
Abstract
To protect against danger, the innate immune system must promptly and accurately sense alarm signals, and mount an appropriate response to restore homeostasis. One endogenous trigger of immunity is tenascin-C, a large hexameric protein of the extracellular matrix. Upregulated upon tissue injury and cellular stress, tenascin-C is expressed during inflammation and tissue remodeling, where it influences cellular behavior by interacting with a multitude of molecular targets, including other matrix components, cell surface proteins, and growth factors. Here, we discuss how these interactions confer upon tenascin-C distinct immunomodulatory capabilities that make this matrix molecule necessary for efficient tissue repair. We also highlight in vivo studies that provide insight into the consequences of misregulated tenascin-C expression on inflammation and fibrosis during a wide range of inflammatory diseases. Finally, we examine how its unique expression pattern and inflammatory actions make tenascin-C a viable target for clinical exploitation in both diagnostic and therapeutic arenas.
Keywords
Introduction
The innate immune system is constantly challenged by danger of diverse origin. Traditionally, its primary function was considered to be the recognition of non-self entities. Indeed, upon infection, the presence of a pathogen is sensed by innate immune cells through membrane-bound and intracellular pattern recognition receptors (PRRs). The identification of highly conserved pathogen-derived molecular motifs, collectively referred to as pathogen-associated molecular patterns (PAMPs), by PRRs triggers cells to activate an inflammatory response. Following the release of chemokines, phagocytes and antigen-presenting cells are recruited to swiftly destroy the interloping pathogen, prevent widespread tissue colonization, and shape targeted adaptive responses to confer long-lasting immunity against recurrent invasion. 1
However, infection is not the sole threat to homeostasis. Inflammatory responses are also activated by entities of non-microbiological origin, for example, by a wide variety of endogenous stimuli generated as a result of cellular stress and tissue damage. These signals include molecules released upon cell death and components of damaged extracellular matrix, which are vital for triggering pathways designed to clear cellular debris and restore tissue integrity.2–4 Furthermore, the immune system can be activated by inorganic irritants, such as asbestos 5 ; by deposits of endogenous molecules, such as amyloid beta and cholesterol6,7; and by tumor-associated antigens. 8 Thus, it appears to be not a simple matter of discriminating between harmful foreign and benign self, but rather detecting any potential threat, regardless of origin.
Many questions around this model of immunity remain unanswered, not least how the identity of endogenous inflammatory molecules is defined, and the mechanisms by which their activity must be tightly regulated to avoid catastrophic autoimmune destruction. Moreover, while the induction of inflammatory responses by PAMPs is relatively well understood, 9 the mode of action of endogenous inflammatory triggers remains elusive. Answering these questions will not only provide insight into an intriguing immunological puzzle but may also open new therapeutic avenues to treat inflammatory diseases of a non-infectious origin, where pathology is driven by endogenous inflammatory stimuli.
Tenascin-C is a large hexameric protein of the extracellular matrix that exhibits limited expression in healthy tissues, but which is rapidly upregulated upon tissue damage. Typically, its expression is transient, with mRNA downregulated and protein cleared by the time tissue repair is complete. 10 However, persistent expression of tenascin-C is associated with a wide variety of pathological conditions, where it accumulates at sites of inflammation, for example, in autoimmune, fibrotic, and metabolic diseases, and in many cancers. 11 Despite this well-established pattern of expression, it was not until comparatively recently that the ability of tenascin-C to trigger inflammation, and directly shape immune responses, was reported. In this review, we present the current understanding of tenascin-C’s involvement in endogenous immunity, and the data emerging that implicate this matrix molecule as a promising biomarker of inflammatory disease status and a tractable target for therapeutic applications.
Tenascin-C Up-Close: A Diverse Interaction Partner
Tenascin-C is characterized by a distinct domain organization. An assembly domain at the N-terminus, responsible for the formation of the six-armed oligomer known as the hexabrachion, is followed by a series of epidermal growth factor-like (EGF-L) repeats, constitutively expressed and alternatively spliced fibronectin type III-like repeats (FNIII), and a C-terminal fibrinogen-like globe (FBG). This complex structure enables tenascin-C to establish a wide variety of molecular interactions (reviewed in Midwood et al. 12 ). Tenascin-C is an important regulator of the matrix itself, modulating its assembly and the biophysical properties of tissue. Within the extracellular matrix, tenascin-C binding partners include collagen, fibronectin, periostin, fibrillin-2, and proteoglycans: perlecan, aggrecan, neurocan, and phosphacan/receptor protein tyrosine phosphatase β/ζ. Tenascin-C also binds to proteins exposed on the cell surface, such as syndecan 4, contactin, glypican, annexin II, and CALEB/CSPG5. It can further directly influence internal cell signaling by binding to cell surface receptors such as epidermal growth factor receptor (EGFR), toll-like receptor 4 (TLR4), and integrins. Moreover, tenascin-C is able to bind numerous growth factors, including members of the fibroblast growth factor family and the transforming growth factor beta (TGF-β) superfamily. This multitude of interactions makes tenascin-C a diverse modulator of cellular behavior, such as proliferation, migration, adhesion, and differentiation, capable of producing highly context- and cell-type-specific responses. Here, we discuss further a selection of these interactions by which tenascin-C mediates, and modulates, inflammatory responses.
