
Abstract
Renal fibrosis is an important component of chronic kidney disease, an incurable pathology with increasing prevalence worldwide. With a lack of available therapeutic options, end-stage renal disease is currently treated with renal replacement therapy through dialysis or transplantation. In recent years, many efforts have been made to identify novel targets for therapy of renal diseases, with special focus on the characterization of unknown mediators and pathways participating in renal fibrosis development. Using experimental models of renal disease and patient biopsies, we identified four novel mediators of renal fibrosis with potential to constitute future therapeutic targets against kidney disease: discoidin domain receptor 1, periostin, connexin 43, and cannabinoid receptor 1. The four candidates were highly upregulated in different models of renal disease and were localized at the sites of injury. Subsequent studies showed that they are centrally involved in the underlying mechanisms of renal fibrosis progression. Interestingly, inhibition of either of these proteins by different strategies, including gene deletion, antisense administration, or specific blockers, delayed the progression of renal disease and preserved renal structure and function, even when the inhibition started after initiation of the disease. This review will summarize the current findings on these candidates emphasizing on their potential to constitute future targets of therapy:
Keywords
Introduction
Renal fibrosis is a major outcome of chronic kidney diseases (CKDs), which affect nearly 10% of the population and contribute to the increased number of deaths and the surcharge of the national health systems worldwide. 1 CKD can originate from different causes, such as diabetes, hypertension, immune, or toxic stimuli; however, it involves common pathological mechanisms like chronic inflammation and development of renal fibrosis that lead to the impairment of renal function. 2
Fibrosis initially appears as a normal response to injury, where activated fibroblasts produce high amounts of extracellular matrix (ECM) in the context of a wound-healing process, in order to assist in the repair of the damaged tissue. In case of a repetitive injury, chronic wound healing leads to an excessive accumulation of ECM which fails to be resolved by the remodeling mechanisms and leads to organ dysfunction. 3 As it is commonly acknowledged that fibrosis plays a major role in the pathology of CKD and the progression to end-stage renal disease, many efforts were made during the last years to identify novel mediators and targets for therapy of renal fibrosis. Great attention was paid to major growth factors or cytokines that were early shown to mediate both inflammation and fibrosis during progression of renal disease in animal models, including angiotensin II (Ang-II), transforming growth factor-β (TGF-β), platelet-derived growth factors (PDGFs), connective tissue growth factor (CTGF), endothelin-1 (ET-1), macrophage chemoattractant protein-1 (MCP-1), and tumor necrosis factor-α (TNF-α).4,5 Despite the fact that drugs targeting some of these mediators were approved for tests in clinical trials and inhibitors of the renin-angiotensin system are currently used in subgroups of CKD patients, no major improvement has been made in targeting specifically renal fibrosis.
Our group has recently identified novel mediators of CKD, focusing on their potency to promote the development of renal fibrosis, namely discoidin domain receptor 1 (DDR1), periostin, connexin 43 (Cx43), and cannabinoid receptor 1 (CB1). This review will summarize the latest advances on the roles of these proteins, with a focus on renal diseases, and emphasize their potential as novel promising therapeutic targets to fight renal fibrosis.
