
Abstract
Focal segmental glomerulosclerosis (FSGS) presents with scar in parts of some glomeruli and often progresses to global and diffuse glomerulosclerosis. Podocyte injury is the initial target in primary FSGS, induced by a circulating factor. Several gene variants, for example, APOL1, are associated with increased susceptibility to FSGS. Primary FSGS may be due to genetic mutation in key podocyte genes. Increased work stress after loss of nephrons, epigenetic mechanisms, and various profibrotic pathways can contribute to progressive sclerosis, regardless of the initial injury. The progression of FSGS lesions also involves crosstalk between podocytes and other kidney cells, such as parietal epithelial cells, glomerular endothelial cells, and even tubular epithelial cells. New insights related to these mechanisms could potentially lead to new therapeutic strategies to prevent progression of FSGS.
Keywords
Around 40% of nephrotic syndrome in adults and 20% in children are diagnosed as focal segmental glomerulosclerosis (FSGS). 1 FSGS is a histological term, which is characterized by the presence of sclerosis in parts (segmental) of some (focal) glomeruli. According to the Columbia classification, FSGS can be histologically subdivided into five variants: collapsing, tip, perihilar, cellular, and NOS (not otherwise specified). 2 Podocyte injury has been recognized as the cause of primary FSGS. However, although all podocytes are equally affected by genetic abnormalities, glomerulosclerosis begins in a segmental manner. Furthermore, these initially segmental and focal lesions will spread to become global and diffuse. In this review, we will discuss mechanisms of initial injury and progression of FSGS.
Podocyte Injury Is the Initial Target in FSGS
Genetic Effects on Podocytes in FSGS
Animal models indicate podocyte injury is an initial event in FSGS. Classical animal models for FSGS rely on either direct or indirect induction of podocyte injury through the use of renal ablation, podocyte-specific toxins (adriamycin, puromycin aminonucleoside), or targeted genetic mutations (NEP25, diphtheria toxin [DT] receptor, or Thy-1.1 transgenic models). 3 As an example, the transgenic mouse strain NEP25 has been engineered to express the human CD25 receptor exclusively on podocytes. LMB2 is an immunotoxin that selectively targets human CD25. Injection with LMB2 causes glomerular injury in a dose-dependent manner. Mice dosed with 0.625 ng LMB2 per gram body weight developed FSGS characterized by progressive nonselective proteinuria, extensive podocyte foot process effacement, vacuolar degeneration, detachment, and downregulation of podocyte markers WT-1, synaptopodin, nephrin, and podocalyxin. 4 More recent development of animal models has focused on manipulating genes involved with specific functions of podocyte metabolism and filtration barrier maintenance with the intent of studying novel genes that could possibly be involved in initial podocyte injury in FSGS. Often, the genes that are manipulated in these models (e.g., ACTN4, NPHS2, WT1, CD2AP) have been identified by familial genetic studies as potentially playing a role in FSGS pathophysiology. 3
Familial FSGS has been linked to a number of different genetic mutations, most of which encode for proteins that facilitate podocyte cytoskeleton dynamics, slit diaphragm integrity, and cellular signaling. 5 NPHS1 and NPHS2 encode for nephrin and podocin, respectively. Nephrin and podocin are podocyte transmembrane proteins that interact with one another to form a key structural component of the slit diaphragm. Children with steroid-resistant nephrotic syndrome are often tested for mutations in NPHS1 and NPHS2, 5 and some case series report that as much as 25% of pediatric FSGS patients exhibit a mutation in NPHS2. 6 ACTN4 encodes α-actinin-4 which interacts with F-actin in the podocyte actin cytoskeleton as well the phosphatidylinositol 4,5-bisphosphate signaling pathway, cell adhesion proteins, and several transcriptional activation pathways.7,8 FSGS-linked mutations in ACTN4 have been found to eliminate its activity on the nuclear receptors for retinoic acid and estrogen and hamper the capacity of the cytoskeleton to adapt to periodic stretching forces, thus promoting podocyte detachment.9,10 Finally, WT1 encodes for Wilms tumor protein, a key podocyte marker and master regulator protein responsible for the differentiation of podocytes from early parietal epithelial cell (PEC) progenitors. Certain mutations in WT1 can lead to Frasier syndrome, which is characterized by histological findings of FSGS in early childhood. 11
