
Abstract
Thrombospondin 1 (TSP1) is a matricellular extracellular matrix protein that has diverse roles in regulating cellular processes important for the pathogenesis of fibrotic diseases. We will present evidence for the importance of TSP1 control of latent transforming growth factor beta activation in renal fibrosis with an emphasis on diabetic nephropathy. Other functions of TSP1 that affect renal fibrosis, including regulation of inflammation and capillary density, will be addressed. Emerging roles for TSP1 N-terminal domain regulation of collagen matrix assembly, direct effects of TSP1–collagen binding, and intracellular functions of TSP1 in mediating endoplasmic reticulum stress responses in extracellular matrix remodeling and fibrosis, which could potentially affect renal fibrogenesis, will also be discussed. Finally, we will address possible strategies for targeting TSP1 functions to treat fibrotic renal disease.
Keywords
Introduction
Matricellular proteins are components of the extracellular matrix (ECM) that interact with cells and other ECM components: Their functions are to regulate cell behavior and ECM organization rather than acting as primary structural elements of the ECM.1 –3 Thrombospondin 1 (TSP1) is a prototypic matricellular ECM protein with diverse and significant roles in tissue remodeling and fibrosis. TSP1 is a disulfide-bonded, homotrimeric secreted glycoprotein and a member of the group A subfamily of five TSP family members. 4 TSP1 is composed of multiple structural domains with distinct intermolecular interactions, cellular receptors, and functional roles. 1 , 5 , 6 First studied as a component of platelet α-granules for its role in platelet aggregation, TSP1 is now known to be widely expressed by diverse cells in a highly regulated manner. 7 TSP1 regulates an array of cellular processes, primarily through its interactions with cellular receptors and also through interactions with other extracellular molecules, including ECM proteins and glycosaminoglycans, matrix-regulating enzymes, and growth factors.1,4 TSP1 has roles in regulating cell adhesion and deadhesion, cell migration, apoptosis, ECM organization, activity of matrix-degrading enzymes such as matrix metalloproteinases and lysyl oxidases, and growth factor activity.
Given these roles in regulating cellular functions, it is not surprising that TSP1 is important in disease processes involving tissue remodeling—both fibrotic and normal reparative states—and in inflammation. 5 , 8 Several functions of TSP1 have been shown to be involved in fibrotic disease. The most prominent role for TSP1 in fibrosis centers on its ability to activate latent transforming growth factor beta (TGF-β), particularly in diabetic nephropathy. 9 Other studies suggest that TSP1 regulates metabolic inflammation and fibrosis. 10 , 11 In addition, there are roles for TSP1 in regulating collagen matrix assembly and crosslinking. 5 , 12 , 13 Moreover, there is an emerging appreciation that intracellular TSP1 might have roles in endoplasmic reticulum (ER) stress and calcium regulation. 14 , 15
In this review, we will first focus on known roles for TSP1 in fibrotic renal diseases, with an emphasis on its role in controlling latent TGF-β activation (Fig. 1). We will also consider some emerging functions of TSP1 in ECM remodeling and discuss how these TSP1 functions could also contribute to dysregulation of ECM in fibrosis. Finally, we will discuss how these TSP1 functions might be regulated for therapeutic interventions.
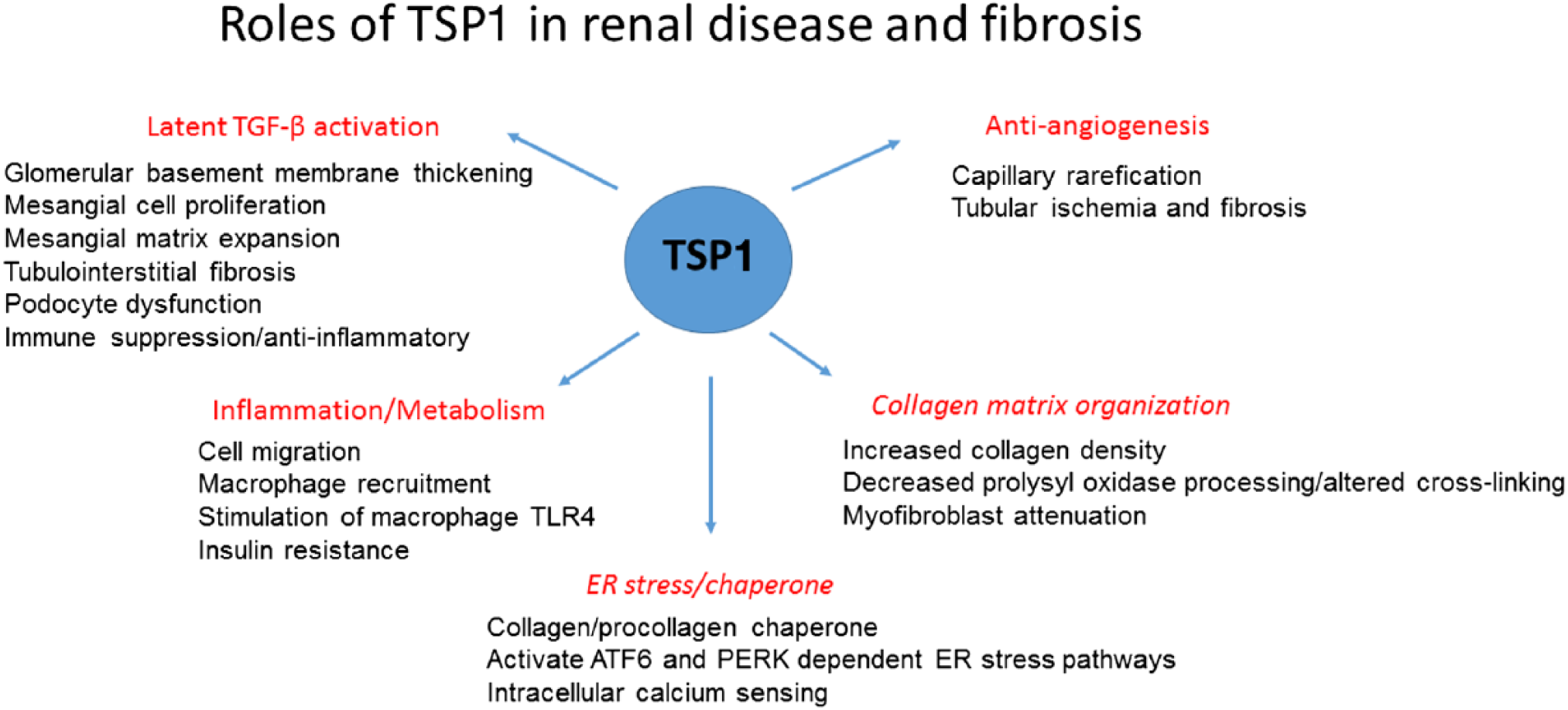
TSP1 has diverse functions in renal disease and fibrosis. Major functions are described in red, with specific implications for renal cells and disease pathogenesis listed below the headings. TSP1’s functions in collagen organization and as an ER stress regulator (headings are italicized) have not yet been investigated in the context of renal disease, although these functions might indeed be relevant to the pathogenesis of some renal diseases. Abbreviations: TSP1, thrombospondin 1; ER, endoplasmic reticulum; TGF-β, transforming growth factor beta.
TSP1 as an Activator of Latent TGF-β