The Proinflammatory Capabilities of Tenascin-C
To date, three main binding partners have been identified to be responsible for the inflammatory effects of tenascin-C: TLR4, and integrins α9β1 and αVβ3. The impact of tenascin-C activation of these receptors is summarized in Table 1.
Tenascin-C as a Modulator of Cellular Behavior in Inflammation.
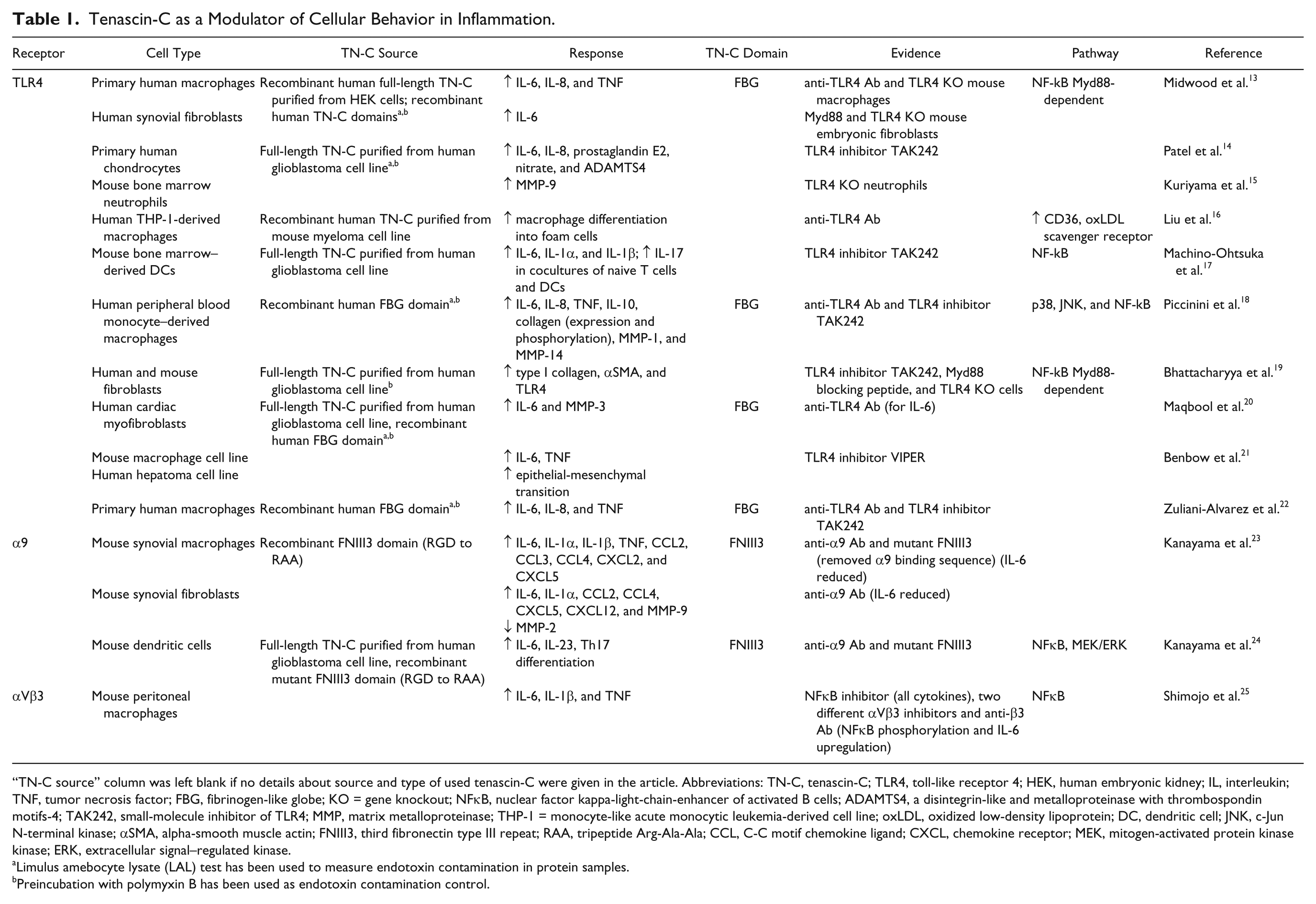
“TN-C source” column was left blank if no details about source and type of used tenascin-C were given in the article. Abbreviations: TN-C, tenascin-C; TLR4, toll-like receptor 4; HEK, human embryonic kidney; IL, interleukin; TNF, tumor necrosis factor; FBG, fibrinogen-like globe; KO = gene knockout; NFκB, nuclear factor kappa-light-chain-enhancer of activated B cells; ADAMTS4, a disintegrin-like and metalloproteinase with thrombospondin motifs-4; TAK242, small-molecule inhibitor of TLR4; MMP, matrix metalloproteinase; THP-1 = monocyte-like acute monocytic leukemia-derived cell line; oxLDL, oxidized low-density lipoprotein; DC, dendritic cell; JNK, c-Jun N-terminal kinase; αSMA, alpha-smooth muscle actin; FNIII3, third fibronectin type III repeat; RAA, tripeptide Arg-Ala-Ala; CCL, C-C motif chemokine ligand; CXCL, chemokine receptor; MEK, mitogen-activated protein kinase kinase; ERK, extracellular signal–regulated kinase.
