
Abstract
Autophagy has been involved in the pathogenesis of various lung diseases. However, it is not yet known whether autophagy plays a role in hypersensitivity pneumonitis (HP). HP is an interstitial lung disease resulting from exposure to a wide variety of antigens that provoke an exaggerated immune response in susceptible individuals. The aim of this study was to explore the localization of autophagy key proteins in lungs from HP patients and controls by immunohistochemistry and analyze their expression levels by immunoblot. Macrophages and epithelial cells were strongly positive for the autophagosome biomarker LC3B (microtubule-associated protein light chain 3 beta) in HP lungs compared with controls. A similar pattern was found for the autophagy receptor p62 and the enzyme ATG4B. Unexpectedly, nuclear p62 signal was also noticed in macrophages from HP lungs. Regarding ATG5 and ATG7 localization, we observed positive staining in neutrophils, vascular smooth muscle cells, and endothelial cells. Our findings provide for the first time evidence that proteins from the autophagy machinery are highly expressed in the lungs of HP patients and describe the specific cellular and subcellular localization of LC3B, p62, ATG4B, ATG5, and ATG7 in HP lungs:
Introduction
Hypersensitivity pneumonitis (HP) is an inflammatory interstitial lung disease resulting from exposure to a wide variety of organic antigens (from fungi, bacterial, protozoa, and animal origin) and non-organic compounds (known as haptens) that provoke an exaggerated immune response in susceptible individuals.1,2 HP may occur in a variety of occupational, home, and recreational environments and has a variety of names based on the source of exposure (e.g., farmer’s lung, pigeon breeder’s disease). Hypersensitivity responses to a given antigen involve a latent period and are developed in two stages: a sensitization phase and an elicitation or effector phase. 3 The clinical behavior is heterogeneous and may present as acute or chronic forms.1,2,4,5 Importantly, patients with chronic HP may evolve to progressive lung fibrosis, and in an advanced stage may be very difficult to distinguish from idiopathic pulmonary fibrosis (IPF). Strong evidence indicates that HP is primarily a T-cell-mediated disease; however, immunocomplexes with specific IgG antibodies also have important regulatory functions.1,2
Autophagy influences the development, homeostasis, and survival of inflammatory cells, including macrophages, neutrophils, and lymphocytes, which play critical roles in the development and pathogenesis of inflammatory processes. 6 Moreover, strong evidence has demonstrated that autophagy is critical in the regulation of innate and adaptive immune responses in the respiratory system through regulation of antigen presentation and lymphocyte development, inflammasome activities, direct elimination of invading pathogens, and modulation of cytokine secretion and signaling, among others. 7 Given that autophagy has a critical role in both inflammatory response and lung homeostasis, we wondered whether it also contributes to the pathogenesis of HP.
Autophagy is a degradation pathway of cytoplasmic components within lysosomes, and the key proteins in this pathway are encoded by AuTophaGy-related (ATG) genes.6 –8 Autophagy is mediated by a unique structure consisting of double bilayer membranes called the autophagosome. Autophagy includes several sequential steps: initiation, elongation, autophagosome maturation, fusion with the lysosome (autolysosome formation), cargo degradation by hydrolases, and recycling of degraded products (Supplemental Fig. 1A). 8 Autophagosome biogenesis involves two ubiquitin-like conjugation systems, the ATG12 and the LC3 systems. In the first conjugation system, ATG12 is covalently conjugated to ATG5 by a series of ubiquitination-like reactions that involve ATG7 and ATG10. The ATG12–ATG5 complex non-covalently binds ATG16L to form a trimeric complex that binds to membranes for the recruitment of LC3 and autophagosome formation (Supplemental Fig. 1B). In the second conjugation system, the ATG4B protease cleaves pro-LC3B protein (microtubule-associated protein 1 light chain 3 beta encoded by the MAP1LC3B gene) to expose a glycine residue at the C-terminus to generate the LC3B-I form, which is then activated by ATG7 and conjugated to ATG3. The ATG3–LCB3-I complex binds to the ATG16L trimeric complex, and the exposed glycine is then conjugated to phosphatidylethanolamine (PE) generating the LC3B-II form. LC3B-II is then incorporated into the outer and inner membranes of the autophagosome, leading to its elongation and maturation. 9 After autolysosome formation, LC3B-II in the outer membrane is deconjugated by the ATG4B protease that removes lipid conjugates and releases LC3B to the cytosol. LC3B interacts directly with the receptor and adaptor protein Sequestosome-1/p62 (encoded by the SQSTM1 gene). The p62 receptor recognizes ubiquitinated cargo and targets it into autophagosomes for lysosomal degradation and recycling (Supplemental Fig. 1C).8,9
Previous studies have shown that autophagy in chronic respiratory diseases can be both protective and detrimental. For example, accumulation of polyubiquitinated proteins and defective autophagy have been described in cystic fibrosis and IPF lungs, suggesting that autophagy could play a role as a protective mechanism.10,11 In addition, we have demonstrated that Atg4b-deficient mice display significantly higher inflammatory and fibrotic responses to bleomycin and tunicamycin treatments.12,13 Moreover, Atg7 deficiency in macrophages impairs host defense against Klebsiella pneumoniae, and Atg5 deficiency exacerbates inflammation and fibrosis after silica and bleomycin challenge.14,15 By contrast, increased protein levels of LC3B-II and p62 as well as a high number of autophagosomes were found in chronic obstructive pulmonary disease (COPD) lungs compared with control tissues, suggesting that dysregulated autophagy drives lung inflammation and injury. 16 Similarly, elevated levels of BECLIN-1 and ATG5 have been shown in ciliated epithelial cells from asthmatic patients, suggesting a potential role of ciliophagy in asthma. 17
Despite the recognition of autophagy in driving inflammatory responses and regulating several mechanisms in chronic lung diseases, its role in HP has not yet been studied. In this study, we explored for the first time the cellular and subcellular localization of key autophagy proteins that belong to the two conjugation systems that are crucial for autophagosome biogenesis (ATG5, ATG7, ATG4B, and LC3B) and for cargo recognition (p62) by immunohistochemistry, as well as their protein expression level by immunoblot in lung tissues from patients with chronic HP and control subjects.