Our lab discovered that human platelet TSP1 purified from releasates of thrombin-stimulated platelets was associated with biologically active TGF-β, also a major component of platelet α-granules. 16 Further studies by our group identified that TSP1 binds to both the small and large forms of latent TGF-β and that this binding interaction can convert the inactive precursor to a biologically active form of TGF-β in a protease-independent manner. 17 , 18 We identified key sequences in both TSP1 and the latent TGF-β complex necessary for latency and for TSP1-dependent activation of the latent complex.19 –22 Small peptides of the TSP1-activating sequence (KRFK) or of the TGF-β latency-associated peptide (LSKL) were used as probes to either mimic or inhibit TSP1-dependent TGF-β activation as a means of assessing the relevance of the TSP1–TGF-β pathway in vitro and in multiple animal models of disease. 5 ,23 –26 This function is specific to the TSP1 family member as thrombospondin-2 (TSP2) can bind to the latent complex but lacks the KRFK activation sequence, and the thrombospondin (TSP) type B subfamily members lack the type 1 repeat region that contains the KRFK and WxxW TGF-β binding sequences. 19 A detailed discussion of the mechanism and cellular variations by which TSP1 activates latent TGF-β was recently published. 26
TSP1 as an Activator of Latent TGF-β in Renal Fibrosis
TGF-β is a key mediator of renal fibrosis in multiple animal models and in diverse human renal diseases.27 –29 With the discovery by our lab that TSP1 can activate latent TGF-β, multiple groups investigated whether TSP1 plays a role in renal fibrosis through its ability to control latent TGF-β activation. 30 TSP1 is widely expressed at low levels under quiescent conditions by most renal cell types, including podocytes, mesangial cells, tubular epithelial cells, and macrophages.31 –34 As detailed in the specific studies described in the following sections, TSP1 is upregulated in human renal fibrotic diseases, in various animal models of renal fibrosis, and in renal cell culture systems exposed to fibrotic or inflammatory stimuli.35 –37
Using multiple different models of renal injury, Hugo et al. showed that TSP1 expression was upregulated in tubular cells and myofibroblasts and by some macrophages and that TSP1 expression predicted the severity of tubulointerstitial fibrosis. 36 , 38 Daniel et al. used antisense oligonucleotides to TSP1 in the anti-Thy-1 antibody rat model of mesangioproliferative glomerulonephritis: Thy-1 is a protein expressed on mesangial cells, and the anti-Thy-1 antibody model is a rapidly developing model of glomerulonephritis which mimics human mesangial cell proliferative glomerulonephritis characterized by mesangial cell lysis with later repopulation by proliferating myofibroblast-like mesangial cells, inflammatory cell infiltration, and glomerular fibrosis with eventual resolution. 39 , 40 Treatment with antisense oligonucleotides to TSP1 decreased TGF-β activity and Smad signaling as well as reduced ED-A fibronectin and collagen I deposition, but there was no effect on macrophage infiltration or glomerular mesangial or endothelial cell proliferation. 40 Further studies from this group used two different peptides that block TSP1 binding to latent TGF-β (LSKL, AAWSHW) to prevent TSP1-dependent activation and observed reduced TGF-β activation, glomerular ECM deposition, and proteinuria in the anti-Thy-1 antibody rat model. 9 The blocking LSKL peptide also reduced renal interstitial fibrosis in rats subjected to unilateral ureteral obstruction (UUO): Myofibroblast accumulation, phosphorylated Smad 2, tubular injury, and collagen deposition were reduced in rats treated with LSKL peptide. 41 In models of chronic kidney disease, mice lacking both Col4α3 and Thbs1 had delayed disease progression with reduced TGF-β activity, fibroblast proliferation, and fibrosis compared with animals lacking only Col4α3, but expressing TSP1. 42 In the absence of TSP1-activated TGF-β, there was a shift to a Th1 type of inflammation with decreased Th17 and FOXP3+ T-regulatory cells, likely due to loss of the anti-inflammatory activity of TGF-β, but also potentially due to loss of direct TSP1 actions. 42