Limulus amebocyte lysate (LAL) test has been used to measure endotoxin contamination in protein samples.
Preincubation with polymyxin B has been used as endotoxin contamination control.
TLR4 is a cell surface receptor present on leukocytes and stromal cells. It was first identified as the PRR responsible for mediating innate immune responses to the potent bacterial virulence factor, lipopolysaccharide (LPS) or endotoxin, but it also has a growing number of endogenous activators; this list includes tenascin-C, biglycan, fibrinogen, and S100 proteins, among many others. 26 The most widely reported effect of TLR4 activation by tenascin-C is the induction of soluble proinflammatory mediators, such as interleukin (IL)-6, IL-8, and tumor necrosis factor (TNF). This has been observed in a number of cell types, including macrophages,13,18,21,22 dendritic cells (DCs), 17 fibroblasts,13,20 and chondrocytes. 14 More recently, tenascin-C signaling through TLR4 has been implicated in inflammasome priming. Inflammasomes are multiprotein, intracellular complexes that drive specific inflammatory responses including the release of IL-1β and IL-18, and induction of pyroptosis, a type of programmed cell death. 27 Typically, inflammasomes require two separate signals for activation: The first “priming” signal comprises PRR-driven procytokine production and synthesis of inflammasome components; this is followed by inflammasome assembly and caspase-1-mediated maturation and release of bioactive IL-1β and IL-18. 28 Stimulation of rat epicardium–derived cells with tenascin-C induced TLR4-dependent pro-IL-1β transcription, implicating elevated tenascin-C expression in rat cardiac tissue following ischemic injury as a potential priming step for inflammasome assembly that can be driven by adenosine or ATP-mediated purinergic stimulation in this cell type. 29 In addition, increased levels of secreted mature IL-1β could be detected in cell culture medium of mouse bone marrow–derived DCs stimulated with tenascin-C, 17 indicating that tenascin-C may be capable of driving IL-1 synthesis in the absence of an exogenous second signal. However, a direct link between tenascin-C activation of TLR4 and purinergic signaling, or the requirement for any other second signal, has not been investigated.
Tenascin-C upregulation therefore would be expected to increase tissue levels of proinflammatory cytokines and chemokines, orchestrating innate immune cell recruitment and activation. In addition, a number of reports suggest that tenascin-C-mediated cytokine release is also important for creating a microenvironment permissive for selective T-cell polarization. DCs derived from tenascin-C-deficient mice exhibited diminished proinflammatory cytokine release in response to LPS stimulation, and a reduced capacity to drive naive T cells toward Th17, but not Th1, Th2, or Treg, cell differentiation. 30 Stimulation of DCs with exogenous tenascin-C promoted their ability to drive the generation of Th17 cells, which effect could be ablated with anti-IL-6 and anti-TLR4 antibodies, suggesting that IL-6 synthesis induced by tenascin-C activation of TLR4 is a key determinant of T-cell subset polarization. 17 Interestingly, tenascin-C also stimulated chondrocyte secretion of prostaglandin E2, 14 a complex mediator of inflammation known to be involved in Th17 polarization.31,32
Wider profiling of the impact of tenascin-C activation of TLR4 in primary human macrophages highlighted functional consequences beyond cytokine synthesis. Comparison of macrophages activated by tenascin-C, or by a pathogenic ligand, LPS, revealed overlapping, but distinct downstream impacts on cell behavior. While both ligands induce classical NFκB and mitogen-activated protein kinase (MAPK) signaling, LPS drove macrophages toward an aggressive phenotype designed to fight infection, characterized by high levels of proinflammatory cytokines and tissue degrading matrix metalloproteinases, while tenascin-C favored the upregulation of extracellular matrix protein expression and phosphorylation, resulting in macrophage behavior more suited to tissue repair. 18 Tenascin-C has also been shown to help drive macrophages to become foam cells. Tenascin-C potentiated oxidized low-density lipoprotein (oxLDL)–induced foam cell formation, in a TLR4-dependent manner. Tenascin-C was also upregulated upon macrophage stimulation by oxLDL, and was capable of inducing the expression of CD36, a scavenger receptor involved in LDL uptake, suggesting the existence of a positive feedback loop promoting the generation of these cells key to the development of atherosclerosis. 16 These data collectively show that tenascin-C profoundly influences macrophage polarization, that it induces a macrophage phenotype quite different to that induced by pathogenic activation of TLR4, and that macrophage subsets created by tenascin-C activation can differ depending on context.
An increasingly wide range of cell types are reported to respond to tenascin-C via activation of TLR4. In murine bone marrow isolated neutrophils, tenascin-C has been shown to upregulate the expression of matrix metalloproteinase (MMP)-9 in a TLR4-dependent manner. 15 Activation of the TLR4/tenascin-C axis has been also reported to induce epithelial-mesenchymal transition and promote migration of human hepatoma cell line. 21 Tenascin-C also induces TLR4-mediated cytokine synthesis in fibroblasts. For example, in synovial fibroblasts it promotes high levels of IL-6 secretion, although it does not induce IL-8 in these cells, in contrast to macrophages. 13 Moreover, other outputs have been reported in tenascin-C-activated stromal cells. TLR4-dependent activation of primary human foreskin fibroblasts by tenascin-C induces synthesis of type I collagen and alpha-smooth muscle actin. 19 These data support a role for tenascin-C not just in triggering immune cell–mediated inflammation but also in stromally driven defense responses, suggesting that this matrix molecule contributes to both inflammatory processes immediately following tissue damage and subsequent tissue remodeling by activating cell-type-specific responses via TLR4.