Materials and Methods
Tissue Samples
Formalin-fixed paraffin-embedded (FFPE) tissue blocks were selected from the National Institute of Respiratory Diseases Biobank. We analyzed n=6 patients diagnosed with chronic HP (four females, two males, mean age = 53 ± 6.5 years, three former smokers) and three controls (one female, two males, mean age = 59 ± 8.5, non-smokers). Diagnosis of HP was based on combined clinical criteria, history of exposure to antigens and laboratory proof of exposure (serum-specific IgG), bronchoalveolar lavage lymphocytosis, and high-resolution computed tomography (HRCT) of the chest compatible with HP, including the presence of poorly formed granulomas, ground-glass attenuation, mosaic in inspiration and air trapping in expiration, and histopathological features compatible with HP. 18 Chronic HP was defined as HRCT displaying in addition to micronodules and ground-glass attenuation, fibrotic lesions (defined as the presence of reticulation, traction bronchiectasis, and/or honeycombing), and the presence of fibrosis and architectural distortion in the histopathological evaluation of the lung biopsy affecting more than 10%. The research protocol was approved by the Ethics Committee of the National Institute of Respiratory Diseases, Mexico.
Immunohistochemistry
Immunohistochemistry was performed in 3-µm-thick FFPE lung tissue sections from six HP patients and three controls. The tissue sections were deparaffinized and then rehydrated and blocked with 3% H2O2 in methanol, followed by antigen retrieval in a microwave in 10 mM citrate buffer, pH 6.0. Tissue sections were treated with universal blocking solution (HK085-5K; BioGenex, Fremont, CA) for 10 min and then incubated overnight at 4C with the following primary antibodies: anti-LC3B (L7543; Sigma-Aldrich, St. Louis, MO), anti-p62 (P0067; Sigma-Aldrich), anti-ATG4B (A2981; Sigma-Aldrich), anti-ATG5 (A0731; Sigma-Aldrich), and anti-ATG7 (A2856; Sigma-Aldrich). A secondary biotinylated anti-immunoglobulin followed by horseradish peroxidase–conjugated streptavidin (HK330-5K; BioGenex) was used according to the manufacturer’s instructions. The reagent 3-amino-9-ethyl-carbazole (HK092-5K; BioGenex) in acetate buffer containing 0.05% H2O2 was used as the substrate. The sections were counterstained with hematoxylin. The primary antibody was replaced by non-immune serum for the negative control slides. Histology slide images were analyzed and captured with an optical microscope using ×20, ×40, and ×100 objectives and NIS-Elements software (Nikon Instruments; Melville, NY). A minimum of five representative, non-overlapping fields from lungs of six HP patients and three controls were evaluated.
Immunoblot
Lung tissue was homogenized in a 20-mM Tris buffer, pH 7.4, containing 150 mM NaCl, 1% Triton X-100 (T8787; Sigma-Aldrich), 10 mM EDTA, and UltraCruz Protease Inhibitor Cocktail (sc-29131; Santa Cruz Biotechnology, Dallas, TX). Tissue and cell extracts were centrifuged at 15,000 × g at 4°C and supernatant fractions were collected. Protein concentration was quantified by bicinchoninic acid technique (BCA protein assay kit 23225; Thermo Fisher Scientific, Waltham MA). A total of 25 µg of protein was loaded on either 8% or 13% SDS polyacrylamide gels. After electrophoresis, gels were electrotransferred onto polyvinylidenedifluoride membranes (IPV H00010; Millipore, Burlington, MA), and then the membranes were blocked with 5% non-fat dried milk in TBS-T (Tris-buffered saline with 0.05% Tween 20) and incubated overnight at 4C with primary antibodies diluted in antibody diluent (003118; Thermo-Fisher Scientific). The same primary antibodies used for immunohistochemistry were used for incubating the membranes, except the anti-GAPDH antibody (sc-47724; Santa Cruz Biotechnology). After three washes with TBS-T, membranes were incubated with the corresponding secondary antibody at 1:3000 dilution in 1.5% milk in TBS-T and developed with Immobilon Western Chemiluminescent HRP substrate (WBKLS0500; Millipore).
Statistics
All experimental data are reported as mean ± SD. Statistical analyses were performed by Student’s t-test using GraphPad Prism Software Version 6.0 (GraphPad Software Inc., San Diego, CA), and p values lower than 0.05 were considered significant.
Results
LC3B and p62 Immunolocalization in Lung Tissue From Controls and HP Patients
Lung tissue sections from controls and HP patients were analyzed for the expression and localization of autophagy proteins. LC3B has been typically characterized as an autophagosome biomarker because it is incorporated into both the inner and outer autophagosome membranes. 8 In control lungs, we observed low LC3B diffuse cytoplasmic staining only in some macrophages, whereas other cell types were negative (Fig. 1A). Conversely, a strong LC3B-positive staining was observed in alveolar and interstitial macrophages and in some neutrophils in the lungs of patients with HP. Almost all accumulated macrophages within alveolar spaces were positive for LC3B and showed a granular cytoplasmic pattern in HP lungs (Figs. 1B, C and 2A and B). A strong cytoplasmic positive staining for LC3B was frequently observed in hypertrophic and hyperplastic alveolar epithelial cells, in the epithelium from respiratory bronchioles between bridging fibrosis, and also in epithelial cells between centrilobular fibrosis (Figs. 1C, D and 2A and B). No LC3B-positive signal was observed in either HP or control tissues in the absence of primary antibody, indicating that the staining was specific.