Diabetes is the most common cause of end-stage renal failure in the United States. 29 , 43 TGF-β is well documented as a major factor in the pathogenesis of diabetic nephropathy, with involvement in the various processes leading to renal failure, including glomerular hypertrophy with mesangial matrix expansion, glomerular basement membrane thickening, renal tubular injury and interstitial fibrosis, and alterations in podocyte slit barrier function, leading to proteinuria. 29 ,44 –46 A myriad of factors in the diabetic milieu, including increased levels of glucose and advanced glycation end products, increased activity of the renin–angiotensin system and hypertension, and increased oxidative stress, are linked to increased TGF-β in diabetic renal disease.47 –53 Evidence from multiple labs has established TSP1 as a major regulator of the inappropriate TGF-β activity in diabetic nephropathy. Renal biopsy samples from patients with both type 1 and type 2 diabetic nephropathy showed corresponding increases in glomerular TSP1 and latent and active TGF-β expression during progression of diabetic nephropathy, and correlation of TSP1 expression, Smad signaling, and proteinuria was seen in patients with type 2 diabetic nephropathy. 37 , 54 TSP1 is also induced by both glucose and angiotensin II, two factors directly linked to increased TGF-β activity in diabetic nephropathy.55 –58 Our lab demonstrated that high glucose or angiotensin II treatment induced TSP1 protein by rat mesangial cells. 56 , 59 Glucose also induces TSP1 mRNA and protein by podocytes. 33 Glycated albumin stimulates increased USF2 expression and tubular TSP1 protein. 60 , 61 Under normal glucose conditions, nitric oxide–dependent activation of cGMP-dependent protein kinase (protein kinase G [PKG]) normally suppresses TSP1 transcription. 62 , 63 In contrast, increased oxidative stress under high glucose conditions decreases PKG activity, resulting in attenuation of transcriptional repression of TSP1 and upregulation of the transcription factor USF2 to mediate increased TSP1 transcription. 64 The importance of USF2 stimulation of TSP1 expression was shown in studies demonstrating that streptozotocin-treated USF2-overexpressing mice had accelerated development of diabetic nephropathy, with increased TSP1, collagen I, and TGF-β protein in glomeruli isolated from these mice. 65 The hexosamine pathway was also shown to induce TSP1 transcription and mRNA levels by high-glucose-treated vascular smooth muscle cells. 66 More direct evidence for the importance of TSP1 in regulating TGF-β activity in diabetic nephropathy comes from studies in two different models of type 1 diabetic nephropathy. The first model showed that streptozotocin-treated Thbs1 null mice are protected from glomerulosclerosis and podocyte loss, 67 and in the second model, treatment of uninephrectomized type 1 diabetic Akita mice with the TSP1 antagonist peptide LSKL reduced proteinuria and tubulointerstitial fibrosis and decreased glomerular TGF-β signaling as measured by phosphorylated Smad 2. 25 The Akita mouse has a mutated cysteine in the insulin gene, which prevents proper protein folding and is in effect an insulin-deficient, non-obese model of type 1 diabetes. 68
In addition to these models of type 1 diabetic nephropathy, Thbs1 null mice are protected from renal dysfunction and had reduced glomerular phosphorylated Smad 2 and 3 in a high-fat diet model of obesity. 11 Recent data showed that the TSP1 blocking peptide LSKL reduced active TGF-β in macrophages exposed to hypoxia. 69 Furthermore, in studies with myeloid cell–specific deletion of Thbs1, but not adipocyte deletion, mice had reduced adipose tissue macrophage infiltration and inflammation, reduced adipose tissue TGF-β signaling and fibrosis, and improved glucose tolerance and insulin sensitivity. 69 Together, these data suggest that TSP1-dependent TGF-β activation is important in obesity-related renal and metabolic diseases, such as hypertension and type 2 diabetes. Moreover, these findings suggest that specific cellular sources of TSP1 can variably affect regulation of TGF-β activation in disease.