DDR1
DDR1 is a transmembrane collagen receptor with tyrosine kinase activity and a predominant expression in epithelial cells. DDR1 is also known with multiple alternative names, most of which reflect the function or the localization of the protein, such as cell adhesion kinase (CAK), epithelial discoidin domain-containing receptor 1 (EDDR1), neuroepithelial tyrosine kinase (NEP), mammary carcinoma kinase 10 (MCK10), and CD167 antigen-like family member A (CD167a). DDR1 is composed of three distinct domains: an extracellular discoidin homology domain with specific binding capacity to collagen, a transmembrane region essential for receptor dimerization, and an intracellular domain that is phosphorylated upon receptor activation.6,7 DDR1 activation promotes mitogen-activated protein kinase (MAPK), PI3 kinase/Akt, or matrix metalloproteinase (MMP) signaling in different epithelial, vascular smooth muscle or cancer cells, thus regulating vital cell functions like migration, proliferation, and survival. 7 De novo expression and activation of DDR1 have been reported in a number of different human diseases, including cancer, atherosclerosis, and fibrosis. DDR1 overexpression was correlated with increased tumorigenesis and poor prognosis and was shown to induce epithelial-to-mesenchymal transition in cancer cells.8,9 In the low-density lipoprotein receptor knock-out (KO) model of atherosclerosis (Lldr-/-), DDR1 deficiency was associated with a reduction in macrophage infiltration and the development of atherosclerotic lesions. 10 Moreover, in the bleomycin-induced lung injury model, DDR1 was de novo expressed by injured epithelial cells and promoted proinflammatory p38 MAPK signaling and pulmonary fibrosis. 11
DDR1 in Renal Fibrosis
We were among the first to investigate the potency of DDR1 deficiency or inhibition to ameliorate the progression of renal disease. In a model of hypertensive nephropathy induced by chronic Ang-II administration, DDR1 was upregulated in vascular smooth muscle cells and mesangial cells of diseased kidneys. DDR1 null mice were protected against perivascular inflammation, arteriosclerosis, glomerulosclerosis and interstitial fibrosis, exhibiting a markedly decreased accumulation of types I and IV collagens. 12 The preservation of renal structure was accompanied by an improvement of renal function, evidenced by the decreased albuminuria in DDR1-/- mice. 12 In this study, DDR1 was proposed to be an amplifier of the initial damage, activated by collagen binding and promoting deleterious proinflammatory and profibrotic pathways, including MAPK pathways, cytokine synthesis and further production of collagens.
In a subsequent study, we found that DDR1 was highly expressed by tubular cells and infiltrating macrophages in the unilateral ureteral obstruction (UUO) model. 13 DDR1-deficient mice showed decreased expression of several proinflammatory cytokines, as well as TGF-β1 and Col3, accompanied by attenuated inflammation and interstitial fibrosis. 13 Apart from promoting collagen production and fibrosis in this model, DDR1 was also directly associated with macrophages activation and migration to the damaged tissue.
Finally, we showed that DDR1 expression is increased in the glomeruli of patients with lupus nephritis and goodpasture syndrome. 14 In a mouse model of severe glomerulonephritis leading to rapid deterioration of renal structure and function, DDR1 was de novo expressed in damaged podocytes. 14 We investigated the role of DDR1 in this context using both KO mice and antisense oligonucleotides specifically targeting DDR1.14,15 Both strategies protected mice from severe proteinuria and uremia, glomerular crescent formation and fibrin deposition, inflammation, and interstitial fibrosis. Interestingly, the administration of antisense oligonucleotides targeting DDR1 was effective in the protection against glomerulonephritis even when it started after the onset of the disease, either at an early or intermediate phase. 15
Other investigators examined the role of DDR1 in Alport syndrome and the remnant kidney model. In mice lacking the Col4α3 gene, a model of Alport syndrome, DDR1-deficiency resulted in a decreased renal expression of proinflammatory and profibrotic cytokines, leading to attenuated inflammation and fibrosis, preserved renal function and increased survival. 16 A recent study in the remnant kidney model showed that mutation in either the collagen binding site or the kinase domain of DDR1 inhibited collagen production by mesangial cells, demonstrating that both domains are indispensable for the receptor-mediated collagen deposition. 17
DDR1 as a Target of Therapy
DDR1, as a membrane receptor, can be easily accessed by potential drugs. DDR1 could be targeted either by blocking its interaction with collagen extracellularly or by inhibiting its tyrosine kinase activity intracellularly, preventing the receptor activation. Known tyrosine kinase inhibitors, which are widely used in cancer (dasatinib, imatinib, nilotinib), were also demonstrated to block DDR1 autophosphorylation induced by collagen. These inhibitors are not specific for DDR1, which increases the possibility for off-target effects. However, a recent publication showed that inhibition of DDR1 and its newly identified substrate breakpoint cluster region (BCR) protein by nilotinib reduces the metastatic potential of patient-derived colorectal cancer cell lines in mice. 18 Interestingly, novel selective tyrosine kinase inhibitors targeting DDR1 have been recently developed. They can efficiently inhibit the receptor activation and the propagation of cancer cells overexpressing DDR1. 6 Pharmacological inhibition of DDR1 with an orally available small molecule inhibitor (7rh) could reduce the tumor progression in orthotopic pancreatic xenografts and KRAS-driven lung adenocarcinomas in mice.19,20 Strikingly, a novel selective DDR1 inhibitor manufactured by Roche-Chugai ameliorated the progression of experimental glomerulonephritis in both preventive and therapeutic regime, demonstrating better results than imatinib, a non-selective tyrosine kinase inhibitor widely used in cancer. 21 These inhibitors, together with other selective compounds that may be identified, could be tested in animal models of renal disease for their efficacy to slow down or even block the development and/or progression of renal fibrosis and CKD. Furthermore, a genetic variant near the DDR1 gene was recently identified in a genome-wide association analysis as one of the 16 loci that predispose to diabetic nephropathy. 22 It remains to be elucidated whether this variant causes a change in DDR1 expression or function associated with the progression of the disease. If that is the case, DDR1 could be also considered as a potential target for the therapy of diabetic nephropathy.