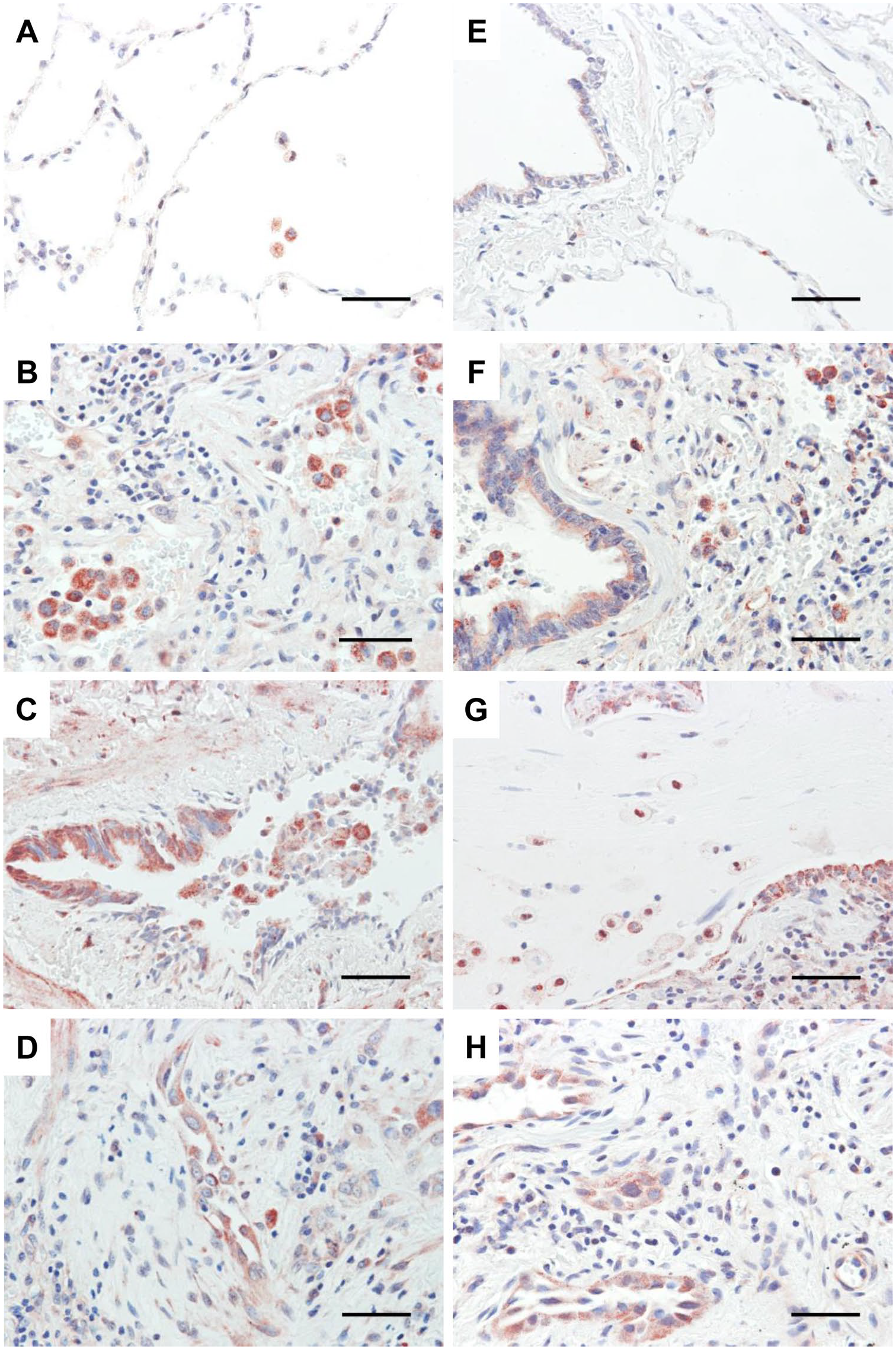
LC3B and p62 immunostaining of lung tissue from controls and HP patients. Representative photomicrographs of LC3B immunohistochemical staining in lung tissue from control (A) and HP patients (B–D). P62 immunohistochemical analysis in lung tissue sections from control (E) and HP patients (F–H). Positive signal is observed in red. All sections were counterstained with hematoxylin (blue). Scale bars in all images represent 50 µm. Abbreviation: HP, hypersensitivity pneumonitis.