That tenascin-C drives very different TLR4-mediated inflammatory signaling than LPS supports the idea that the immune system is able to distinguish different alarm signals using common sensors, to activate distinct, tailored responses to particular threats, that is, injury versus infection. However, it is not yet clear on the molecular level how different outputs arise from activation of the same PRR. We still do not fully understand how tenascin-C binds to and activates TLR4. What we do know is that the binding site comprises three distinct epitopes within the FBG domain of tenascin-C, 22 and that the classical LPS coreceptors, myeloid differentiation protein-2 (MD-2) and CD14, are not essential for TLR4 activation by tenascin-C. 13
In addition to TLR4, tenascin-C activation of two other cell surface receptors has been shown to drive inflammatory responses. Integrins α9β1 and αVβ3 are transmembrane receptors capable of binding to a number of matrix proteins, including fibronectin and tenascin-C. The binding of tenascin-C by both integrins induces proinflammatory cytokine synthesis, with the interaction site responsible for the observed effects pinpointed to the third FNIII repeat for α9β1. 23 Although αVβ3 is known to bind to two different sites within tenascin-C, the third FNIII repeat 33 and the FBG domain, 34 it is still unknown which of them, if any, is responsible for conferring the proinflammatory activity. In murine synovial macrophages and fibroblasts, tenascin-C-induced α9 integrin–mediated signaling upregulates a wide array of proinflammatory molecules, including IL-6, IL-1α, CCL2, CCL4, and CXCL5. 23 α9 integrin activation by tenascin-C is also involved in Th17 cell differentiation through induction of IL-6 and IL-23 in DCs. In this study, stimulation of IL-6 secretion by tenascin-C was shown to be a synergistic effect of both α9 integrin- and TLR4-mediated signaling. 24 The outcome of αVβ3 activation by tenascin-C was studied in peritoneal macrophages, where it resulted in the upregulation of IL-6, IL-1β, and TNF. This effect of tenascin-C on cytokine secretion could be ablated with an NFκB inhibitor. 25 These data show that tenascin-C can induce cytokine synthesis by activating more than one receptor family; how this interplay is mediated on the cellular level remains to be elucidated, but such receptor redundancy may illustrate the importance of tenascin-C as a danger signal, ensuring that it does not go unnoticed at times of cellular stress.
In addition to its influence through binding to membrane receptors, tenascin-C may also indirectly modulate intracellular inflammatory signaling. In a human glioblastoma cell line, tenascin-C downregulates the Wnt inhibitor DKK1, resulting in increased classical Wnt signaling by stabilization of β-catenin. 35 Tenascin-C also interacts with Wnt3a, and human adenocarcinoma cell line cells plated on tenascin-C in the presence of Wnt3a exhibit enhanced classical Wnt signaling. Interestingly, when cells were incubated with soluble tenascin-C, the signaling was markedly reduced. 36 This might suggest that while soluble tenascin-C would sequester Wnt ligands, within the ECM it could be responsible for accumulating ligands close to the cell surface enabling signaling activation. Due to the fact that a complex crosstalk exists between Wnt and NFκB pathways, 37 regulation of Wnt signaling by tenascin-C could exert both positive and negative modulation of the inflammatory response. Furthermore, evidence is emerging that supports a role for tenascin-C in PRR-mediated responses to pathogen invasion. In murine bone marrow–derived macrophages, rapid upregulation of tenascin-C upon LPS detection by TLR4 controls the maturation of the micro-RNA miR-155. Impaired maturation of miR-155 in the absence of tenascin-C prevents effective translation of LPS-induced proinflammatory cytokines including TNF. 38 Finally, tenascin-C is capable of interfering with HIV transmission. Tenascin-C present naturally in human breast milk was shown to bind HIV-1 Envelope protein, neutralizing the infectious properties of this virus in reporter cell lines and peripheral blood mononuclear cells. 39 This cooperative impact of microbiological and endogenous alarm signals demonstrates an amazing complexity of signaling within the innate immune system.
