice were infected with influenza virus, T-cell function was restored. The authors of this seminal work concluded that versican V0/V1 accumulation impedes migration of CD8+ T-cells from draining lymph nodes to the periphery, which is critically important for the establishment of full effector function and eventual clearance of the viral pathogen. 27
Versican controls the release of inflammatory cytokines by myeloid cells. Versican acts as a danger-associated molecular pattern (DAMP) molecule that interacts with Toll-like receptors (TLRs), such as TLR2 on alveolar macrophages, to promote the production of inflammatory cytokines, including tumor necrosis factor-α (TNFα), interleukin (IL)-6, and other proinflammatory cytokines.28–31 Lipopolysaccharides (LPS) and poly I:C, two TLR agonists, stimulate versican expression in both murine bone marrow–derived macrophages in vitro and in murine alveolar macrophages32,33 and stromal cells in vivo. 34
Macrophages constitute a major source of versican production in the inflammatory milieu. Versican gene is differentially expressed in M1 macrophages, as opposed to M2 macrophages. 35 Versican produced by macrophages can form complexes with matrix metalloproteinases (MMPs), 36 such as MMP-9, implying possible roles for versican in controlling the activity of matrix-degrading enzymes. Such activity suggests that versican could assist myeloid cells in shaping their own microenvironment. 17 Versican can also alter the inflammatory milieu through chemokine regulation. Versican expression is elevated in CD14+ monocytes isolated from patients with systemic sclerosis, and this elevated expression is accompanied by increased expression of CCL2. 37 Earlier studies had also shown that CCL2 binds to versican and impacts inflammation in a model of neuronal inflammation hyperalgesia. 38
In the setting of acute lung injury, versican and HA are increased during inflammation associated with Escherichia coli pneumonia. 32 The accumulation of a versican-enriched ECM coincides with invasion and retention of leukocytes within different compartments of the lung during early stages of infection. 34 Versican is also a crucial mediator of chronic inflammation. Versican accumulates in chronic lung diseases that involve persistent inflammation such as pulmonary fibrosis, acute respiratory distress syndrome, asthma, and chronic obstructive pulmonary disease.39–42 Versican, which is mainly secreted by fibroblasts throughout the airway tree, contributes to airway remodeling in asthma, leading to persistent airway obstruction and subsequent decline in lung function. 43
Versican proteolysis can also drive new blood vessel formation (angiogenesis) as part of inflammatory events associated with tissue repair (see subsequent section regarding tumor neoangiogenesis). Injection of an adenoviral vector expressing VEGF164 (vascular endothelial factor) into the skin induces a robust angiogenic response by increasing ADAMTS-1 and versikine’s proteolytic fragment, versikine. 44 In addition, proteoglycan accumulation is a hallmark of aorta medial degeneration, contributing to thoracic aortic aneurysm and dissection. Tissue swelling imposed by versican is deleterious to aortic wall mechanics and smooth muscle cell homeostasis, predisposing to type A aortic dissections. 45 Mice lacking an ADAMTS versicanase cleavage site have accelerated wound healing, suggesting that accumulation of intact versican facilitates wound repair. 46 Additional evidence points to a role for versican in resolution of inflammation, suggesting that the roles of versican in regulating inflammatory responses may be context- and tissue-dependent: Xu et al. 47 reported that mice lacking versican V1 have a more severe inflammatory response to LPS-induced acute lung injury. Taken together, the above studies suggest that versican accumulation vs proteolytic degradation may be pathological or protective depending on the specificities of the injury.
Versican Sources in the Tumor Microenvironment
There are at least four sources of versican production in the tumor bed: tumor cells, stromal cells, tumor-associated myeloid cells, and possibly tumor-infiltrating lymphoid cells, as low-level versican expression was detected in certain activated B-cells. 48 Versican sources are often context-dependent and not necessarily mutually exclusive.