Periostin
Periostin, also known as POSTN, PN or osteoblast-specific factor 2 (OSF-2), is a 90 kDa secreted matricellular protein, highly expressed in bone and dental tissues. 23 Although the expression of periostin is high in development, it is absent from most adult tissues. However, it is de novo induced in injury and remodeling conditions. 24 The protein structure of periostin is composed of a N-terminal and a C-terminal region separated by a tandem repeat of four fasciclin-I domains, showing homology to the homonymous insect domain, which mediates neuronal adhesion. Periostin interacts via its different domains with ECM molecules like collagen 1, fibronectin, BMP-1, tenascin-C, or different cell-surface integrin receptors. It can thus mediate signals to both extracellular and intracellular environments and control processes like cell adhesion, migration, proliferation, and differentiation.25–28
Periostin was shown to promote inflammatory and fibrotic processes during progression of chronic or acute diseases affecting a plethora of different organs, including myocardial infarction,29,30 cardiac hypertrophy,31,32 asthma,33,34 pulmonary fibrosis,35,36 skin sclerosis, 37 hepatic fibrosis,38,39 muscular dystrophy,40,41 and cancer.42,43 Strikingly, inhibition of periostin in animal models of these diseases was able to prevent the progression of the pathology.
Periostin in Renal Fibrosis
Over the last decades, an increasing number of studies has implicated periostin in the progression of animal and human renal diseases. Periostin is overexpressed in biopsies from patients with various CKD etiologies, including focal segmental glomerulosclerosis, 44 IgA nephropathy,45,46 diabetes, 46 lupus nephritis, 47 polycystic kidney disease (PKD), 48 and transplant rejection. 49 In these studies, periostin was localized in areas of glomerular and interstitial fibrosis and its expression levels were positively correlated with disease progression. Interestingly, several studies reported an elevated urinary level of periostin in CKD patients, which reflected a more advanced pathological stage and was associated with an aggravated renal outcome.45,46,50
We were among the first to identify periostin as a novel potential therapeutic target in animal models of renal disease. First, in a model of L-NAME-induced hypertensive nephropathy, periostin emerged as one of the most highly upregulated genes. It was localized in fibrotic regions, around sclerotic vessels and injured tubules. Moreover, its expression level was inversely correlated with the drop in renal function. 51 Finally, periostin level was also highly correlated with the progression or regression of the disease when the animals were treated with losartan, an Ang-II receptor blocker. 51
In subsequent studies, our group demonstrated that periostin is a central mediator of different aspects of renal disease. We showed that mice lacking periostin were protected against renal inflammation and fibrosis accompanied by preservation of renal structure in the models of UUO and nephrotoxic serum (NTS)-induced glomerulonephritis.52,53 Strikingly, in both preventive and curative pharmacogenetic approaches in the L-NAME and NTS models, respectively, the administration of antisense oligonucleotides against periostin was capable to block the progression of the disease.52,53
Periostin was also found to play an important role in models of PKD and lupus nephritis. Periostin was overexpressed in cyst-lining epithelial cells of PKD patients and a genetic mouse model of PKD. It accumulated in the matrix around cysts, where it was associated with their proliferation. Mice lacking periostin were protected against PKD progression, displaying decreased number and size of cysts, reduced fibrosis and preserved renal function. 54 Conversely, mice conditionally overexpressing periostin in collecting duct cells demonstrated accelerated cyst formation, interstitial fibrosis and decline of renal function. 55 In mice with lupus nephritis, periostin was produced by mesangial cells of the glomerulus, where it stimulated their proliferation and excess production of ECM. 56 A recent study in a chronic renal failure model induced by 5/6-nephrectomy demonstrated that inhibition of periostin with shRNA improved renal and cardiac dysfunction and attenuated organ fibrosis through negative regulation of the PPARα pathway. 57