Tenascin-C and the Resolution of Inflammation
The induction of tenascin-C expression is a transient process during the progression of inflammation; it is not detectable following the completion of tissue repair. However, persistent tenascin-C expression is associated with chronic inflammation, where the proinflammatory properties of tenascin-C mediated by interactions with both TLR4 and the integrins might offer an interesting insight into the mechanism of its involvement in inflammatory diseases. Upregulated as a result of tissue damage, cellular stress, and inflammatory mediators, and subsequently exacerbating the inflammatory response, tenascin-C is likely to activate a positive feedback loop driving disease. At this point, the vital importance of strict control over tenascin-C expression becomes evident, and the factors regulating tissue levels of tenascin-C have recently been reviewed.11,12,40
The question arises whether tenascin-C plays an active role during the resolution of inflammation, or whether its disappearance from tissues simply creates a less proinflammatory environment. Tenascin-C has been shown to bind to TGF-β, 41 a factor that plays a key role in the resolution of inflammation, 42 and interactions with tenascin-C might increase local concentrations or activity of this immunosuppressive factor. Moreover, tenascin-C has been shown itself to exert immunosuppressive effects on lymphocytes. A number of studies found tenascin-C to be capable of arresting T-cell activation induced by various stimuli.43–47 The blocking of anti-CD3 MAb/fibronectin-induced activation of human peripheral blood T cells by tenascin-C was shown to be mediated through the alternatively spliced repeats A1 and A2 within FNIII domain and was caused by a blockade of TCR/CD3 complex internalization. 46 In addition, in murine CD4+ and CD8+ splenocytes activated with a cocktail of IL-2 and anti-CD3 and anti-CD28 beads, tenascin-C-mediated reorganization of the actin cytoskeleton was implicated in regulating T-cell behavior. 47 On the whole, these data add to the evidence supporting tenascin-C as a sophisticated regulator of the inflammation, capable of conveying both pro- and anti-inflammatory responses.
The Wider Picture: Conclusions From Animal Models
The intricate inflammatory functions of tenascin-C described in vitro prompt questions about the significance of its contribution to inflammation in vivo. The macroscopic effects of tenascin-C gene deletion have been examined using two independently generated knockout mice.48,49 Surprisingly, few gross phenotypic differences could be detected in the tenascin-C knockout (TNC KO) animals, compared with wild type. They were born alive and fertile, normal in size, and exhibited standard development and lifespan. Abnormalities in TNC KO mice were observed during tissue repair following corneal50,51 and myocardial injury, 52 including reduced macrophage infiltration, delayed myofibroblast recruitment, and reduced TGF-β, fibronectin, and collagen synthesis, resulting in delayed healing. These phenotypes stimulated further interest in the protein’s relevance during pathological conditions. Indeed, investigation of inflammatory responses in TNC KO mice challenged with experimental disease has provided considerable insight into the role of tenascin-C in immunity in vivo (summarized in Table 2).
Tenascin-C in Mouse Models of Inflammatory Diseases.
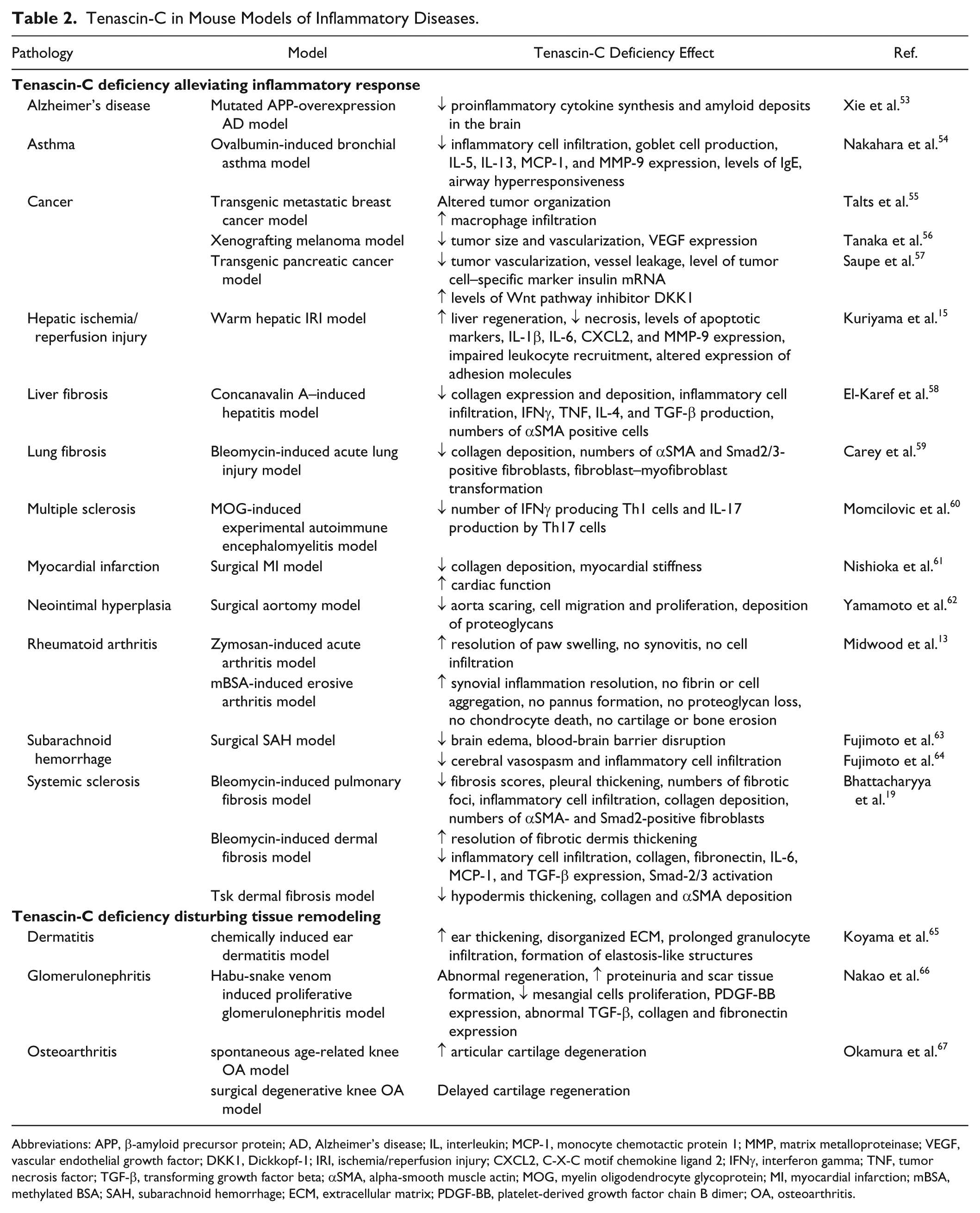
Abbreviations: APP, β-amyloid precursor protein; AD, Alzheimer’s disease; IL, interleukin; MCP-1, monocyte chemotactic protein 1; MMP, matrix metalloproteinase; VEGF, vascular endothelial growth factor; DKK1, Dickkopf-1; IRI, ischemia/reperfusion injury; CXCL2, C-X-C motif chemokine ligand 2; IFNγ, interferon gamma; TNF, tumor necrosis factor; TGF-β, transforming growth factor beta; αSMA, alpha-smooth muscle actin; MOG, myelin oligodendrocyte glycoprotein; MI, myocardial infarction; mBSA, methylated BSA; SAH, subarachnoid hemorrhage; ECM, extracellular matrix; PDGF-BB, platelet-derived growth factor chain B dimer; OA, osteoarthritis.