A major source of versican production in the tumor microenvironment (TME) is the myeloid compartment. Versican originating from myeloid cells promotes tumor metastasis in breast cancer, 49 whereas versican expressed by CD11b+Ly6Chigh cells promotes lung metastasis in a transforming growth factor β (TGF-β)-dependent manner. 50 A recent study raised the possibility of “cross-talk” among different cell subsets: Coculture of myeloid cells with bladder carcinoma cells in vitro resulted in upregulation of versican in the myeloid cells. 51 In patients with acute myeloid leukemia (AML) post-cord blood stem transplantation, macrophages were the major versican-producing cells in the bone marrow (BM). 52 Consistent with the latter observation in the hematopoietic context, our group has demonstrated that macrophages are the major source of versican in the bone marrow of patients with multiple myeloma (MM). 53
In other cancer types, the main source of versican secretion is the tumor cell itself. For instance, in Lewis lung carcinoma (LLC), an experimental murine non–small cell lung cancer model, versican secreted by the tumor cells promotes metastasis to lung, liver, and adrenal gland, a process that depends on TLR2-mediated myeloid cell activation and TNF-α production. 31 Tumor cells also show an increased expression of versican in ovarian cancer, 54 leiomyosarcoma, 55 hepatocellular carcinoma, 56 colon carcinoma, 57 glioma, 58 and bladder cancer. 51 Several of these studies find a direct correlation between tumor versican expression and tumor grade.
In other contexts, stromal cells constitute the main source of versican production, such as in breast cancer,59–62 colon cancer, 63 pharyngeal cancer, 64 ovarian cancer, 65 and prostate cancer.66–68 Stromal versican is often accompanied by increased HA in the TME: High HA levels were seen in peritumoral stroma of serous ovarian carcinomas, correlating with CD44 and versican expression. 69 Peritumoral versican expression is induced in stromal cells by factors secreted by carcinoma cells. Versican secretion by prostatic fibroblasts is regulated by prostate cancer cell mediators, principally TGF-β1. 67 Moreover, in a breast cancer cohort consisting of 58 nodal metastasis-negative patients, versican was deposited in peritumoral stroma by mammary fibroblasts, and its secretion appeared to be regulated by breast cancer cell mediators. 61 Strong versican expression was also observed in primary pharyngeal tumors, whereas in metastatic tumors, stromal versican staining in the metastatic site was shown to be significantly more intense compared with the primary tumor. 64 Finally, in endometrial and cervical cancers, both tumor and stromal cells can be source of versican production. The combination of tumor and stromal expression of versican correlates with shortened disease-free survival and overall survival (OS). 70
Versican in Tumor Cell Survival, Proliferation, and Apoptosis
Versican is highly expressed in proliferating tumor and tumor-associated stromal tissues. Versican can stimulate cell proliferation via two mechanisms: through two EGF-like motifs in the G3 domain that play a role in stimulating cell growth71–73 and through the G1 domain that destabilizes cell adhesion and facilitates cell growth. 74 The EGF motifs were also shown to mediate breast cancer cell self-renewal. 75 Overexpression of the versican G3 domain augmented breast cancer self-renewal through epidermal growth factor receptor (EGFR)/Akt/GSK-3β signaling and conferred enhanced resistance to chemotherapeutic drugs. Of interest, G3-overexpressing tumors not only presented high levels of 4B6, pEGFR, pAKT, and GSK-3β (S9P), all of which were related to tumor invasiveness, but also expressed high levels of tumor stem cell markers Sox2, Sca-1, and ALDH1. 75 Silencing versican in a human uterine leiomyoma cell line (UtLM-hTERT) reduced the mRNA expression of both estrogen receptor 1 and progesterone receptor A, hormone receptors linked with leiomyoma growth control. Knockdown of versican may be a promising therapeutic approach for symptomatic leiomyoma. 76
Versican also controls the proliferation of important tumor-accessory components. For instance, platelet-derived growth factor (PDGF) upregulates versican expression in arterial smooth muscle cells and promotes the expansion of the pericellular ECM, required for the proliferation and migration of these cells.77–79
Genetic and epigenetic alterations regulate tumor cell survival pathways, a key hallmark of cancer. 80 Versican V1 overexpression has been reported to confer either selective apoptotic resistance or selective apoptotic sensitization to tumor cells. This paradoxical combination of selective apoptotic resistance and sensitivity is often seen in cancer cells. In particular, versican V1–transfected cells have elevated resting levels of tumor suppressor p53, which confers apoptotic sensitivity, and also Mdm2, which is a crucial negative regulator of p53. 81 Moreover, versican has been demonstrated to exert an antiapoptotic effect by protecting cells from oxidative stress–induced death through enhancement of cell–matrix interactions, increased cell attachment, and expression of beta1 integrin and fibronectin. 82 However, versican has also been implicated in proapoptotic signaling. Small interfering RNA (siRNA)-mediated versican knockdown prevented G3-modulated cell apoptosis in human breast cancer cell lines. 83 The somewhat contrasting roles of versican in promoting cancer cell survival and apoptosis underline the complexity of apoptosis regulation in tumor development and progression.