Epigenetic Effects on Podocytes in FSGS
Several different microRNAs (miRNAs), which are short, noncoding RNAs that bind to complementary sequences in coding RNAs, can inhibit translation and have been implicated in FSGS pathogenesis. One such example is miRNA-193A, which is upregulated in podocytes of FSGS patients when compared with minimal change disease (MCD). miRNA-193A specifically targets a region of the mRNA transcript that encodes the key podocyte regulator WT1. The binding of miRNA-193A inhibits the translation of the mRNA transcript, thus downregulating WT1 activity in podocytes and leading to extensive foot process effacement and rapid onset of FSGS. 12
Another miRNA of interest is miR-30, which inhibits translation of Notch1 and p53, both of which have been implicated in podocyte injury. Knockdown of this miRNA acts to sensitize podocytes to cytoskeletal damage and apoptosis in vitro. 13 Similarly, miR-135 acts to upregulate the activity of Wnt/β-catenin signaling through inhibition of GSK3β, thus leading to severe podocyte injury. 14 Other miRNAs of interest include miR-125b and miR-186, both of which declined markedly in FSGS patients with complete remission, but not in those patients with no response to steroid treatment. 15
Finally, recent findings have begun to illuminate the importance of other noncoding RNAs in FSGS pathogenesis. Long noncoding RNA LOC105374325 was elevated in the podocytes of individuals with FSGS by an in situ hybridization assay. Subsequent miRNA PCR array and apoptosis antibody array analyses revealed that LOC105374325 most likely functions to outcompete inhibitory miRNAs for binding sites on transcripts of proapoptotic genes, thus acting to increase the overall level of podocyte apoptosis. 16 Of note, podocytes cannot replicate, and thus, podocyte loss is strongly linked to progression of glomerulosclerosis.
Circulating Factor Effects on Podocytes in FSGS
Primary FSGS appears to be caused by a circulating factor that injures podocytes, based on its rapid recurrence in 30% to 40% of transplant patients. Furthermore, a patient with early recurrent FSGS in the transplant kidney, manifest by extensive foot process effacement but no segmental sclerosis at this early stage, underwent transplant nephrectomy after unsuccessful attempts to treat his marked proteinuria. The removed kidney was then placed into an end-stage kidney disease patient whose disease was not FSGS. The kidney recovered completely when removed from the unidentified circulating factor in the first patient. 17
The most extensively studied candidate circulating factors is soluble urokinase–type plasminogen activator receptor (suPAR). suPAR is the soluble, circulating form of urokinase-type plasminogen activator receptor, a glycosylphosphatidylinositol-anchored three-domain membrane protein that is expressed in several different cell types, including podocytes. 18 However, several studies have failed to validate the putative effects on podocyte integrin. Furthermore, increased suPAR was noted in patients with low estimated glomerular filtration rate, regardless of whether they had primary FSGS or not.19–22 suPAR is an acute-phase reactant, and its increase in chronic kidney disease (CKD) patients is thus not specifically linked to primary FSGS.
CD40 is a costimulatory protein that is expressed by antigen-presenting cells, and along with its ligand, it plays a crucial role in the adaptive immune response. The presence of anti-CD40 antibodies in the sera of postrenal transplant patients is significantly predictive of the recurrence of FSGS. Isolated anti-CD40 antibody is highly toxic to cultured podocytes, suggesting it may play a role in FSGS injury. 23
Cardiotrophin-like cytokine factor 1 (CLCF1) is a cytokine that functions in B-cell stimulation. CLCF1 was postulated as a possible injurious circulating factor in primary FSGS. Notably, injection of isolated rat glomeruli with recombinant human CLCF1 increased the glomerular permeability to albumin, reduced the intensity of stress fibers, and strengthened the intensity of lamellipodia in podocytes. In vivo models injected with CLCF1 demonstrated significantly increased albuminuria. Based on further studies, the group postulated that the mechanism of CLCF1 in podocyte injury relies on the JAK/STAT signaling pathway. 24 However, none of these factors discussed above are proven to be causal in human primary FSGS. It is even possible that different or multiple factors are key in individual patients.