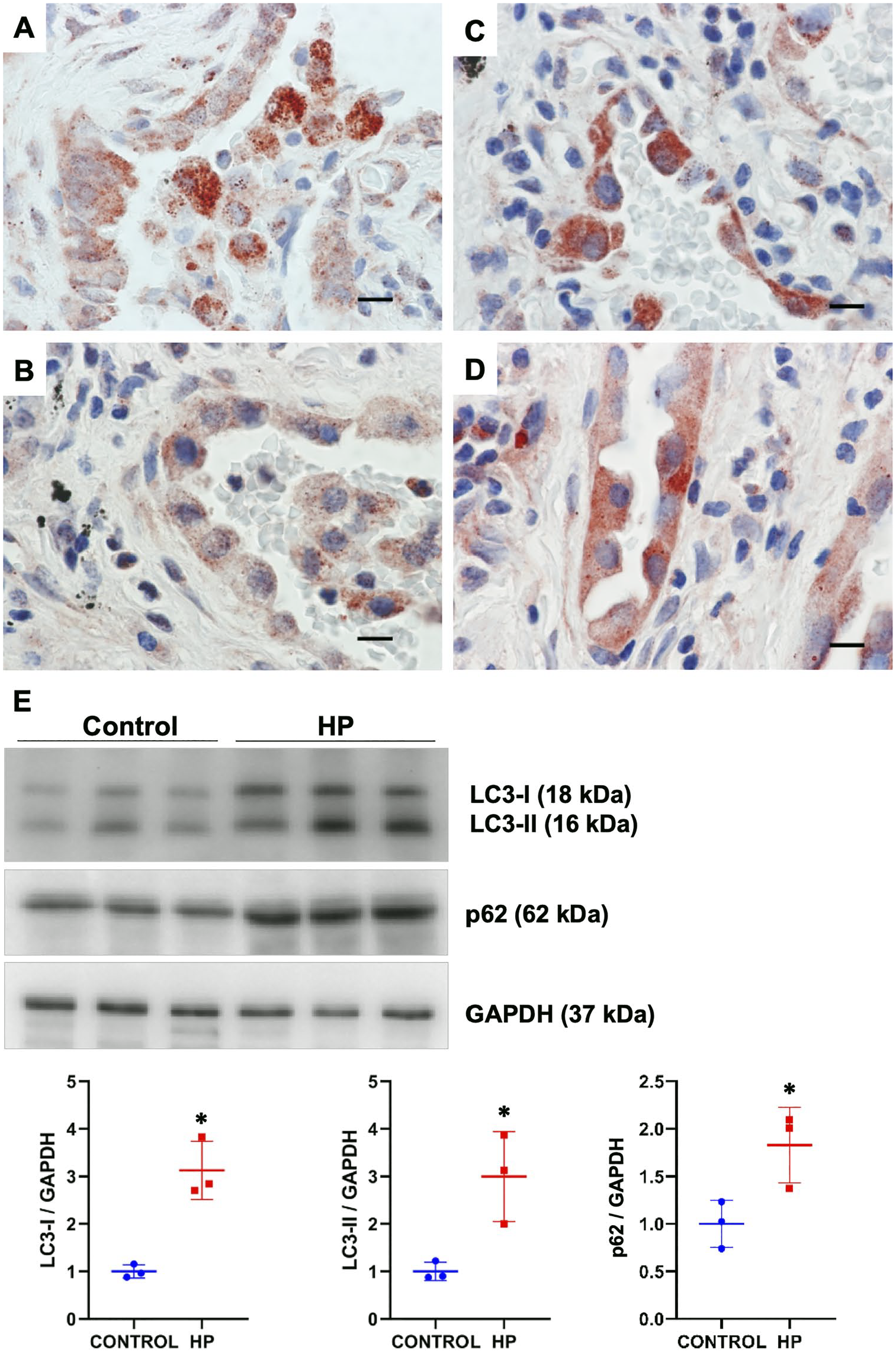
Granular cytoplasmic positive staining for LC3B (A and B) and p62 (C and D) in epithelial cells in lung tissue sections of HP patients. Scale bars in all images represent 50 µm. Representative immunoblots of LC3B-I/II and p62 in total lung tissue extracts from controls and HP patients (E). GAPDH was used as loading control. Densitometry analysis (bottom panels). Results are shown as mean ± SD. Statistical significance was determined by Student’s t test (*p < 0.05). Abbreviations: HP, hypersensitivity pneumonitis; GADPH, glyceraldehyde-3-phosphate dehydrogenase.
Because of its direct interaction with LC3B, the p62 receptor was also investigated. In control lungs, low p62 staining was observed in some macrophages and bronchial epithelial cells. In HP lungs, a strong p62 positive staining, quite similar to the LC3B granular cytoplasmic pattern, was observed in interstitial and alveolar macrophages and also in neutrophils (Fig. 1F and G). Similarly, bronchial and alveolar epithelium showed a granular cytoplasmic staining (Figs. 1F–H and 2C and D). Interestingly, we also found a positive nuclear localization of p62 in alveolar macrophages in HP lungs, whereas nuclear staining in macrophages from control lungs was uncommon (Fig. 1G). In addition, by immunoblot, we observed an increased LC3B and p62 protein level in lung tissue extracts from HP patients compared with control tissues, confirming the findings obtained by immunohistochemistry (Fig. 2E).
Immunolocalization of ATG4B, ATG5, and ATG7 in Lung Tissue From Controls and HP Patients
ATG4B is a cysteine protease that belongs to the LC3 conjugation system and mediates the cycle of LC3-lipidation and delipidation, thereby representing a central node in the regulation of the autophagic flux.12,13 Control lungs exhibited a low level of ATG4B staining in macrophages, whereas other cell types were negative (Fig. 3A). In the lungs from HP patients, ATG4B-positive staining was identified mainly in alveolar macrophages and in bronchial and alveolar epithelial cells lining areas of fibrosis (Fig. 3B–D).