Versican in Tumor Angiogenesis
Versican is a central player in the tumor-associated angiogenesis network. In the context of the well-vascularized tumor glioblastoma, versican appears to facilitate new blood vessel formation. The versican G3 domain enhanced angiogenesis both in vitro and in vivo. Tumors formed by G3-expressing tumor cell implants contained very high levels of fibronectin and VEGF. Endothelial cell adhesion, proliferation, and migration were significantly promoted by versican, fibronectin, and VEGF. Blocking of these factors reversed these processes. 84 Consistent with the observation that G3 domain binds fibronectin, the versican V2 splice isoform promoted extensive vasculature formation by upregulating and binding to fibronectin in glioblastoma. 85 Silencing fibronectin expression by siRNA abrogated the effects of versican V2 in enhancing vascular tube-like structure formation. 85 In the context of breast cancer, versican interacts with the surrounding stromal components, fibronectin, and VEGF to promote neovascularization. 86 Moreover, increased expression of versican has been linked with elevated levels of HA in the vascular and perivascular elastic structures in malignant tumors, and versican is considered to play a key role in HA-mediated angiogenesis by boosting recruitment of host stromal cells. 87 A recent study demonstrated that stromal-derived versican in B16F10 (melanoma) and LLC tumors localized preferentially to the vicinity of tumor vasculature and macrophages, whereas versican proteolytic fragments uniquely localized to the endothelium. Implantation of cancer cells in versican-haploinsufficient Vcanhdf/+ mice led to reduced tumor volumes and impaired vascularization compared with wild-type recipients. 88
Versican in Tumor Cell Adhesion, Migration, and Invasion
Versican is a proliferative, antiadhesive, and promigratory molecule that promotes cancer cell motility. 89 G1 and G3 domains appear to possess different properties in modulating cancer cell motility. Versican enhances the motility of cancer cells and reduces cell adhesion through its G1 domain. Versican enhances locomotion and reduces cell adhesion of astrocytoma cells through the binding of its G1 domain to HA and link protein (HAPLN1),74,90 whereas the versican G3 domain appears to be more important in local and systemic tumor invasiveness of breast cancer. 86 The formation of an HA–versican pericellular matrix enhanced prostate cancer motility in Boyden chamber motility assays using fibronectin as a chemoattractant. Thus, prostate cancer cells in vitro recruited versican produced by prostatic stromal cells to promote their motility. 89 These findings suggest that the formation of a pericellular sheath in vivo by prostate cancer cells using versican deposited by prostate stromal cells may contribute to the development of locally invasive disease. In glioma, versican V0 and V1 expression in tumor vessels was increased and was postulated to promote local invasion. 91 In chondrosarcoma, V1 isoform transfection enhanced tumor cell motility and migration. 92
Elevated versican expression in tumor-associated stroma resulted in enhanced cancer cell local invasion in cervical cancer, 93 whereas increased expression of CD44 and versican was associated with reduced expression of E-cadherin in endometrial cancer. 94 Interestingly, intraepithelial CD8+ T-cells were reduced in cervical cancers overexpressing versican 93 (see also section on tumor immunity below).