Susceptibility Genes Related to Progression of Podocyte Injury in FSGS
Individuals of African ethnicity are at an increased risk of developing CKD when compared with non-African individuals. Genetic studies of African populations with CKD have revealed polymorphisms in several susceptibility genes, including ApoL1, ApoE, and eNOS. 25 Studies of mice humanized for variant APOL1 expression in podocytes showed podocyte injury and FSGS, but further studies have suggested that APOL1 may not be directly toxic and that all variants have only mild, if any, effects without additional hits. 26 More recent studies suggest a role for APOL1 risk allele RNA, but not protein, to induce injury in mice. 27 Thus, although ApoL1 allele variants G1 and G2 have been linked to increased risk of FSGS development, as of yet there is no definitive evidence that has elucidated the mechanism by which ApoL1 may function in podocyte injury in FSGS. 28
A recent case study of a 51-year-old Japanese man with hyperlipidemia and FSGS who exhibited the rare apolipoprotein E (ApoE) variant ApoE5 lends credence to the involvement of ApoE regulation of lipid metabolism in the pathogenesis of FSGS; however, more studies are necessary to corroborate this hypothesis. 29
Parietal Epithelial Cells, Glomerular Endothelial Cells (GECs), and Tubular Epithelial Cells (TECs) Contribute to Progression of FSGS
Currently, FSGS is classified into five variants according to morphology. Of note, none of the morphological patterns is pathognomonic for so-called primary FSGS. Clinical studies showed that collapsing FSGS has shortened renal survival versus tip variant, supporting the rationale for this classification. However, within a single FSGS biopsy, glomeruli show varying lesions or may appear normal by light microscopy (LM), even though there is extensive foot process effacement by electron microscopy. In primary FSGS, this effacement is extensive in all glomeruli, even in those without segmental lesions. Our data show that these normal-appearing glomeruli by LM had more claudin-1 and CD44 and less WT-1 in visceral epithelial cells and more CD44 in PECs compared with normal glomeruli in normal kidneys or MCD. 30 Glomeruli without LM lesions in cases classified as collapsing variant had more claudin-1 and CD44+ cells in PECs, whereas non-affected glomeruli from tip cases had more WT1+ cells. Furthermore, the segments without sclerosis in glomeruli with a segmental sclerosis lesion had more claudin-1 and CD44 but less WT-1 in the tuft area versus normal or MCD. Non-lesional areas from collapsing glomeruli had more CD44 and WT-1 positivity in PECs, whereas non-lesional areas from tip glomeruli had more WT-1+ cells in valvular endothelial cells. These data suggest that derangements in glomerular epithelial cell phenotype in glomeruli without evident sclerosis by LM or non-sclerotic areas in those with segmental lesions precede and could cause the subsequent structural FSGS lesion. Glomeruli without sclerosis by LM and NOS glomeruli in different FSGS variants have heterogeneity in podocyte and PEC phenotypes, suggesting that glomeruli may be affected by the adjacent microenvironment.
In animal models, sclerosis can spread from a segmental lesion area to the adjacent area or from glomeruli to glomeruli. We have found that injured podocytes can injure remaining initially normal podocytes. We studied the NEP25 chimeric mouse, where only some podocytes expressed the toxin receptor (human CD25). 31 However, podocyte injury spread from receptor-positive to receptor-negative podocytes over time. We propose that three types of cells could contribute to such podocyte injury and progression of FSGS, namely, PECs, GECs, and TECs.