Renin–angiotensin antagonists such as angiotensin-converting enzyme inhibitors or angiotensin receptor blockers are the current standard of care for diabetic kidney disease, and these drugs reduce proteinuria due to normalization of glomerular hyperfiltration, but do not reverse or halt progression to end-stage renal disease. 70 , 71 Nearly 20% of treated patients progress to end-stage renal disease. 72 Interestingly, we showed that the angiotensin II type 1 receptor antagonist valsartan reduces angiotensin II stimulation of TSP1 expression by rat mesangial cells. 59 Synergy between renin–angiotensin system blockers and TGF-β antagonists has been demonstrated in animal models of diabetic nephropathy despite some overlap in their targets.73 –75 It remains to be determined whether TSP1-TGF-β antagonists might have synergy with renin–angiotensin blockers.
Interestingly, drugs that tightly regulate glucose have unexpectedly shown cardiovascular and renal benefit in clinical trials and in rodent models of nephropathy in obese and type 2 diabetic animals: Those drugs that target glucose transporters, such as the sodium glucose cotransporter 2 (SGLT2) inhibitors, empagliflozin and canagliflozin, slow renal decline, independent of their effects on glycemia.76 –79 A recent study investigating possible mechanisms of renoprotection showed that SGLT2 inhibitors decrease TSP1 expression in two human proximal tubular cell lines. 80
TGF-β-Independent Functions of TSP1 in Renal Fibrosis
It should be noted that other important functions of TSP1 potentially contribute to renal fibrosis, independent of its role in activating latent TGF-β. TSP1 binding to its receptor CD36 is involved in podocyte apoptosis. 81 TSP1 has a pro-inflammatory effect on macrophages by stimulating increased toll-like receptor 4 expression, which in turn induces tumor necrosis factor-α production in a manner blocked by peptides that prevent TSP1–CD36 binding. 82
TSP1 is a known inhibitor of angiogenesis through its ability to downregulate nitric oxide signaling downstream of binding to CD47, and the TSP1–CD47 pathway is well documented to exacerbate ischemia–reperfusion injury. 83 , 84 TSP1 signaling through CD47 has been shown to inhibit tubular epithelial repair, potentially predisposing to later development of chronic kidney disease. 84 TSP1 can also antagonize FGF2, also known as basic fibroblast growth factor (bFGF), and vascular endothelial grown factor (VEGF) binding to cellular receptors to impede angiogenesis while promoting platelet-derived growth factor (PDGF) binding to mesenchymal stromal cells. 85 , 86 A decrease in peritubular capillary density, termed capillary rarefaction, promotes hypoxia and progression of chronic kidney disease with fibrosis and tubular atrophy. 87 In the UUO model, TSP1 short hairpin RNA increased VEGF protein levels and peritubular capillary density and reduced tubulointerstitial fibrosis. 88 A peptide (peptide 545) from the type 1 repeat of TSP containing the WSXW sequence, but lacking the TGF-β-activating KRFK sequence, blocked glomerular endothelial cell proliferation stimulated by bFGF or PDGF in vitro and in vivo in the anti-Thy1 model. 89 Increased TSP1 in the aging kidney has been associated with decreased peritubular and glomerular capillary density and concomitant tubulointerstitial fibrosis and glomerulosclerosis. 90 It is possible that under these conditions, both the TGF-β-activating and the antiangiogenic functions of TSP1 drive fibrogenic progression. 87 , 91
TSP2 shares many structural and functional similarities with TSP1. 8 However, TSP2 lacks the TGF-β-activating sequence of TSP1 and can actually antagonize latent TGF-β activation by TSP1. 19 Daniel et al. explored the role of TSP2 in the anti-Thy-1 model of glomerulonephritis and showed that Thbs2 null mice had increased cell proliferation, inflammation, and matrix metalloproteinase 2 activity. 92 Forced overexpression of TSP2 in this mouse model reduced endothelial cell proliferation, inflammation, TGF-β activation, and ECM deposition, although prolonged TSP2 overexpression resulted in capillary rarefication in a model of chronic allograft nephropathy. 93 , 94
TSP1 Actions in Repair and Fibrosis in Non-renal Systems
In the following sections, we will discuss additional functions of TSP1 in regulating ECM remodeling in repair and fibrosis. Many of these actions have been studied in non-renal models. However, given the complexity and context dependence of TSP1 actions, it is useful to have a broad perspective on the diverse abilities of this molecule. Hopefully, a more comprehensive view of the multiplicity of its actions will lead to new insights into its possible roles in renal fibrosis.