In most animal models of renal disease, periostin was shown to mediate both renal inflammation and fibrosis. This is in accordance with the potency of both pro-fibrotic and pro-inflammatory factors to upregulate periostin in vitro or in vivo in different pathological contexts. For example, TGF-β1, Ang-II, and PDGF-B were shown to induce the expression of periostin in cardiac fibroblasts, 58 vascular smooth muscle cells, 59 renal tubular 52 or mesangial cells. 56 On the other hand, interleukins IL-4 and IL-13 have been associated with induction of periostin in alveolar epithelial cells and fibroblasts. 60 NF-κB and other pro-inflammatory transcription factors induced periostin expression in glomerulonephritis, 53 while most recently, the IL-13/STAT6 pathway was associated with induction of periostin in PKD. 61 Since inflammation precedes or progresses along with fibrosis in many cases of CKD, periostin may be a potent inhibitor of renal diseases, as it is implicated in both processes.
Periostin as a Target of Therapy
Periostin presents several advantages as a target of therapy against CKD: (1) it is silent in adult tissues, while it is induced at the sites of injury in a variety of renal diseases and (2) it is secreted, which makes it easily accessible to potential drugs. Interestingly, inhibition of periostin with different techniques was efficient in arresting or preventing the progression of renal disease in animal models. Genetic deletion of periostin delayed the progression of UUO, 52 NTS, 53 and PKD pathologies, 54 while administration of antisense oligonucleotides targeting periostin was efficient in arresting the progression of severe glomerulonephritis, even after the initiation of the disease. 53 Most recently, neutralization of periostin by systemic administration of a blocking anti-periostin polyclonal antibody or a periostin-binding DNA aptamer in UUO and diabetic nephropathy in mice, respectively, attenuated the progression of renal fibrosis.62,63 The next step toward a periostin-based therapy would be the creation and validation of periostin-targeting drugs with increased stability applicable in humans, which is the subject of further investigation.
Cx43
Cx43, also known as gap junction alpha-1 protein (GJA1), is a member of a large family of 20 proteins called connexins which form gap junctions, specialized structures allowing direct communication between adjacent cells via exchange of small molecules like ions (Na+, Ca2+) or second messengers (cAMP, ATP, IP3, etc.).64,65 Gap junctions are composed of two intercellular hemichannels, the connexons, each of which constitutes a homo- or heterohexamer of individual connexins. Apart from intercellular communication, single connexons can allow the interaction of cells with ECM. 66
Cx43 can affect cell signaling events, mediating changes in gene expression, cytoskeletal rearrangements, vesicle release, and cellular stress. 67 Cx43 is abundant in the heart and is required for normal heart development and function, since Cx43 deletion is lethal. 68 In vessels, Cx43 plays an important role in the regulation of the myogenic tone, proliferation, and migration of vascular smooth muscle cells. 69 A partial decrease in vascular Cx43 expression protected from progression of atherosclerosis possibly though anti-inflammatory effects. 70 Cx43 is highly expressed in brain astrocytes, while there is controversy on whether it exerts a neuroprotective or a deleterious role. 71 Cx43 is also upregulated in several cancers including liver, prostate, and breast tumors. Again, it could exert both pro- and anti-metastatic functions, indicating a context-dependent role in carcinogenesis.72,73
Cx43 in Renal Fibrosis
In the kidney, Cx43 localizes in the renal vasculature, mesangial cells and collecting ducts under normal conditions. 74 Soon after its first detection in the kidney, Cx43 was found upregulated in patient biopsies with different inflammatory renal diseases, where it was highly expressed by infiltrating cells, damaged tubular epithelial cells, and at interstitial sites of injury. 75
In a first attempt to study the role of Cx43 in CKD, we used a mouse model overexpressing renin, which causes a stable increase in blood pressure leading to hypertensive nephropathy and a progressive deterioration of renal function. 76 Cx43 was highly induced in peritubular and glomerular capillaries in this model, while Cx43 heterozygous mice (Cx43+/-) were protected in terms of albuminuria, inflammatory infiltration, and interstitial fibrosis. 77 The protection from renal disease was primarily linked to a reduced interaction between endothelial and inflammatory cells.