Phenotype Change of PECs and Contribution to FSGS
PECs line Bowman’s capsule. These cells can proliferate in response to various stimuli, including podocyte loss, and have the capacity to replace injured podocytes. A subset of PECs, located at the urinary pole, has been identified as progenitor cells with coexpression of CD24 and CD133, markers of adult tissue stem cells.32,33 PECs located at the vascular pole express podocyte markers (nestin and podocalyxin) without the expression of CD24/CD133. Other PECs express both progenitor cells and podocyte markers. 34 During glomerulogenesis, PECs and podocytes originate from common mesenchymal progenitors. Lineage tracing studies indicate that podocytes can be recruited from PECs that migrate along Bowman’s capsule to the vascular stalk and onto the tuft during development. 35 In disease conditions, the fate of PECs is more controversial. Several studies showed that PECs with CD44 positivity appeared within sclerotic tuft areas after glomerular injury.36–38 These CD44+ cells undergo hypertrophy and hyperplasia and then migrate from Bowman’s capsule to the glomerular tuft, a process which could be regulated by several signaling pathways such as Wnt-β-catenin, CXCR4/SFD1, angiotensin II/AT2 receptors, and Notch 1.39–41 This type of PEC activation and migration could appear during the initial phases of FSGS. 37 The markedly increased CD44 expression in PECs might distinguish FSGS from MCD. 42 A recent study using CD44 knockout mice and overexpression of CD44 in cultured PECs revealed that CD44 is involved in cell adhesion, cell matrix interaction, and cell migration. 43 In this activated state, PECs produce increased specific extracellular matrix (LKIV69), which initially accumulates along Bowman’s capsule basement membrane and in the glomerular tuft when the PECs transit. 44 This PEC recruitment can occur via two distinct pathways, involving vacuolization leading to glomerulosclerosis or PEC proliferation resulting in proliferative pseudocrescents, which can also be found in human biopsies.44,45 However, PECs may also express podocyte markers after glomerular injury and potentially could contribute to podocyte regeneration.46,47 In a genetically engineered mouse study, labeled PECs entered onto the glomerular tuft after podocytes were depleted. After a couple of weeks, these PECs lost CD44 expression, gained podocyte markers, and showed activated extracellular-signal-regulated kinase. 48 The study suggests that there can be two results of activated PECs: consistent CD44 expression, contributing to matrix and glomerulosclerosis; and in the reparative phase, loss of CD44 expression and differentiation to podocytes. In fact, podocyte generation from PECs in mouse models is confined to development and acute glomerular injury, but little is found in aging kidneys or in response to chronic nephron loss.49,50 A recent study subgrouped PECs into three types according to the morphology: flat, cuboidal, and intermediate. Cuboidal and intermediate PECs were more easily activated and contributed to the formation of sclerotic lesions. Furthermore, the epithelial cells seen in FSGS patient biopsies with tip lesions expressed markers of intermediate PECs. 51 These observations suggest varying plasticity of PECs in response to injury.