Versican in Tumor Metastasis
Versican accumulation has been linked with tumor metastasis to distant organs. Michael Karin’s group has demonstrated that by activating TLR2/TLR6 complexes and inducing TNF-α secretion by myeloid cells, versican strongly enhanced metastatic growth in the murine lung cancer model, LLC. TLR2 was absolutely necessary for metastatic growth, as no metastatic enhancement was seen in Tlr2−/− mice. 31 TNF-α was one of the major prometastatic factors produced by host myeloid cells. TNF-α can suppress cancer cell apoptosis and stimulate proliferation through nuclear factor kappa B activation. 95 As TLR2 is absolutely necessary for versican’s metastatic potential and TNF-α is secreted by versican-stimulated myeloid cells, either or both of these targets could provide useful tools for antimetastatic therapeutic intervention.
In mouse mammary tumor virus-polyoma middle T-antigen (MMTV-PyMT)-induced cancers, versican derived from the CD11b+ Ly6Chigh monocytic fraction is crucial in promoting metastasis to the lung in a TGF-β-dependent manner. 50 Consistently, versican knockdown in the bone marrow significantly impaired lung metastases in vivo, without impacting their recruitment to the lungs or altering the immune microenvironment. 50 In accordance with these observations, a recent study using 4T1 murine mammary carcinoma demonstrated that versican expression in the lung correlated with tumor-associated macrophage (TAM) accumulation and increased the numbers of pulmonary metastatic nodules. 96
Complex and Conflicting Roles of Versican in Tumorigenesis
Several already presented studies alluded to complex regulation of the apoptotic machinery by versican (see earlier section on tumor apoptosis). A small body of intriguing literature further suggests that versican might be antitumorigenic through diverse activities. Fanhchaksai et al. 97 showed that versican V3 limited fibrosarcoma tumor growth and tumor angiogenesis. Moreover, versican V3 overexpression reduced melanoma cell growth. 98 Yet a study by Miquel-Serra et al. 99 showed in the same model that versican V3 overexpression promoted lung metastasis. Contradictory effects of versican V1 were highlighted by two additional studies: Fujii et al. 100 reported that although versican overexpression conferred lymphoid cells with increased migratory capacity, it also rendered them hypersensitive to activation-induced cell death and certain chemotherapeutics. In another contrast, V1 cells (cultured fibroblasts overexpressing versican V1) induced tumor formation in nude mice but were also found sensitive to a wide range of cytotoxic agents. 81 These examples highlight the complexity of understanding versican’s role in cancer.
Versican in Adaptive Antitumor Responses
Versican acts in the interface between innate inflammatory and adaptive immune responses in cancer.30,101 Dendritic cells (DC) play a crucial role in the regulation of the balance between CD8+ T-cell immunity vs tolerance to tumor antigens. The important cross-presenting Batf3-DC subset has been shown to be essential for intratumoral effector T-cell response and immunotherapy efficacy. 102 Batf3-DC are indispensable for “innate sensing” of tumors and priming of the adaptive immune response. 103 Nevertheless, DC-mediated cross-presentation of tumor antigens in tumor-bearing hosts often induces T-cell tolerance instead of immunity. There is strong evidence that the TME modulates tumor-infiltrating DC and other antigen-presenting cells, such as macrophages, leading to impairment of antitumor immunity and even promotion of tumor progression.104,105
Tumor-derived versican leads to DC dysfunction through TLR2 activation. TLR2 ligation not only triggered the secretion of autocrine IL-10 and IL-6 but also led to sustained elevation of the cell-surface receptors for these cytokines, which decreased the threshold concentration required to activate STAT3. This positive feedback loop rendered DCs tolerogenic, producing high amounts of IL-10 rather than IL-12 and IL-1β when stimulated with LPS, a classic TLR4 agonist. Thus, versican impeded immunogenic DC maturation and conceivably downstream Th1 and cytotoxic lymphocyte differentiation.106,107
In MM, versican is robustly expressed and processed in the bone marrow. 53 Our group has suggested a model in which versican activates myeloma-associated monocytes/macrophages through TLR2/6 signaling, thus triggering trophic IL-1β and IL-6 upregulation. 53 The significance of versican pathway for human MM was further highlighted by two recent reports: first, high-resolution analysis of the human immune microenvironment in MM showing that myeloid-derived versican transcription was very strongly correlated with MM progression and loss of protective T-cell stem-like (Tcf1+) memory in favor of dysfunctional/exhausted T-effectors; 108 and second, the demonstration that immunosuppressive macrophages (expressing versican, ENTPD1, and STAB1) were associated with persistence of minimal residual disease (MRD) post–autologous stem cell transplant (ASCT) for MM, thus leading to relapse. 109
In mesothelioma, versican enhanced tumor progression through protumoral macrophage polarization. Mice harboring versican-deficient tumors presented fewer tumor/pleural macrophages and neutrophils, and fewer pleural T-regulatory cells, compared with control animals. 110 In conclusion, the multipartite immune cell cross-talk orchestrated by versican leads to an immunosuppressive TME that favors cancer progression and metastasis.