Overexpression of Cx43 in podocytes during progression of experimental glomerular renal disease was first observed in rat models of puromycin aminonucleoside nephrosis (PAN) and type-2 diabetes.78,79 Subsequently, we demonstrated that Cx43 was de novo expressed by suffering podocytes in the model of NTS-induced glomerulonephritis in mice, through binding of c-Jun and STAT1 on Cx43 promoter. 80 Interestingly, Cx43+/- mice showed preserved renal function and structure with decreased podocyte apoptosis, inflammatory cell infiltration, and renal fibrosis. Mechanistically, TGF-β1 treatment of cultured podocytes induced expression of Cx43. Blockade of either Cx43 or purinergic receptors mediating ATP signaling partially reversed the TGF-β1-induced expression of mesenchymal and migratory markers. The crosstalk between Cx43 and ATP signaling could thus promote deleterious podocyte depolarization. 80 Other in vitro studies demonstrated that blockade of Cx43 gap junctions or siRNA treatment against Cx43 restored cell viability and inhibited podocyte apoptosis and reactive oxygen species (ROS) production after PAN- or aldosterone-induced podocyte damage.81,82
In contrast to the increased podocyte expression of Cx43 in animal models of glomerulonephritis, there is controversial evidence regarding the expression and function of Cx43 in mesangial cells. Both in vivo in the anti-Thy 1.1 rat model 83 and, in vitro, in high glucose- or aldosterone-treated mesangial cells, Cx43 was downregulated. Its overexpression reversed the cytoskeletal rearrangements and proliferation induced by high glucose 84 and aldosterone, 85 respectively. On the other hand, increased levels of extracellular ATP induced by Cx43 could control mesangial matrix expansion through a TGF-β1-purinergic receptor pathway. 86
TGF-β1 was also shown to induce expression of Cx43 accompanied by ATP release and increased levels of IL-6 and fibronectin in cultured tubular epithelial cells. 87 In the mouse UUO model, Cx43 was strongly upregulated in tubules, while partial inhibition of Cx43 expression using heterozygous Cx43+/- mice or antisense oligonucleotides blunted the inflammatory response and interstitial fibrosis. Blockade of Cx43 gap junctions was able to reverse TGF-β1-induced collagen production and mitogen-activated protein kinase signaling in cultured tubular cells. 77
Cx43 as a Target of Therapy
Reduced expression of Cx43 was sufficient to protect against hypertensive nephropathy, obstructive nephropathy, and glomerulonephritis;77,80 this probably due to the role played by Cx43 in the progression of both inflammatory and fibrotic processes in animal models of CKD. Therapeutic strategies to prevent Cx43 function include the use of either antisense oligonucleotides or Cx43 blocking peptides. Interestingly, exogenous dermal application of a gel containing Cx43 antisense oligonucleotides was efficient for the treatment of skin wounds and foot ulcers in clinical trials. 88 In our studies, administration of antisense oligonucleotides against Cx43 after the establishment of proteinuria or renal damage efficiently inhibited the progression of the disease,77,80 indicating that Cx43-based treatments may constitute a potent therapeutic approach against renal pathologies.