Endothelial Cell Injury and Contribution to FSGS
As part of the glomerular filtration barrier, endothelial cells have special fenestrae and a luminal glycocalyx layer. 52 FSGS patients have elevated circulating endothelial cells and endothelial dysfunction markers, such as soluble thrombomodulin and von Willebrand factor. 53 These changes were predicted by changes in cholesterol and serum albumin levels and were largely related to the disease activity. In an acute mouse model, podocyte loss caused intracapillary thrombi, a consequence of endothelial cell injury. 54 In puromycin aminonucleoside nephrosis, an in vivo imaging study showed that thrombosis occurred in situ shortly after podocyte detachment. 55 In some steroid-resistant FSGS patients, increased endothelin expression in glomeruli was demonstrated. 56 In patients with membranous nephropathy, more GEC injury, with nuclear and cytoplasmic swelling and loss of fenestrae, was found in those patients who, in addition to the membranous lesions, had secondary segmental sclerosis. 57 These findings suggest podocyte injury in FSGS may lead to endothelial cell dysfunction. On the contrary, endothelial cell dysfunction preceded podocyte injury in adriamycin-induced nephropathy in endothelial nitric oxide synthase–deficient mice. 58 There are several mechanisms to explain the crosstalk between podocytes and endothelial cells. One is the endothelin-1 (EDN1)/EDN1 receptor type A (EDNRA) system. Podocytes release EDN1 after injury, which mediates mitochondrial oxidative stress and dysfunction. Endothelial cells express EDNRA and show increased mitochondrial injury after adriamycin-induced podocyte injury. Inhibition of EDNRA or scavenging of mitochondrial-targeted reactive oxygen species prevented podocyte loss, albuminuria, and glomerulosclerosis in this FSGS model. 59 Another potential mechanism for this crosstalk is the vascular endothelial growth factor A (VEGFA)/Flk-1 system. Normal podocytes are the source of glomerular VEGFA and endothelial cells express its receptor Flk-1. Conditional knockout of VEGFA in podocytes results in endothelial cell death and thrombotic microangiopathy, whereas overexpression of VEGFA in podocytes leads to collapsing lesion in glomeruli.60,61 Our unpublished data showed that glomerular VEGFA levels were reduced to ~30% of normal in an FSGS mouse model induced by 5/6 nephrectomy, accompanied by loss of podocytes.
The Crosstalk of Glomeruli and Tubules Contributes to FSGS
Focal tubulointerstitial fibrosis has largely been viewed as a consequence of glomerular scarring in FSGS, reflecting hypoxia downstream from the scarred glomeruli. Classically, a vicious cycle with glomerular scarring leading to increased stress on remnant nephrons, then causing progressively more glomerulosclerosis, has been the focus for intervention in CKD. 62 We showed that preexisting, even mild, and functionally recovered tubulointerstitial injury sensitizes glomeruli to subsequent podocyte-specific injury. 63 These findings have important implications for progressive kidney disease, beyond the classic model of hyperfiltration stress on remnant nephrons perpetuating increased sclerosis. 64
The proximal tubule is particularly vulnerable to injury due to its high workload, such as that induced by proteinuria in FSGS. Tubular injury shows a spectrum of mild injury with temporary dedifferentiation, mitochondrial derangement related to hypoxia, necrosis, apoptosis, and autophagy, resulting in regeneration versus epithelial cell growth arrest and impaired repair.65,66 Tubular cells can produce matrix and contribute to interstitial fibrosis. However, the major source of interstitial matrix is the activated fibroblast, and interstitial fibrosis could also contribute to tubular injury in several ways. Increased interstitial matrix can increase pressure to tubular basement membranes. Fibroblasts can be activated from the initial tubular injury and in turn secrete profibrotic growth factors that perpetuate tubular injury. Interstitial fibrosis can also decrease peritubular capillary density, thus increasing hypoxia. 67 Hypoxic injury is associated with induction of hypoxia-inducible factors, which regulate oxygen delivery and cellular adaptation to oxygen deprivation. 68 PT epithelial cells, especially at S1, are sensitive to hypoxia, followed by mitochondrial dysfunction. 69 All of these could induce PT atrophy, resulting in stenosis of the glomerulotubular junctions, with concomitant remodeling of Bowman’s capsule, resulting in atubular glomeruli, that is, glomeruli without patent connection to the PT. In many human and experimental disease conditions including FSGS and diabetic nephropathy, atubular glomeruli are associated with decreased glomerular filtration rate (GFR) and disease progression.70–73 Atubular glomeruli do not significantly contribute to GFR, and thus may increase workload on remnant glomeruli, promote sclerosis in these connected glomeruli, and also increase susceptibility of podocytes to any additional insult.