TSP1 as a Regulator of Collagen Matrix Organization
A key characteristic of matricellular proteins is that in vitro surfaces coated with these ECM proteins do not support full cell adhesion with focal adhesion and stress fiber formation. 1 , 95 We identified a heparin-binding sequence in the N-terminal domain of TSP1 and TSP2 (aa 17–35) that stimulates the disassembly of focal adhesions in fibroblasts and endothelial cells. 96 , 97 TSP and peptide mimetics of this sequence bind to a cell-surface form of the ER stress and calcium regulatory protein, calreticulin, to signal focal adhesion disassembly in a phosphoinositide 3-kinase–dependent manner.98 –100 This peptide (hep I) also triggered both random and directed cell migration of fibroblasts and endothelial cells. 101 As calreticulin lacks a transmembrane domain, TSP1 or hep I peptide bound to calreticulin triggers signaling by inducing calreticulin association with LDL receptor–related protein 1 (LRP1) to activate G protein signaling and transiently downregulate Rho activity. 100 , 102 , 103 In further studies, we showed that the N-terminal TSP1 domain or the hep I peptide prevented anoikis of non-adherent fibroblasts through binding to calreticulin/LRP1. 104
Increased cell migration and persistence of fibroblasts are frequently observed in fibrosis. 105 , 106 To address the in vivo effects of TSP1 signaling through the calreticulin/LRP1 pathway, we developed a model of the foreign body response using sponges with forced local expression of the secreted TSP1 calreticulin-binding sequence. 13 In animals implanted with collagen-loaded sponges overexpressing this secreted calreticulin-binding sequence, we observed the formation of a dense and highly organized collagen capsule surrounding the foreign body in comparison with animals implanted with control sponges expressing an inactive, mutated TSP1 sequence, despite no observed differences in the number or types of cells infiltrating into the foreign body sponge. In vitro studies confirmed the ability of the TSP1 calreticulin-binding sequence to stimulate increased fibrillar collagen protein expression in conditioned media and incorporation of collagen I into a deoxycholate-insoluble ECM. N-terminal domain regulation of collagen matrix occurred independent of TSP1’s TGF-β-activating ability and was dependent on signaling through Akt. 13 The increased collagen expression and ECM deposition induced by the TSP1 calreticulin-binding sequence does not involve changes in collagen I transcript or altered N- and C-terminal collagen propeptide processing. 13 The specific mechanism by which TSP1 binding to the calreticulin/LRP1 complex increases collagen expression and matrix organization remains unclear, but could possibly involve regulation of posttranscriptional secretory processes, as well as of factors involved in collagen fibril assembly.
The N-terminal domain of TSP1 can be released by multiple proteases involved in tissue remodeling, including ADAMTS1, cathepsin G, and neutrophil elastase, as well as by a collagenase used clinically for wound debridement (Santyl), suggesting roles in tissue repair responses.107 –111 The protein receptors, calreticulin and LRP1, for this sequence of TSP1 have variable roles in fibrosis. Calreticulin is upregulated in the UUO model of renal fibrosis and in bleomycin-induced lung fibrosis, and downregulation of calreticulin attenuates fibrosis. 112 , 113 Furthermore, we showed that calreticulin in the ER regulates collagen transcription, processing, and assembly into the ECM, as well as TGF-β-mediated ECM transcription through calcium-dependent activation of nuclear factor of activated T cells (NFAT). 114 , 115 However, these studies focused on intracellular ER calreticulin rather than on calreticulin at the cell surface, where it would be a ligand for TSP1 and a co-receptor with LRP1. In fact, exogenous extracellular calreticulin promotes scarless wound healing. 116 , 117 However, there is a recent report that cell surface calreticulin on HK-2 proximal tubule cells binds to surfactant protein-A to induce oxidative stress, type 1 collagen expression, and cellular apoptosis and to downregulate matrix metalloproteinases, suggesting a possible role in renal fibrosis. 118 In addition, both profibrotic and antifibrotic actions have been reported for LRP1 through both its signaling and endocytic functions. Cellular responses appear to be cell type specific and possibly vary depending on which ligand engages LRP1: For example, tissue plasminogen activator binding to LRP1 engages signaling through integrins that has profibrotic effects on kidney interstitial fibroblasts.119 –123 Ultimately, direct examination of TSP1 N-terminal domain binding to calreticulin–LRP1 on collagen organization in fibrotic remodeling remains to be addressed using TSP-binding calreticulin peptides that block TSP1–calreticulin interactions in animal models. 13 , 99
The TSP family has been long known to play a role in the regulation of collagenous ECM fibril organization. Thbs2–/– mice have irregular collagen fibril formation, and it was recently shown that TSP2-regulated miR-29 is decreased in these mice, leading to reduced lysyl oxidase and defective collagen crosslinking. 124 , 125 Cartilage oligomeric matrix protein (COMP), also known as thrombospondin-5 (TSP5), is an important regulator of collagen fibrillogenesis and acts to increase the rate of fibrillogenesis. 126 TSP1 is well known to bind to various fibrillar collagens, and altered collagen matrix has been noted in Thbs1–/– mice. 127 , 128 New data provide interesting evidence for direct TSP1–collagen interactions in regulating collagen fibril formation. 12 In this work, Rosini et al. 12 identified TSP1 binding sites in the C-propeptide domain of intracellular pools of collagen and in a KGHR containing conserved sequence in crosslinking sites in the triple helical region of mature, secreted collagen. Interestingly, both COMP and thrombospondin-4 (TSP4) were shown to bind to the GXKGHR motif in collagen type II, suggesting a conservation of this interaction with fibrillar collagens among these TSP family members. 129 Thbs1–/– dermal fibroblasts have altered collagen crosslinking, a finding that was recently confirmed in both Thbs1- and Thbs2-deficient female bone diphyses. 12 , 130 TSP1 binds prolysyl oxidase and prevents prolysyl oxidase activation by BMP-1. The TSP1–KGHR interaction is important in fibroblast homeostasis as peptides that disrupt this interaction increase alpha-smooth muscle actin (α-SMA)–positive myofibroblasts in a manner dependent on signaling through TGF-β receptor I. 12 These findings are intriguing and suggest that TSP1–procollagen/collagen interactions potentially regulate collagen trafficking, processing, and fibril assembly. These data also suggest that TSP1 bound to collagen in the ECM potentially “normalizes” collagen matrix and prevents myofibroblast differentiation, thus indicating a possible antifibrotic role for collagen-bound TSP1. This would be distinct from various profibrotic activities of secreted TSP1 (which is presumably soluble or receptor bound) in promoting TGF-β activation, cell migration, or dense collagenous capsule formation. It is not known whether these TSP1–collagen interactions are functionally related to the observations of increased collagen organization in response to extracellular stimulation with the calreticulin binding N-terminal domain sequence. These multiple collagen regulatory functions of TSP1 suggest that we do not yet fully understand the breadth of TSP1 functional complexity in the regulation of the collagenous ECM. Studies that potentially address alterations in collagen crosslinking and myofibroblast induction in Thbs1-deficient mice in models of wound healing and renal fibrosis would be useful in confirming these functions in pathology.