Connexin blocking peptides are already available and have been used in pre-clinical models of diseases. They function by either inhibiting the hemichannel opening or the gap junction formation. GAP19 and GAP26 efficiently prevented the progression of muscular dystrophy, 89 myocardial infarction, 90 and spinal cord injury 91 in animal models. Topical application of another selective Cx43 gap junction blocker, GAP27, improved corneal wound healing in a rat model, associated with increased granulocyte infiltration. 92 The authors concluded that GAP27 may facilitate epithelial wound healing in an early phase, but prolonged usage may provoke an unwanted stromal inflammatory response. On the other hand, more recent publications demonstrated that blocking Cx43 hemichannel opening and ATP release protects from inflammatory disease progression. The Cx43 mimetic peptide P5 efficiently attenuated the hemichannel activity in vitro and ameliorated the inflammatory response in in vivo models of septic microbial infection and hepatic ischemia/reperfusion injury. 93 Moreover, an interesting study in a rat model of retinal and choroidal photodamage demonstrated that intravitreal administration of nanoparticles gradually releasing a Cx43 mimetic peptide was more efficient in maintaining retinal structure and function and reducing inflammation than a single dose of the native peptide in solution. 94 We have demonstrated that in vitro blockade of gap junction function of Cx43 with the GAP26 peptide hampered pro-inflammatory and pro-fibrotic cell responses in renal cells.77,80 It remains to be elucidated whether the existing Cx43 blockers can confer protection against the progression of renal disease in vivo in animal models.
CB1
The cannabinoid system is mainly composed of two membrane G protein-coupled receptors, cannabinoid receptor 1 (CB1 or CB1R, encoded by the CNR1 gene) and cannabinoid receptor 2 (CB2, encoded by the CNR2 gene). Their endogenous lipid ligands are called endocannabinoids. The cannabinoid pathway was first identified in the central nervous system and has major physiological roles in the regulation of pain, appetite, behavior, memory, and metabolism. CB1 expression is high in the brain, while it is less abundant in other organs like lung, liver, and kidney. 95
Deregulation of CB1 expression has been implicated in a plethora of diseases, including neuroinflammation and brain injury, cancer, liver fibrosis, gastrointestinal, and cardiovascular diseases. 96 In some disorders, such as neuropathic pain, brain injury, and hypertension, upregulation of CB1 is thought to alleviate the symptoms and inhibit the progression of the disease. In other cases, alterations in the receptor’s expression are maladaptive. For example, CB1 upregulation in liver fibrosis promotes hepatocyte lipogenesis and fibrogenesis, while CB1 downregulation in colorectal cancer leads to enhanced colorectal tumor proliferation. 96 Thus, the regulation of cannabinoid receptor expression using specific agonists or antagonists is of therapeutic interest.
CB1 in Renal Fibrosis
In normal kidney, CB1 is found in the vasculature 97 where it can stimulate the vasodilatation of efferent arterioles. 98 Accumulating evidence indicates a potential role for CB1 in various renal pathologies. CB1 mediates high glucose- or toxicity-induced endoplasmic reticulum stress and apoptosis in podocytes, mesangial cells and tubular epithelial cells.99-101 Moreover, CB1 agonism in renal tubular cells was recently shown to induce mitochondrial fission, associated with increased ROS and reduced ATP production and a decline in mitochondrial biogenesis. 102 Several studies demonstrated the implication of CB1 in diabetic nephropathy or obesity-induced CKD. In Zucker rats and db/db mice, two models of diabetic nephropathy, CB1 blockade decreased albuminuria, tubulointerstitial lesions, mesangial expansion, and fibronectin expression.103–106 In a model of streptozotocin-induced type 1 diabetic nephropathy, podocyte-specific deletion of CB1 protected against tubular dysfunction and fibrosis in addition to preventing podocyte injury and loss. 107 Another study on the same model demonstrated that miR-29a negatively regulated CB1 expression and protected against renal hypertrophy, inflammation, and fibrosis. 108 Moreover, CB1 deletion in renal proximal tubular cells attenuated inflammation, fibrosis, renal dysfunction, and lipid accumulation in obesity-induced CKD, by inducing activation of the liver kinase B1/AMP-activated protein kinase pathway, resulting in enhanced fatty acid β-oxidation. 109