Tubular injury, especially impaired PT absorption, could activate tubuloglomerular feedback and reduce GFR, as seen in acute kidney injury.74–76 Our previous data showed that DT-induced tubular injury resulted in a prominent increase in neuronal nitric oxide synthase protein, which is mainly expressed in the macula densa within the kidney, indicating that tubular injury can activate the tubuloglomerular feedback through macula densa. 63 Cells of renin lineage have remarkable plasticity which could be regulated by tubulointerstitial injury, which may be involved in crosstalk between macula densa cells and juxtaglomerular apparatus (JGA) cells. Recent studies demonstrated that renin-producing cell recruitment is dependent on nitric oxide (NO) availability and the NO-guanylate cyclase signaling pathway. 77 Renin lineage cells, located at the JGA, along the afferent arterioles and interstitial pericytes, represent additional potential podocyte progenitor cells. Renin lineage cells can migrate to Bowman’s capsule or to the glomerular tuft, replacing PECs, podocytes, and mesangial cells after podocyte injury.78–80 This repair capacity is remarkable because podocytes are normally derived from cap mesenchyme cells, whereas renin lineage cells derive from Foxd1+ stromal cells. Similar to PECs, renin+ progenitor cells can differentiate either to podocytes, contributing to repair, or to activated mesangial cells, contributing to matrix secretion. 81 Renin lineage cells may migrate directly to mesangial or podocyte locations or to the PEC location, differentiate to PECs, and then migrate to the glomerular tuft. Once present at the glomerular tuft, these PECs may contribute to podocyte repair or become profibrotic-type visceral epithelial cells, contributing to sclerosis. The signal(s) that triggers renin lineage cells to migrate and transdifferentiate into podocytes or mesangial cells has not yet been identified.
Clinical Implications
Due to the low prevalence of FSGS, many clinical trials did not recruit large case numbers. Indeed, not only the morphology but also genetic or epigenetic factors may influence the heterogeneous response to treatment. Three randomized clinical trials in FSGS using calcineurin inhibitors showed extremely different remission rates: 82% in an Indian study, 72% in a Toronto study, and 46% in a North American study. 82 One subset of FSGS patients showed increased tumor necrosis factor-α (TNF-α). Whether these patients could be more sensitive to adalimumab, a TNF-α inhibitor, has been postulated. 83 Precision medicine approaches with subgrouping of FSGS patients, according to etiology, tissue and cellular morphology, and molecular profile, may be necessary to optimize tailored and targeted treatment approaches.
As a first-line treatment, immunosuppressive treatment with prednisolone has been a valuable option with good remission rates. In patients with steroid dependence or steroid resistance of their nephrotic syndrome, calcineurin inhibitors have been considered. Possible mechanisms likely go beyond immunosuppression with its inhibition of the nuclear factor of activated T-cell pathway, and include stabilization of the podocyte cytoskeleton. Angiotensin-converting enzyme inhibitors and angiotensin receptor blockers have antiproteinuric effects beyond hemodynamic effects and are associated with better renal survival and a slower progression of FSGS, perhaps due to direct effects on podocytes and fibrosis pathways. Other treatments, such as removal or neutralization of the putative circulating injurious factors by plasmapheresis or galactose, modification of the melanocortin receptors by Acthar, altered cellular immunity by rituximab or abatacept, and targeting fibrosis by transforming growth factor beta or TNF-α antibodies, are ongoing in clinical trials. None of them yielded dramatic success so far. Regulating the interaction between podocytes and other nephron components could be an additional future direction for consideration both in FSGS and in other causes of CKD. The biggest challenge of clinical therapy is to correctly define the detailed phenotype of the patient population, match treatments with patient-specific derangements, and also identify patients at an optimal time point when therapy can be effective. FSGS characteristically progresses to end-stage renal disease. After initial podocyte injury, the progression of FSGS involves local cellular events, including PECs, endothelial cells, TECs, and adverse crosstalk among these cells (Fig. 1). Increased understanding of the mechanisms can lead to major advances, resulting in focused interventions to target both the podocyte and other nephron components of injury and potentially prevent progression in FSGS.