In line with the proinflammatory capabilities of tenascin-C reported in vitro, tenascin-C persistently emerges as a key player in chronic inflammation in vivo. The reduction of proinflammatory cytokine secretion and subsequent diminished inflammatory cell recruitment as a result of tenascin-C deficiency have been shown to exert a protective effect in a number of inflammatory disease models. Tenascin-C ablation in transgenic mice overexpressing mutated amyloid precursor protein resulted in suppression of Alzheimer’s disease–specific inflammation. Lower levels of proinflammatory mediators, such as TNF, IL-1β, and IL-6, and higher levels of anti-inflammatory mediators, such as IL-10, MRC1, and CCL2, in the central nervous system were associated with reduced amyloid deposition and ameliorated Alzheimer’s disease pathology. 53 Knockout of tenascin-C was reported to reduce immune cell infiltration and prevent brain edema and blood–brain barrier disruption following insult in a surgical subarachnoid hemorrhage model.63,64 Remarkable anti-inflammatory effects of tenascin-C deficiency were also reported in models of joint inflammation. During acute inflammation induced by zymosan, the resolution of synovial inflammation was significantly quicker in TNC KO animals compared with wild type. Furthermore, tenascin-C-deficient mice were protected from prolonged synovitis, and cartilage and bone destruction, in a model of erosive arthritis induced by methylated BSA. 13 During this erosive arthritis, tenascin-C deficiency resulted in decreased cytokine synthesis in the joint, including attenuated IL-6 and IL-23 expression, and this altered cytokine profile created a microenvironment that could not support Th17 polarization, manifesting in significantly reduced IL-17 expression. 30 TNC KO mice also exhibited a decrease in the production of IL-17 by Th17 cells during experimental autoimmune encephalomyelitis, a mouse model of multiple sclerosis, accompanied by a reduction in the number of interferon gamma (IFNγ)–producing Th1 cells. 60 Moreover, during ovalbumin (OVA)-induced bronchial asthma, TNC KO animals exhibited significantly attenuated allergic inflammation, with reduced eosinophil infiltration and reduced levels of IgE levels. This dampening of the inflammatory response was shown to be a result of impaired differentiation of Th2 cells, leading to a decrease in the expression of the classical type 2 cytokines, IL-5 and IL-13. 54 These studies support in vitro data suggesting that tenascin-C can modulate T cell polarization, but highlight that tenascin-C exerts complex and highly context-specific effects, affecting T-cell subsets shown to orchestrate particular pathologies: Th2 in asthma, 68 Th17 in rheumatoid arthritis (RA), 69 and Th17 and Th1 in autoimmune encephalomyelitis. 70
In addition to exacerbating inflammatory responses, tenascin-C has been shown to play a particularly deleterious role promoting tissue fibrosis. For example, in the cardiovascular system, tenascin-C deficiency relieves postsurgical neointimal hyperplasia62,71 and improves cardiac function after myocardial infarction. 61 Moreover, in a concanavalin A–induced hepatitis model, a reduction in collagen expression and deposition was observed in the livers of TNC KO mice. This was associated with reduced secretion of inflammatory mediators IFNγ, TNF, and IL-4, and lower lymphocyte and neutrophil infiltration into the liver, as well as a significant downregulation of TGF-β expression compared with wild-type mice. 58 Impaired TGF-β signaling, with diminished nuclear translocation of Smad-2 and Smad-3, was also shown to protect tenascin-C-deficient animals from lung fibrosis in a bleomycin-induced acute lung injury model. 59 Moreover, a decrease in TGF-β expression and attenuation of collagen deposition caused reduced dermal and pleural thickening, as well as accelerated resolution of inflammation, in the absence of tenascin-C in bleomycin-induced mouse models of systemic sclerosis. 19 Consistent with in vitro studies, these data indicate that tenascin-C impacts both the immune and the stromal compartment during inflammatory disease models, and suggest that enhanced fibrosis may be mediated at least in part by raising tissue levels of TGF-β, enabling this factor to exert fibrogenic effects by promoting fibroblast activation, and stimulating excessive production of extracellular matrix. 72 Finally, liver tissue regeneration was enhanced as a result of tenascin-C ablation in a model of hepatic ischemia/reperfusion injury. In the absence of tenascin-C, the secretion of IL-6, IL-1β, and CXCL2 was decreased and the recruitment of leukocytes impaired. This inflammatory phenotype was accompanied by attenuated catabolic processes, with depleted necrosis, lower levels of apoptotic markers, and decreased expression and activation of MMP-9, an extracellular matrix–degrading enzyme, 15 suggesting tenascin-C can impact tissue fibrosis by both promoting matrix deposition and inhibiting matrix removal.