Intracellular TSP1 as an ER Stress Regulator of ECM
Given the diversity of the TSP1 interactome, 6 it seems almost intuitive that TSP1 might play intracellular roles in regulating protein trafficking in the ER. However, until recently, possible intracellular TSP1 functions have been largely unexplored. 1 , 131
The unfolded protein response (UPR) is an adaptive cellular response, called ER stress, to ensure proper proteostasis under challenging conditions such as increased oxidative stress, exposure to high glucose levels as in diabetes, or altered nutrient conditions. 132 Under acute stress conditions, mechanisms are activated to attenuate protein translation, to increase chaperone expression to assure proper protein folding, and to degrade misfolded proteins. However, under prolonged stress, these responses become detrimental and lead to apoptotic cell death and activation of fibrosis pathways.133 –135 This is analogous to acute tissue repair responses that are active in fibrotic processes due to continued inappropriate stimuli. There is now a significant body of literature that links the ER stress response with fibrosis and enhanced TGF-β activity in various organ systems. 113 ,135 –141 It is well established that ER stress pathways are activated in fibrotic renal disease, including in diabetic nephropathy and in ischemia, and that chemical chaperones such as 4-phenylbutyrate and tauroursodeoxycholic acid have been shown to reduce ER stress and result in fibrosis in animal models of renal disease.142 –149
It is unclear how TSP1 and other TSP family members might be participating in the UPR and other branches of the ER stress response and whether intracellular TSP1 functions are involved in fibrogenic processes. Studies on mutant COMP (TSP5) in models of pseudoachondroplasia showed a more severe phenotype in animals in which the mutant protein was expressed than in knockout mice. 150 , 151 It was also observed that mutant COMP associated with other ECM proteins and chaperones such as calreticulin in the ER, suggesting the formation of intracellular ECM vesicles. 152 Indeed, Rosini et al. 12 reported intracellular vesicles of TSP1 and procollagen. Moreover, TSP1 and TSP4 bind to the ER luminal domain of activating transcription factor 6 (ATF6), a branch of the UPR that increases chaperone expression and downregulates protein translation. 15 TSP–ATF6 binding occurs through the type 3 repeats of TSP as demonstrated in studies with recombinant TSP4 fragments. 15 TSP1, similar to TSP4, binds to ATF6 specifically in the Golgi compartment and increases nuclear shuttling of ATF6 and chaperone expression to activate ER stress protective mechanisms, particularly in cardiac myocytes and in skeletal muscle cells. 15 Further studies showed that TSP1 prevents pancreatic β-cell death from lipotoxic palmitate by activating the ER stress pathway PKR-like ER kinase (PERK)–NRF2 antioxidant signaling. 153 It is interesting that this cell-protective effect was only induced by intracellular TSP1 engineered with an ER retention KDEL sequence and was not observed with conditioned medium TSP1. 153 , 154 In this pancreatic β-cell system, ER TSP1 colocalized with protein disulfide isomerase. Further studies showed that TSP1 was also cytoprotective to β cells acutely exposed to the inflammatory cytokines interleukin-1β or interferon-γ or to thapsigargin-induced ER stress, although prolonged exposure (4–24 hr) resulted in downregulation of TSP1 expression and proteosomal degradation, leading to cell death. 154 Under these conditions, TSP1’s cytoprotective effect was dependent on the expression of mesencephalic astrocyte-derived neutrotrophic factor rather than ATF6 or modulation of oxidative stress. 154 These studies suggest that in acute or under limited stress or injury, TSP1 might have cytoprotective effects that could be lost upon chronic stimulation. The conditions that determine ER retention of TSP1 are not entirely clear, although low calcium levels have been shown to retain TSP1 in the ER of renal carcinoma cells. 155 In addition, given TSP1’s complex disulfide bond arrangement and its glycosylation, it is reasonable that TSP1 could be improperly folded under conditions of ER stress and thus retained in the ER or targeted for degradation through the endoplasmic reticulum–associated degradation pathway. Proper O-glycosylation of TSP1 type 1 repeats stabilizes proper folding and promotes transit from the ER. 156 This suggests a scenario in which TSP1 is initially a cytoprotective protein in cellular homeostasis. However, TSP1 message and protein levels are increased in many fibrotic processes: One scenario might be that during prolonged ER stress, an increasing fraction of TSP1 with altered glycosylation or defective protein folding could escape the ER, which could potentially enhance extracellular interactions with profibrotic receptors or factors such as latent TGF-β.