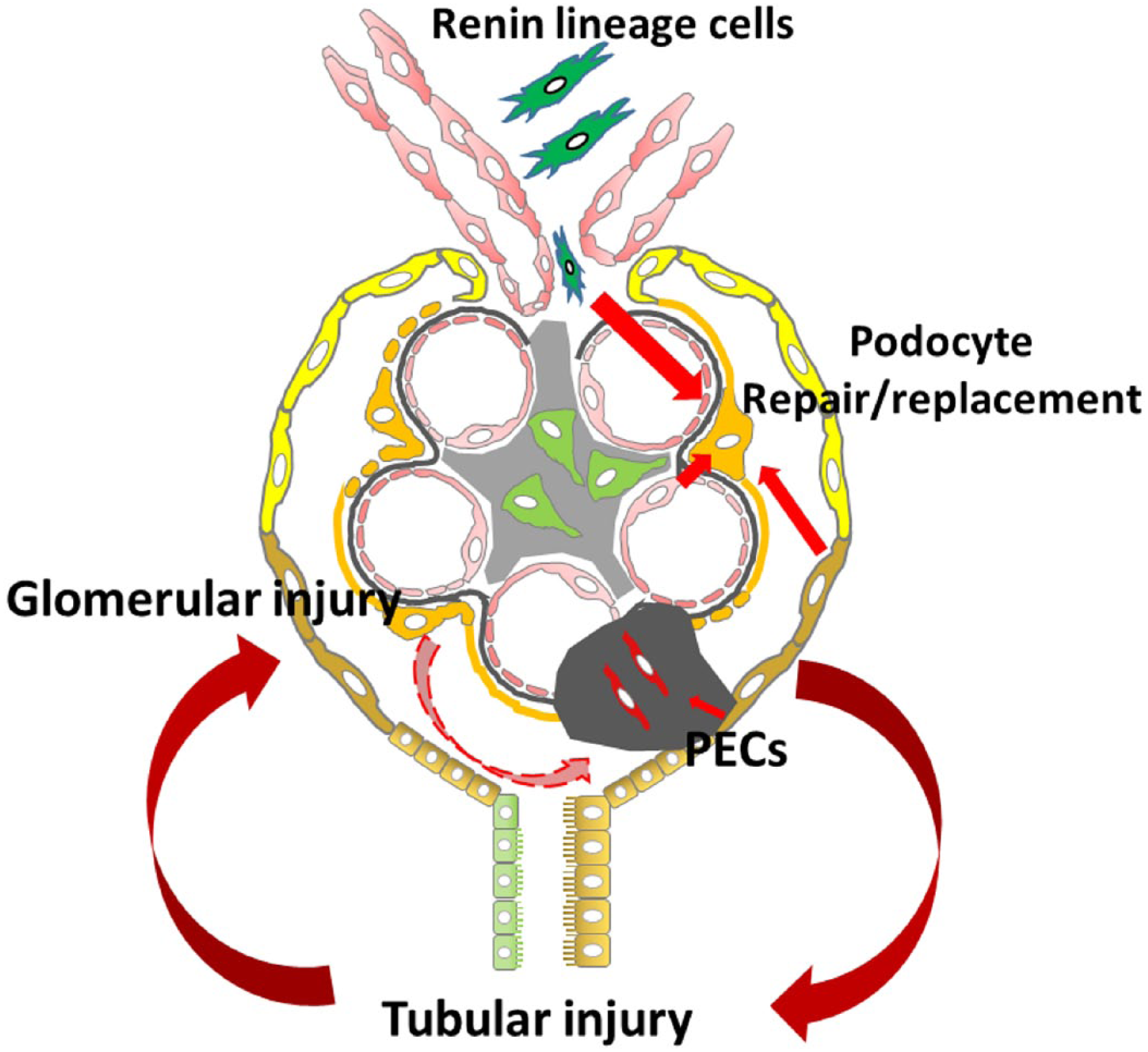
Adverse crosstalk among podocyte and other local cells in FSGS.
Footnotes
Competing Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
H-CY and ABF designed the study. JZ, JBW, and H-CY drafted the manuscript. All authors have read and approved the final manuscript.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by NIH NIDDK DK56942-09 (ABF) and DK114809 (ABF).