These data indicate that tenascin-C expression in experimental models contributes to prolonged inflammation and fibrosis during disease pathogenesis. However, a number of studies provide evidence supporting a positive role for tenascin-C during inflammation in vivo. Following renal injury in a snake venom–induced model of proliferative glomerulonephritis, an inflammatory kidney disease, TNC KO animals showed drastically impaired renal regeneration, with increased proteinuria and scar tissue formation, decreased mesangial cells proliferation, and aberrant patterns of TGF-β, collagen, and fibronectin expression. The severity of observed abnormalities resulted in death of all knockout animals within 4 months, despite the injury being reversible in wild-type controls. 66 Tenascin-C has been also reported to attenuate inflammation in chemically induced dermatitis. Tenascin-C-null mice exhibited increased swelling and prolonged polymorphonuclear cell infiltration. During postinsult regeneration, a disorganization of extracellular matrix and formation of elastosis-like structures were observed. 65 A significant delay in tissue regeneration was also observed in tenascin-C-deficient mice in a surgical degenerative knee osteoarthritis model. In contrast to controls, the injured cartilage could not be regenerated in TNC KO animals, and the defects were filled with fibrous tissue. Moreover, age-related spontaneous articular cartilage degeneration was accelerated in knockout animals. 67
Collectively, these studies confirm tenascin-C is a potent regulator of inflammation and fibrosis in vivo. However, these data also highlight that tenascin-C displays a remarkable functional dichotomy; in some models, it positively influences tissue repair, protecting from excessive inflammation and remodeling, and preventing fibrotic tissue formation. In contrast, in other models, it is capable of driving chronic inflammation and promoting profibrotic responses often to the detriment of the organism. Temporal control of tissue levels of tenascin-C might be of importance here; where expression is downregulated tenascin-C mediated inflammation and repair can proceed in a controlled manner, while persistent expression of tenascin-C would fuel proinflammatory and profibrotic responses. Another key influencing factor might be the tissue microenvironment where expression of distinct tenascin-C splicing variants, differential proteolytic processing, and the presence of different tenascin-C receptors exposed on the cell surface would also be expected to shape the context of tenascin-C-mediated responses.
In considering the role of tenascin-C in inflammation in vivo, more intriguing questions continue to emerge. Tenascin-C has been shown to contribute to the immune response to LPS-induced sepsis. TNC KO animals were protected from systemic inflammation following the cytokine storm caused by LPS administration, supporting a role for tenascin-C in host defense against infection in vivo. Interestingly, this phenotype in tenascin-C-deficient mice could be partially rescued by bone marrow transplant from wild-type littermates, 38 highlighting that both myeloid and stromally derived tenascin-C participate in these immune responses. Within the bone marrow compartment, a decrease in cell colony–forming capacity has been reported in TNC KO animals, yet the steady-state hematopoiesis was not affected. 73 However, tenascin-C-deficient mice did exhibit impaired hematopoietic recovery as a result of a bone marrow ablation by irradiation. 74 Tenascin-C was also shown to regulate bone marrow–mediated angiogenesis. 75 In addition, a recent study found that tenascin-C expressed in the bone marrow, but not the myocardium, was responsible for the attenuation of fibrosis in a mouse model of cardiac hypertrophy. TNC KO animals showed an exacerbation of inflammation and impaired cardiac function following the transverse aortic constriction surgery. This phenotype could be rescued by a bone marrow transplant from a wild-type donor, and simultaneously, a transplant from a tenascin-C-null donor to a wild-type recipient resulted in a declined cardiac function. 76 Together, these data pose questions about the site of action of tenascin-C during inflammation, suggesting a role not only locally, at the site of inflammation, but also distally, impacting inflammatory cell behavior at sites some distance from pathology. These data also raise a cautionary note that it will be important to take into account the background strain of mice used in studies, as different strains of mice are known to exhibit different immune compartment biases that could define the ultimate impact of tenascin-C deletion in vivo.
Tenascin-C in the Clinic
In humans, a missense mutation in the TNC gene is responsible for autosomal dominant hear loss, caused by irreversible abnormalities in developing cochlea. 77 Polymorphisms in the gene have been also associated with increased risk of asthma, rhinoconjunctivitis, and Achilles tendinopathy.78–80 However, it is the wild-type protein that attracts most interest as a potential target for diagnostics and therapy.