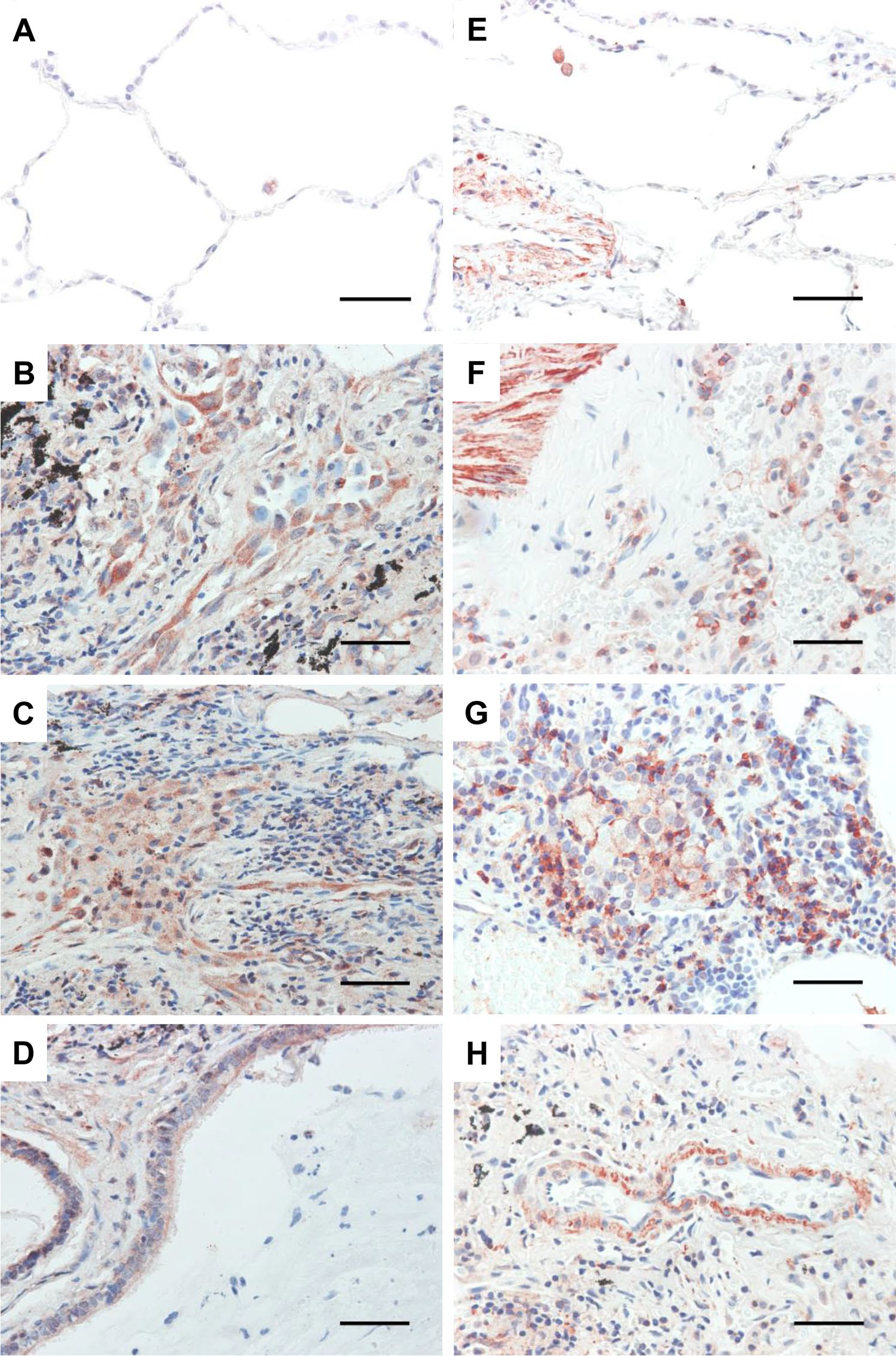
ATG4B and ATG5 immunostaining of lung tissue from controls and HP patients. Representative photomicrographs of ATG4B immunohistochemical staining in lung tissue sections from control (A) and HP patients (B–D). ATG5 immunohistochemical staining in lung tissue sections from control (E) and HP patients (F–H). Positive signal is observed in red. All sections were counterstained with hematoxylin (blue). Scale bars in all images represent 50 µm. Abbreviation: HP, hypersensitivity pneumonitis.
ATG5 belongs to the ATG12 conjugation system, whereas ATG7 participates in both ATG12 and LC3 conjugation systems.8,9 The expression of ATG5 was found in both control and HP lungs, mainly in bronchial and vascular smooth muscle cells (Fig. 3E). However, strong staining of ATG5 was also identified in endothelial cells, macrophages, and some neutrophils in alveolitis areas in HP lungs (Fig. 3F–H). Particularly, we observed an ATG5 accumulation near to the plasma membrane in some macrophages and mast cells in HP lungs (Fig. 5C). Regarding ATG7, control lungs exhibited a low level of staining in some macrophages and other cell types were negative (Fig. 4A), whereas strong positive signal was observed in macrophages and bronchial epithelial cells and in some neutrophils from HP lungs (Fig. 4B–D). By immunoblot, we found a significant upregulation of ATG4B and ATG5, and a non-significant increase in ATG7 levels in HP lungs compared with control tissues (Fig. 4E).