It has also been suggested that TSP1 might regulate calcium signaling through binding of the C-terminal signature domain of TSP1 to the N-terminal ER luminal domain of the ER membrane calcium sensor, STIM1 (stromal interaction molecule 1). 14 STIM1 senses low calcium levels and activates store operated calcium entry (SOCE) mechanisms to restore ER calcium levels. 157 Although it is unknown whether TSP1 affects STIM1 function, overexpression of COMP attenuates ER calcium release–activated calcium channel activity, whereas it increases arachidonate calcium channels at the plasma membrane and results in increased thapsigargin-induced cytosolic calcium levels. 14 Interestingly, extracellular TSP1 binds to plasma membrane STIM1 on platelets. 158 Currently, there are no data to directly link ER TSP1 to fibrosis through regulation of SOCE mechanisms, although there are data from cardiac fibrosis models showing that SOCE and STIM1 can induce fibrosis through downstream activation of NFAT and TGF-β signaling. 159 In renal models, TGF-β signals transcription of fibronectin and collagen type 1 in mesangial cells in an NFAT-dependent manner,160 –162 and an NFAT inhibitor reduces renal fibrosis in mouse models of diabetic nephropathy. 163 Forced TSP1 overexpression in cardiomyocytes increases expression of the ER chaperone and calcium regulatory protein, calreticulin, which our lab showed is critical for TGF-β-mediated transcription of fibronectin and collagen in fibrosis models through stimulating calcium-dependent activation of NFAT. 15 , 114 , 115 , 164 , 165 Moreover, the NoC1 calreticulin-binding fragment of TSP1 increases calreticulin expression in fibroblasts (unpublished data). TSP1 is also a transcriptional target of NFAT, 166 , 167 representing a possible feedforward mechanism. However, indirect TSP1 activation of NFAT might be cell type or cell context specific, because TSP1 binding to CD36 activates both JNK2 and NFκB, which block NFAT activation and inhibit angiogenesis. 168 Nonetheless, possible intracellular TSP regulation of cytosolic calcium levels in modulation of transcriptional pathways such as NFAT downstream of TGF-β in fibrosis warrants further exploration.
TSP1 as a Therapeutic Target
This review highlights some of the diverse functions of TSP1 in the pathogenesis of fibrosis in the kidney (Table 1). Given the multiple roles of TSP1 in the pathogenesis of fibrosis, it will be important to identify which specific interactions and functions of TSP1 are important in a particular disease entity and target only those specific actions. This focused approach could avoid side effects from general blockade of all TSP1 functions. For example, studies that induced renal overexpression of TSP2 to competitively antagonize TSP1-dependent TGF-β activation in a model of chronic allograft nephropathy not only decreased TGF-β activity as predicted, but also induced glomerular and peritubular capillary rarefication due to excessive antiangiogenic activity of TSP2. 94
Diverse Interactions and Roles of Thrombospondin 1 in Renal Disease.
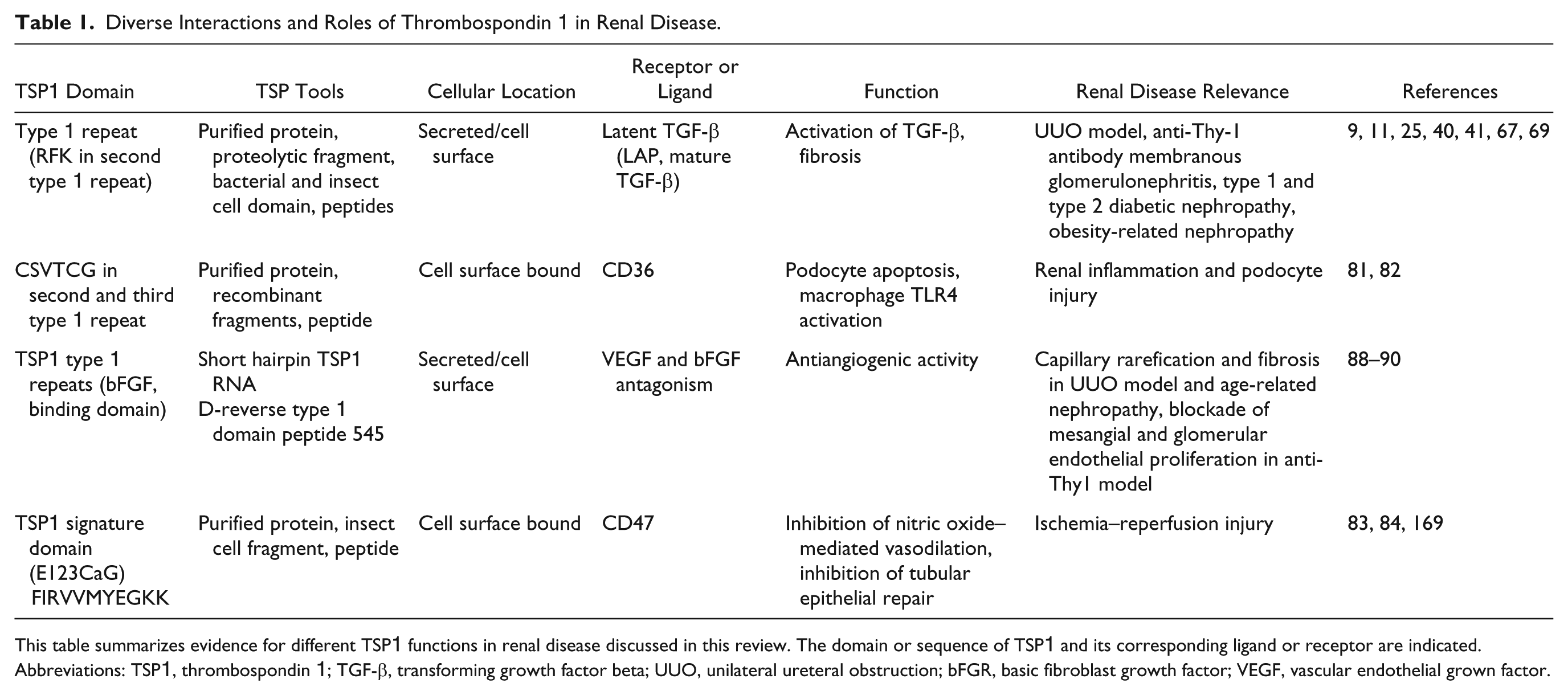
This table summarizes evidence for different TSP1 functions in renal disease discussed in this review. The domain or sequence of TSP1 and its corresponding ligand or receptor are indicated. Abbreviations: TSP1, thrombospondin 1; TGF-β, transforming growth factor beta; UUO, unilateral ureteral obstruction; bFGR, basic fibroblast growth factor; VEGF, vascular endothelial grown factor.