Versican Proteolysis and Versican-matrikines in Tumor Immunity and Immunotherapy
Immunotherapeutic interventions show effectiveness across a wide range of cancer types, but only a subset of patients shows clinical response to therapy.111,112 Response to immunotherapy has been linked to the presence of a preexisting “inflamed” TME. However, immune cell cross-talk with the extracellular matrix has been a relatively understudied facet of intratumoral immune regulation. 113
Regulated proteolysis of versican by ADAMTS proteases at the Glu441–Ala442 bond of the V1 isoform is associated with robust CD8+ infiltration in MM bone marrow114,115 and solid tumors. 116 The association between versican proteolysis and CD8+ infiltration may reflect the intratumoral depletion of versican (compared with findings in Gorter et al. 93 ) and/or the novel functions of the released bioactive versican fragments (versican-matrikines). Matrikines have been defined as ECM-derived fragments that regulate cell activity, often in a manner distinct from that of their parent macromolecule. 117 Proteolytic processing of versican V1 at Glu441–Ala442 generates a bioactive N-terminal matrikine, versikine (Figs. 1 and 2). Versikine has important and non-redundant roles in organ development, summarized in state-of-the-art reviews by the Apte group 3 as well as by Timms and Maurice. 118
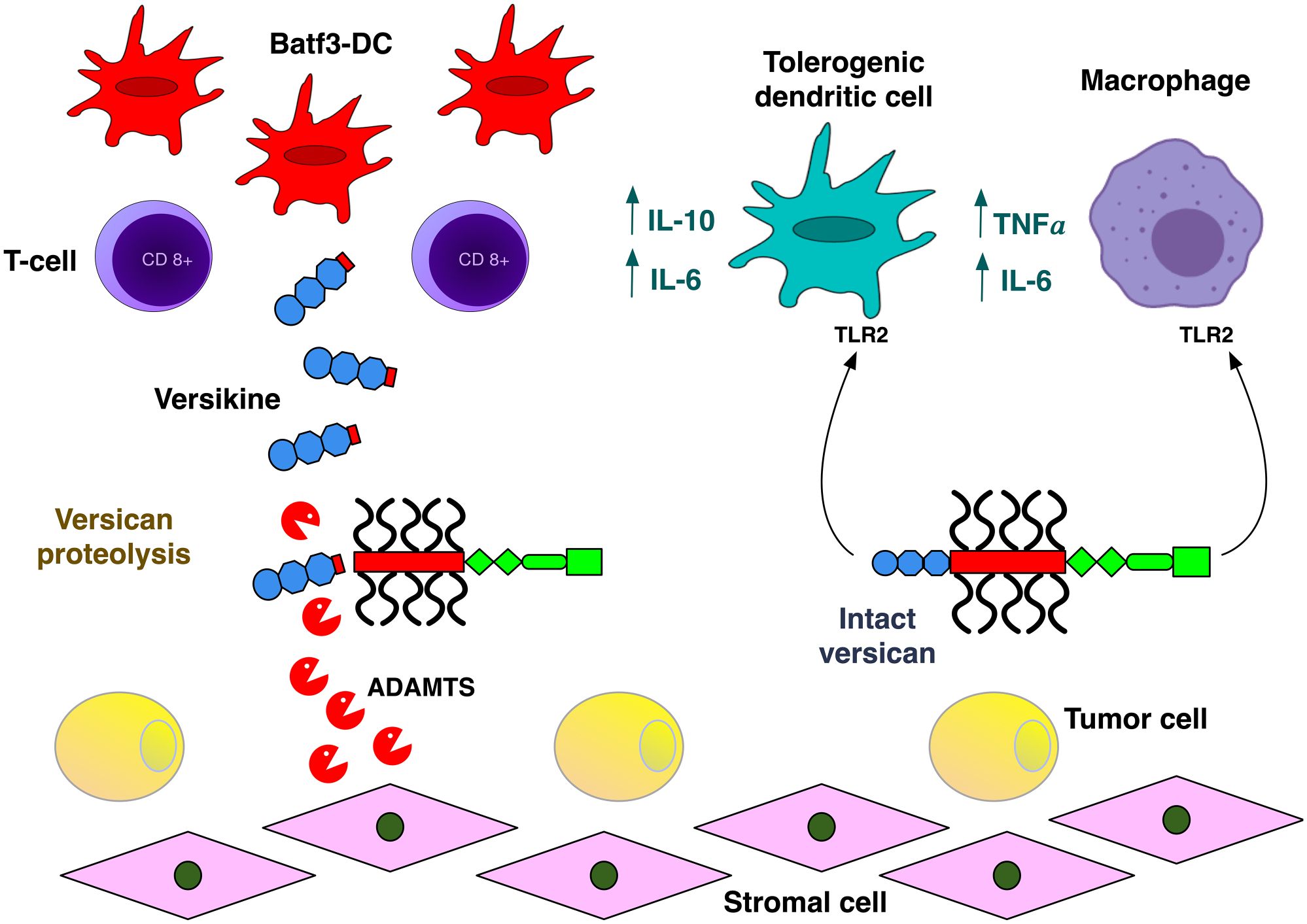
Concerted actions of versican and its proteolytic product, versikine, in tumor-immune cell cross-talk. Intact versican acts through TLR2 on antigen-presenting and other tumor-associated myeloid cells to influence their polarization toward protumoral roles. Versican’s proteolytic product, versikine, promotes Batf3-DC in the tumor bed and thus could act to mitigate the detrimental effects of parental versican in antitumor immunity. Versican proteolysis is associated with T-cell