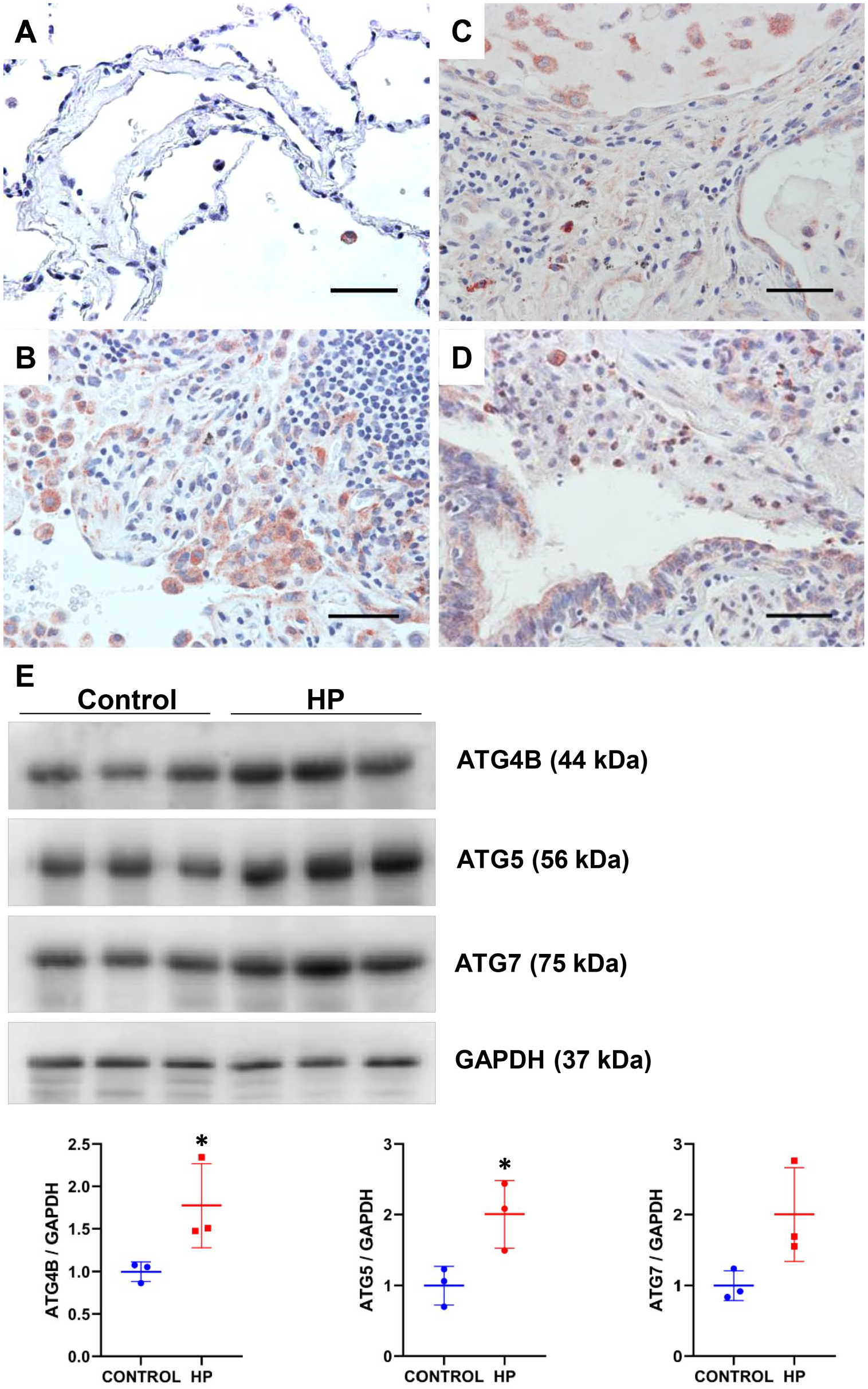
Immunohistochemical analysis of ATG7 in lung tissue sections from control (A) and HP patients (B–D). Scale bars in all images represent 50 µm. Representative immunoblots of ATG4B, ATG5, and ATG7 in total lung tissue extracts from controls and HP patients (E). GAPDH was used as loading control. Densitometry analysis (bottom panels). Results are shown as mean ± SD. Statistical significance was determined by Student’s t-test (*p < 0.05). Abbreviations: HP, hypersensitivity pneumonitis; GADPH, glyceraldehyde-3-phosphate dehydrogenase.
Different Subcellular Localization Patterns for Autophagy Markers in Macrophages From HP Lungs
Our results demonstrate that all autophagy markers analyzed in this study are expressed in macrophages from both controls and HP patients, although much stronger in HP lungs, suggesting that autophagy is constitutively active in macrophages under homeostatic conditions, but inducible during lung injury and inflammation. We also found a specific nuclear p62 signal in alveolar macrophages from HP lungs, but this finding was not confirmed with another anti-p62 antibody clone (Fig. 5B). In addition, an accumulation of ATG5 at the plasma membrane was observed in some macrophages in HP lungs (Fig. 5C). Table 1, summarizes the immunohistochemical findings for each autophagy marker and the specific lung cell type.
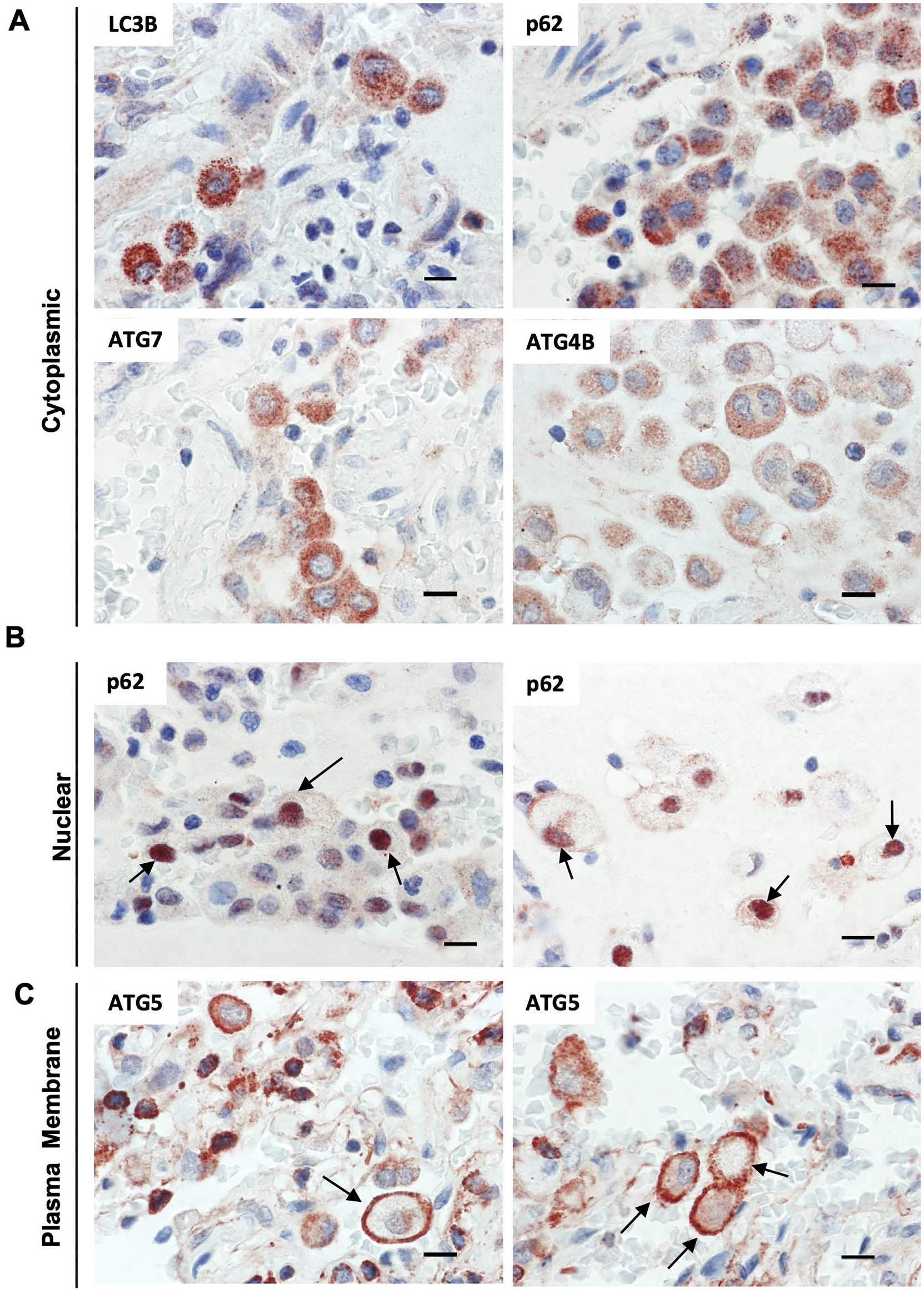
Differential staining patterns for autophagy markers in macrophages from HP lungs. Granular cytoplasmic immunostaining for LC3B, P62, ATG4B, and ATG7 in alveolar macrophages (A). Nuclear immunostaining for p62 in alveolar macrophages (arrows) (B). ATG5 accumulation in plasmatic membrane from alveolar macrophages and mast cells (arrows) (C). Scale bars in all images represent 10 µm. Abbreviation: HP, hypersensitivity pneumonitis.
Semiquantitative Analysis of Immunohistochemical Findings in Controls and Hypersensitivity Pneumonitis Patients.

Intensity staining score: negative (−), weak (+), moderate (++), and strong (+++). Abbreviations: C, control; HP, hypersensitivity pneumonitis.
Together, our results demonstrate an increased expression of LC3B, p62, ATG4B, and ATG5 in HP lungs compared with control tissues, suggesting that autophagy is involved in the pathogenesis of HP, and describe for the first time the cellular and subcellular localization of key autophagy markers in the lungs of patients with HP.
Discussion
In this study, we have shown that the expression of different components of the autophagy pathway is upregulated, suggesting that this process could be involved in HP pathogenesis. We evaluated proteins that belong to the two main conjugation systems that are crucial for autophagosome formation and maturation. In the first system, ATG7 and ATG10 catalyze the conjugation of ATG12 to ATG5 to form an ATG12–ATG5 complex.8,9 In the second system, the protease ATG4B activates proLC3B to LC3B-I, which is then conjugated to PE to form LC3B-II, a reaction catalyzed by ATG7 and ATG3. ATG4B has also an esterase activity that hydrolyzes the phospholipid link in LC3B-II, recycling LC3B to its cytoplasmic free form.12,13 We also chose the autophagy receptor p62 that directly interacts with LC3B and functions as a key cargo adaptor for the degradation of several substrates.6 –8