As described above, genetic and biochemical approaches to blocking TSP1-dependent TGF-β activation have been effective in reducing TGF-β activity, fibrosis, and renal dysfunction in a number of preclinical animal models of renal fibrosis, particularly in models of diabetic nephropathy. To date, the biochemical approaches have involved use of peptide antagonists or antisense oligonucleotides. Because of the short half-life of peptides and the need for injectable delivery routes, longer lived, orally available small molecules would represent more useful drugs for treating patients with chronic kidney disease. Such oral small molecule antagonists are currently under development.
TGF-β has long been a target for the treatment of fibrosis, and in particular, diabetic renal disease. However, results from clinical studies with anti-TGF-β neutralizing antibodies have been disappointing.170 –173 In addition, there are cardiotoxicity concerns with general TGF-β ligand or signaling antagonists. 174 It is becoming more widely appreciated that interventions that block disease-specific mechanisms of latent TGF-β activation, such as blocking TSP1 or integrin-dependent activation, are likely to have a greater therapeutic index than more broadly targeted antagonists. 26 , 173 , 175 Interestingly, some reports suggest that certain integrins can regulate TSP1 expression. In a corneal wound model that deals with fibrosis, knockout of the β6 integrin delayed initial healing and fibrosis and reduced TSP1 levels; however, TSP1 expression increased later in healing and was associated with α-SMA-positive cells. 176 Furthermore, an antibody to the αvβ3 integrin that reduces proteinuria also decreases TSP1 and TGF-β expression in diabetic rats. 177 These intriguing observations suggest that there might be synergy between integrin and TSP-dependent modes of latent TGF-β activation.
Although in vitro studies show that TSP1, TGF-β, and TGF-β activation are regulated by angiotensin II and glucose and that the renin–angiotensin system and sodium glucose cotransporter antagonists downregulate TSP1 and TGF-β expression in renal cells, 59 , 80 data from animal studies using combinations of TSP1-TGF-β antagonists with antagonists of these pathways are lacking. It will be interesting to determine whether there is synergy in using drugs that inhibit multiple related, but distinct, pathways to treat chronic kidney disease in diabetes and hypertension.
Approaches that target more than one action of TSP1, such as blocking both TGF-β activation and angiogenesis inhibition or growth factor binding through combining function-specific inhibitory molecules, could be of benefit. Anti-CD47 antibodies and small molecules that mimic the TSP binding site for bFGF are being tested in clinical trials, in preclinical models, or are under development for various diseases.169,178 –182 Antagonists of these antiangiogenic TSP1 functions have not been tested with antagonists of TSP1’s TGF-β activating function. Combined use of a variety of TSP1 functional antagonists could represent a multipronged approach to treating the complex pathogenesis of fibrosis.
Footnotes
Acknowledgements
The author acknowledges all of the outstanding trainees and collaborators who contributed to the original studies described in this review.
Competing Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Dr. Murphy-Ullrich is a coinventor on several patents to develop antagonists of Thrombospondin 1-dependent transforming growth factor beta activation. There are no other conflicts to report.
Author Contribution
JEM-U wrote the manuscript.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Dr. Murphy-Ullrich’s lab is currently supported by National Institutes of Health grants 1R01 CA175012 and 1R01 EY027924 and by funds from the Alabama Drug Discovery Alliance.