infiltration in many cancers, both solid and hematopoietic. For simplicity, only the versican V1 isoform is depicted. Abbreviations: ADAMTS, a disintegrin and metalloproteinase with thrombospondin motifs; DC, dendritic cell; IL, interleukin; TNF-α, tumor necrosis factor-a; TLR2, toll-like receptor 2.
Versikine has intriguing roles in inflammation and cancer in the adult organism. In contrast to versican, versikine triggers type I interferon–dependent transcription in myeloid cells. 115 In addition, versikine promotes IRF8-dependent Batf3-DC119,120 generation from Flt3L-mobilized BM in vitro 116 and Batf3-DC density in vivo. 121 Batf3-DCs within the TME are necessary for the effector phase of the antitumor T-cell response by orchestrating chemokine networks that enhance intratumoral CD8+ infiltration. 102
To dissect the effects of versikine in DC intratumoral composition in vivo, we used a genetically engineered Ras-driven MM model (VQ)122,123 and transplantable solid tumor models (LLC lung carcinoma and 4T1 mammary carcinoma). Tumor cells were stably engineered to secrete versikine vs empty-vector controls, and they were implanted into syngeneic recipients. Versikine altered the DC milieu in the tumor bed by increasing the density of intratumoral Batf3-DC and depleting the cDC2 (CD11c+ CD11b+) subset. Versikine synergized with STING agonist to augment antigen-specific T-cell immunity in LLC tumors. 121
In recently published data from our laboratory, we observed that intense versican proteolysis in the bone marrow of patients who underwent ASCT for MM correlated with adverse outcomes despite robust CD8+ infiltration. 114 Versican accumulation in this context is likely to generate an intensely immunosuppressive microenvironment that promotes effector dysfunction and impaired antitumor responses, despite the potential moderating effects of versikine signaling (Fig. 2).