A basal level of autophagy is required for lung homeostasis, but it can be activated in response to cellular stress.7,8,12 We observed low p62 staining in bronchial epithelial cells of control lungs, but the other autophagy markers were undetectable in the bronchial and alveolar epithelium. As there is a basal autophagy level in all cell types, probably immunohistochemistry is not a method sensitive enough to detect autophagy markers in the epithelium under physiological conditions. By immunoblot, we were able to detect a basal expression level of all autophagy markers in lung extracts from control subjects, but we have to consider the contribution of all cell types. By contrast, strong positive staining of LC3B, p62, ATG4B, and ATG7 proteins was observed in bronchial and alveolar epithelial cells of HP lung tissues. Therefore, we hypothesized that the expression of autophagy proteins is increased in epithelial cells from HP lungs after exposure to organic or inorganic antigens.
A growing body of evidence supports our hypothesis and suggests that autophagy could be increased in the lung epithelium in response to infectious and non-infectious insults to maintain its integrity. 19 For example, Mycobacterium tuberculosis and Pseudomonas aeruginosa induced autophagosome formation in A549 alveolar epithelial cells, indicating that this degradation pathway participates in pathogen clearance. 20 Streptococcus pneumoniae infection or pneumolysin toxin overexpression in A549 alveolar epithelial cells increased the autophagic flux. 21 Also, inorganic insults, such as air pollution particulate matter, induced ATG5 and BECN1 mRNA expression in A549 cells compared with unexposed control cells. 22
However, several studies have documented that autophagy is a harmful process involved in epithelial apoptosis, ciliophagy (selective removal of cilia components), and inflammatory response, contributing to the pathogenesis of COPD, asthma, and cystic fibrosis. 19 LC3B has been identified as a positive regulator of epithelial apoptosis induced by cigarette smoke, through interactions with CAV1 and FAS. Map1lc3b-deficient mice had significantly decreased levels of apoptosis after cigarette smoke exposure and were protected from emphysema compared with wild-type littermate mice. 23 In addition, in human bronchial epithelial cells, increased autophagy has been associated with mucus hypersecretion after particulate matter exposure. 24 Thus, we do not know whether increased expression of autophagy proteins in epithelial cells in HP lungs is either cytoprotective or deleterious and whether it depends on the antigen nature and exposure. Therefore, functional studies are required to understand the role of this pathway in HP.