Recently, our group showed that CB1 was among the 10 most upregulated genes in the UUO model, and that both its genetic and pharmacological inhibition markedly reduced inflammation and fibrosis. 110 CB1 was highly upregulated in renal myofibroblasts in this model, where it mediated TGF-β1-induced collagen expression. Interestingly, we also found increased CB1 expression in kidney biopsies from patients with diabetic nephropathy, IgA nephropathy and acute interstitial nephritis. CB1 was upregulated in tubular cells, interstitial cells, podocytes, and mesangial cells in these patients, and its expression correlated with renal function. 110
CB1 as a Target of Therapy
CB1 has recently emerged as a potential target of therapy in renal diseases. Since CB1 is involved in the regulation of appetite and metabolism, it was early proposed as a prominent target in diabetes and obesity-induced metabolic syndrome.103–107,109 However, one of the first blockers of CB1 tested in human metabolic syndrome, rimonabant, was withdrawn from the market due to side-effects on central nervous system. 111 On the other side, CB1 antagonists that do not cross the blood–brain barrier were shown to efficiently delay the progression of diabetic nephropathy and albuminuria.111–113
Increased CB1 expression was reported in most cell types during non-metabolic renal disease in animals and humans, while gene deletion or pharmacological blockade of CB1 inhibited accumulation of renal fibrosis and disease progression. 110 Although more animal models are necessary to establish the role of CB1 in non-metabolic renal disease, the potential testing of already available CB1 antagonists seems an interesting possibility for targeting renal fibrosis. A recent study demonstrated that CB1 antagonism can attenuate cardiac hypertrophy and fibrosis secondary to CKD. 114 Given the efficiency of CB1 antagonists in animal models of diabetes and metabolic syndrome103–106 and the improvement of metabolic profiles of patients, 115 favorable outcomes on cardiovascular complications could also be achieved.
In conclusion, renal fibrosis represents a major axis of CKD development, which has reached pandemic proportions and is a considerable cause of death. Despite the significant progress made in our understanding of the complex mechanisms driving renal diseases, current therapeutic interventions are insufficient, the only options being dialysis or transplantation in advanced stages. We identified DDR1, periostin, Cx43 and CB1 as novel mediators of renal diseases. All these proteins are expressed at low levels in normal kidney, highly activated after renal damage, where they localize primarily at the sites of injury. Moreover, their expression level is well correlated with the progression of the disease. Most importantly, inhibition of these proteins by gene deletion, or administration of antisense oligonucleotides or specific blockers efficiently protects from the development of renal fibrosis and preserves renal function in animal models, even when the treatment starts after the initiation of the disease. A summary of the role of these proteins during progression of renal disease is depicted in Fig. 1 and Table 1. Thus, novel therapies based on targeting either of these new candidates may constitute efficient future therapeutic treatments against renal fibrosis and CKD. When it comes to therapy, any potential adverse effects of targeting these proteins should also be considered. For DDR1 and periostin, no undesirable results of targeted treatments in animal models have been described to date, which is also supported by the fact that the KO mice of both proteins are viable and develop similarly to their wild-type littermates. On the contrary, the vital role of Cx43 gap junctions for heart development and function 68 might hamper the clinical applications of Cx43 inhibitors. In this regard, blockers targeting the hemichannel rather than the gap junction function of Cx43 may represent a good alternative, since they are expected to show less adverse effects. Similarly, CB1 antagonists that do not cross the blood–brain barrier should be preferred for targeted treatments against CB1, because of the known physiological functions of CB1 in central nervous system. 110 In any case, targeted therapeutics based on these proteins would require development of validated drugs for use in humans or testing of the already available blockers for some of these candidates in clinical studies, which will hopefully lead to a more efficient or targeted treatment of CKD.