Versican-matrikines other than versikine may have roles in regulating tumor immune responses. A proteolytic fragment of versican detected in neural tissue, glial hyaluronan-binding protein (GHAP), is such a candidate. 124 GHAP is a matrikine generated through ADAMTS proteolysis of versican V0 and versican V2 at a consensus cleavage site (Glu405–Gln406) distinct from the site cleaved to release versikine. 125 In preliminary unpublished data from our laboratory, GHAP was also shown to stimulate type I interferon–dependent transcripts in human macrophages. Further research is needed to investigate whether versikine and GHAP exert similar effects or whether their actions are wholly or partially non-overlapping.
Versican-matrikines (versikine, GHAP, and maybe other less well-characterized entities) are released through the actions of MMPs, including MMP and ADAMTS family members. A full discussion of the regulation of multiple families of these proteases is beyond the scope of this review; however, the reader is referred to excellent review articles on this topic.118,126
Versican in Cancer Biomarker Development
Versican molecular signatures may generate novel prognostic and predictive biomarkers in cancer. A combination of comparative genomic expression profiling and immunohistochemical staining of tissue microarrays from patients followed by multivariate analyses highlighted versican as an unfavorable independent indicator of metastasis-free and disease-free survival in gastrointestinal cancers. 127 In hepatocellular carcinoma (HCC), versican expression correlated with poor prognosis, increased TAM infiltration, poor tumor differentiation, and a higher tumor-grade metastasis (TNM stage).56,128 In colon cancer, versican expression by reverse transcriptase PCR was significantly upregulated (3-fold) compared with normal tissues. 129 Versican was shown to be a stronger prognostic factor than tumor grade in early-stage prostate cancer, and patients with low versican concentration in the peritumoral stroma had significantly better progression-free survival (PFS) than patients with high levels of versican. 66 Increased stromal versican expression correlated with both increased risk of disease recurrence and shortened survival in primary oral squamous cell carcinoma (OSCC). 130 Our group recently published the first set of data ascribing prognostic significance to the versican proteolysis immunoregulatory pathway in a hematological malignancy. We observed the somewhat paradoxical association between intense versican proteolysis and high CD8+ T-cell infiltration with poor post-ASCT survival in MM. Patients with low versican proteolysis compared with moderate/high versican proteolysis had better 2-year PFS (72% vs 29%, p=0.018) and 2-year OS (83% vs 35%, p=0.006). 114 It is important to note that in the post-ASCT bone marrow microenvironment, versican proteolysis paralleled versican accumulation, but this correlation is not universal in other tumor and microenvironmental contexts. Thus, versican expression and/or proteolysis detection may provide powerful prognostic, and in certain cases predictive (e.g., association of versican proteolysis with CD8+ T-cell infiltration and potential response to immune checkpoint inhibitors), cancer biomarkers. 113
There is growing understanding of the multiple roles of versican in tumor progression. Central pathogenic processes such as tumor cell proliferation, tumor cell survival and apoptosis, and tumor cell adhesion and migration have been found to be regulated by versican. Versican promotes tumor vasculature formation, local invasion, metastasis, and chemoresistance. Both intact versican and its proteolytic derivatives modulate the tumor immune milieu. These observations provide a rationale for testing versican accumulation, versican molecular signatures, and versican proteolysis as potential biomarkers to predict patient outcomes. Moreover, tumor matrix–targeting strategies may enhance traditional cancer treatment modalities and novel immunotherapies to improve patient survival.
Footnotes
Competing Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
AP and FA wrote the manuscript. GA, AC, and JW edited the manuscript. AP and FA produced the unpublished data quoted in the manuscript.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Work in the authors’ laboratory is supported by the National Institutes of Health/National Cancer Institute (R01CA252937), the American Cancer Society (RSG-15-045-01-LIB), the Leukemia and Lymphoma Society (6551-18), and the Robert J. Shillman Foundation.