We also observed a strong granular cytoplasmic staining for LC3B and p62/SQSTM1 in macrophages in the lung from patients with HP, whereas a very weak signal was identified in macrophages from control lungs. It is important to note that a granular cytoplasmic staining pattern does not necessarily indicate high levels of active autophagy. These granules, which might represent autophagosomes, could indicate an increased autophagic flux in these cells and also autophagy inhibition because of impaired autophagosome clearance by lysosomes. 25 The autophagic activity not only involves increased synthesis or lipidation of LC3B but also, most importantly, the turnover and recycling of cargo into the autophagosomes.25,26 As HP is derived from inhalation of a variety of antigens such as fungi, thermophilic bacteria, bird proteins, and others, it is possible that xenophagy (a specialized form of autophagy for selective removal of pathogens) could be active in macrophages of HP lungs for the clearance of invading pathogens and other non-host entities. 27
However, it is worth mentioning that LC3 or p62 granular staining could be falsely interpreted as autophagosome formation. Emerging evidence indicates that components of the autophagy apparatus, such as LC3B and p62, could mediate non-autophagic and non-degradative functions. 28 LC3B can be incorporated in non-autophagic structures, for example, during LC3B-associated phagocytosis (LAP), a specialized form of endocytosis. LAP uses proteins of the canonical autophagy machinery, following ligation of surface receptors that recognize a variety of cargos including pathogens or dying cells. 28 LAP has been involved in the elimination of apoptotic cells by macrophages in a process known as efferocytosis. 29 Mice that are LAP-deficient within inefficient clearance of dying cells can result in the accumulation of apoptotic bodies in their tissues and inflammation.30,31 In our study, we found HP macrophages strongly stained for LC3B, suggesting that clearance of dead cells by LAP could be active; however, in vitro or in vivo models are needed to test this hypothesis.
The p62 receptor is a multifunctional protein involved in several cellular processes in addition to autophagy, such as antioxidant response, inflammation, and apoptosis. 32 An important finding in our study was the nuclear localization of p62 in macrophages within HP lungs, mainly those macrophages accumulated in the alveolar spaces. One limitation of our study is that we did not confirm this finding with another anti-p62 antibody clone. However, previous studies have revealed nuclear p62 translocation, as a possible mechanism for oxidative stress sensing, through stabilization and activation of NRF2 (nuclear factor, erythroid derived 2, like 2), a key redox homeostasis transcription factor. 33 Two nuclear localization signals have been identified in p62, and its nuclear translocation is modulated by phosphorylation. 34 Nuclear autophagy is also a mechanism for the clearance of nuclear aggregates that induce cellular stress and could be another possibility to explain p62 nuclear localization. 35 However, further studies are necessary to confirm this finding and to determine what stimulates p62 nuclear translocation in macrophages during HP pathogenesis. Regarding ATG5 and ATG7, we found these proteins were highly expressed in bronchial and vascular smooth muscle cells in HP lungs. Defective autophagy associated with deletion of Atg5 or Atg7 in vascular smooth muscle cells leads to accelerated senescence, neointima formation, and atherogenesis, suggesting that expression of these ATG proteins in this cell type is critical for vascular integrity during development and after injury.36 –38
Another limitation of our study is that we did not colocalize autophagy targets in lung tissues by immunohistochemistry. Although it was expected to find expression of all autophagy markers in the same cell types, as we summarized in Table 1, we found that some autophagy proteins were more abundant in certain cell types than others, probably due to different cell-type-specific autophagic and non-autophagic functions. This is an important issue for future research.
In summary, this study describes for the first time the specific cellular and subcellular localization of LC3B, p62, ATG4B, ATG5, and ATG7 in the lungs from HP patients compared with controls subjects. In addition, we found an increased protein level of LC3B, p62, ATG4B, and ATG5 in HP lungs compared with control lungs. However, this does not necessarily indicate high levels of active autophagy; instead, it could reflect defective autophagy and autophagosome accumulation during HP pathogenesis. Our study is merely descriptive, and additional complementary studies are needed to monitor autophagy in HP pathogenesis given that we are showing only a steady-state image of this dynamic process.
Supplemental Material
2020-00048R1_Production_Supplemental_Figure_1_online_supp – Supplemental material for Identification of Autophagy-related Proteins in Lungs From Hypersensitivity Pneumonitis Patients
Supplemental material, 2020-00048R1_Production_Supplemental_Figure_1_online_supp for Identification of Autophagy-related Proteins in Lungs From Hypersensitivity Pneumonitis Patients by Sandra Cabrera, Carolina Rodríguez-Bobadilla, Dulce Vázquez-Morales, Miguel Gaxiola, Mariana Maciel, Moisés Selman and Annie Pardo in Journal of Histochemistry & Cytochemistry
Footnotes
Acknowledgements
We would like to acknowledge the patients who generously donated lung tissue for this study. We also thank Dr. José Cisneros-Lira for his assistance in preparing figures and LANSBioDyT (Laboratorio Nacional de Soluciones Biomiméticas para Diagnóstico y Terapia, Facultad de Ciencias, UNAM).
Competing Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
SC and MM designed the study, performed experiments, and analyzed the results; MS and AP analyzed the results; CR-B performed the immunohistochemistry; DV-M performed immunoblot experiments; MG performed the histological classification of the lesions, and analyzed and recorded the images; SC, MS, and AP were involved in writing. All authors approved the manuscript.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by UNAM-PAPIIT IN211818 Research Program and SEP-CONACYT Research Program Project Number 235891.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.