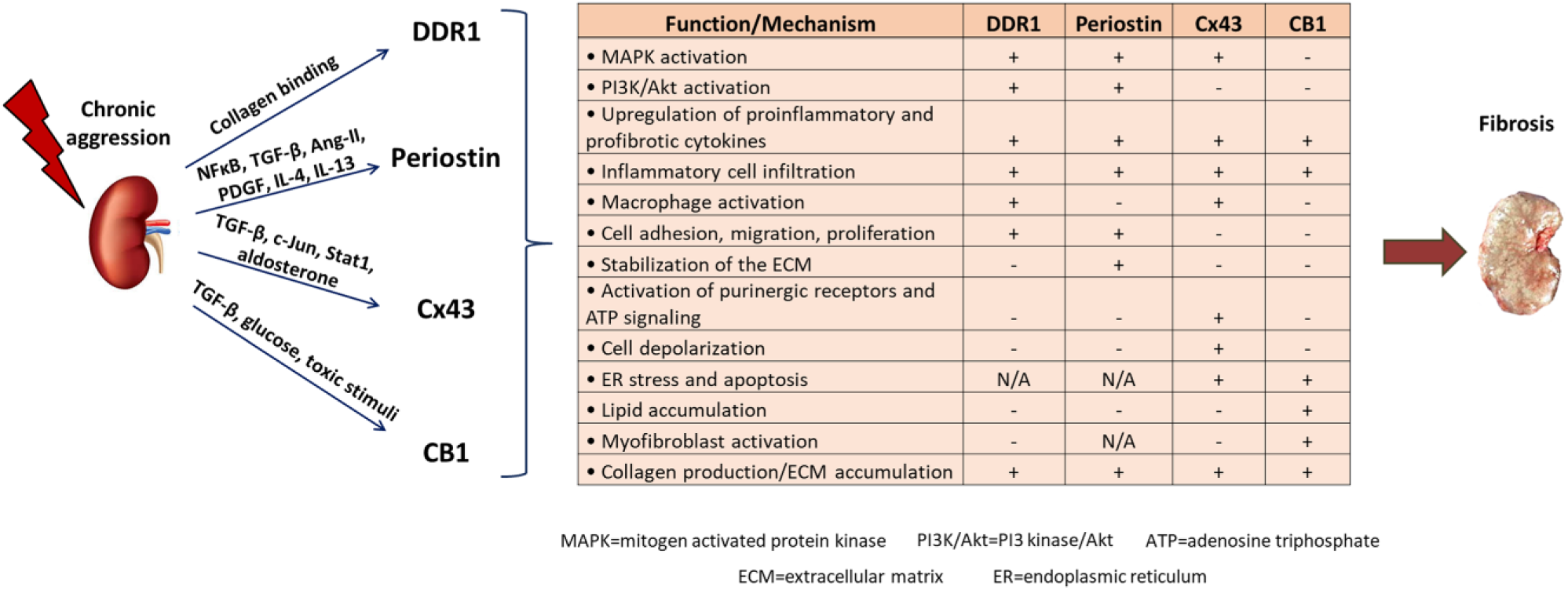
Schematic illustration of the role of DDR1, periostin, Cx43, and CB1 during progression of renal disease. After an initial aggression of the kidney, different factors, cytokines, or signaling pathways upregulate the expression of DDR1, periostin, Cx43, and CB1. Each protein activates downstream receptors and pathways that lead to production of proinflammatory and profibrotic cytokines, inflammation, macrophage activation, energy depletion, apoptosis, and excess matrix production, which cumulatively contribute to renal fibrosis and CKD development. Abbreviations: DDR1, discoidin domain receptor 1; Cx43, connexin 43; CB1, cannabinoid receptor 1; CKD, chronic kidney diseases.
Promising Novel Candidates for Therapy of Renal Fibrosis.

Abbreviations: DDR1, discoidin domain receptor 1; UUO, unilateral ureteral obstruction; NTS, nephrotoxic serum; NEP, neuroepithelial tyrosine kinase; Cx43, connexin 43; CB1, cannabinoid receptor 1; CKD, chronic kidney diseases.
Footnotes
Competing Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
NP reviewed the literature and wrote the manuscript, JH and CC revised and approved the final manuscript.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the Agence Nationale de la Recherche (ANR) and INSERM.