Successful treatment of a disease must be preceded by a correct diagnosis. This is often challenging, as signs and symptoms can be nonspecific, or their severity highly subjective. Hence the widespread use of biomarkers, easily measurable objective factors indicating the medical state of the patient. Biomarkers can be employed to predict disease incidence and severity, treatment outcome, or surgical intervention effects. Although tenascin-C is upregulated in a number of human pathologies, it has been most thoroughly studied in the context of cancer, and was shown largely to correlate with metastasis and indicate poor prognosis. So far its prognostic significance has been confirmed in breast, lung, and bladder cancer, glioma, squamous cell carcinoma, melanoma, and colorectal and hepatocellular carcinoma (reviewed in Lowy and Oskarsson. 81 ). Recently, tenascin-C levels have been investigated as a diagnostic factor in preeclampsia, a hypertensive disorder of pregnancy and one of the most common cause of maternal deaths worldwide. 82 Consistent with its involvement in driving inflammation, tenascin-C has also been widely considered as a biomarker of patient inflammatory status in a number of chronic inflammatory diseases.
A screen of tenascin-C in sera from patients suffering diverse conditions, including cancer, malaria, and sepsis (from a routine chemistry laboratory of a large hospital), found a positive correlation between the levels of tenascin-C and C-reactive protein (CRP), highlighting that elevated serum tenascin-C is associated with pathological inflammation, but is not disease specific. 83 Subsequent studies have shown that circulating tenascin-C levels correlate with disease activity in inflammatory conditions including ulcerative colitis, Crohn’s disease, and systemic lupus erythematosus (SLE).84,85 The clinical relevance of tenascin-C was particularly evident in SLE, as it was found to be a better predictive factor of disease aggravation (so-called “flare”) than conventionally used biomarkers, potentially allowing for quicker identification of the need to escalate immunosuppressive therapy. The diagnostic value of tenascin-C was also investigated in arthritis, revealing upregulation in the synovial fluid of osteoarthritis patients, with levels correlating to disease severity. 86 Similarly, tenascin-C was elevated in RA, although here tenascin-C did not correlate with the levels of CRP in synovial fluid. 87 Upon further investigation, markedly raised circulating levels of tenascin-C in RA patients correlate with joint erosion. 88 Moreover, in RA, a subset of autoantibodies recognizing a citrullinated epitope in the FBG domain of tenascin-C could be detected years before any manifestation of the disease and thus could help in early clinical diagnosis, 89 as well as predicting among people presenting with early synovitis, those whose inflammation will not resolve but who will go on to develop RA. 90 Tenascin-C has been also considered an attractive candidate biomarker in cardiac pathologies (reviewed in Franz et al. 91 ). For example, tenascin-C has been proven a useful biomarker in a retrospective study on Kawasaki disease. Serum levels of tenascin-C were shown to predict the risk of acquiring resistance to the high-dose intravenous immunoglobulin therapy, implemented in Kawasaki disease to reduce pathological chronic inflammation, and also subsequent development of coronary artery lesions, one of the most dangerous complications of the acute form of the disease. 92 Moreover, in line with in vitro data highlighting the profibrotic activity of tenascin-C, this matrix molecule could also be useful in the stratification of patients with conditions caused by pathological cardiac tissue remodeling. High tenascin-C levels in patients suffering from hypertrophic cardiomyopathy correlated with high incidence of heart failure, a potential cause of premature death that is hard to predict with echocardiographic examination. 93 In addition, in systemic sclerosis, a disease driven by persistent fibrosis, significantly elevated tenascin-C expression was found in skin biopsies from patients with inflammatory subset of the disease. What is more, tenascin-C levels were also markedly higher in circulation of patients with both early- and late-stage disease. 19 Together, these data highlight tenascin-C as a prospective diagnostic target in a number of inflammatory and fibrotic conditions, useful in predicting disease progression and determining patient response to treatment.
Tenascin-C is an immunomodulatory protein that plays a key role in physiological tissue repair, but which can also drive pathological inflammation and fibrosis (Fig. 1). While an increasing body of data support a role for tenascin-C in influencing inflammation, this field is still in its infancy. Although tenascin-C impacts immune signaling in a wide range of cell types, the details of these signaling pathways remain to be fully defined in each target cell, from distinct immune populations to stromal cell lineages, and the molecular basis of the cell-type-specific responses reported so far to be dissected. Furthermore, there may be as yet uncovered roles for tenascin-C in impacting other effector cells that may provide a fuller picture of how this matrix molecule orchestrates inflammation. For example, myeloid-derived suppressor cells (MDSCs), a heterogeneous population of immature myeloid cells capable of inhibiting T cell–mediated inflammation, are emerging as key players in immune responses underlying tissue repair 94 as well as cancer 95 and autoimmune diseases such as RA, inflammatory bowel disease, or SLE. 96 Due to their unique immune suppressive potential, MDSCs are emerging as potential cellular immunotherapeutic agents. 97 The expansion of MDSCs can be stimulated by activation of the TLR4 pathway, 98 for example, in the lungs where MDSCs induced by activation of TLR4 signaling by LPS inhibit Th2 effector functions. 99 In a mammary carcinoma mouse model, S100A8/A9 proteins, endogenous TLR4 activators, have been shown to induce MDSCs accumulation in tissue, albeit this was triggered by binding to glycoprotein receptors on the cell surface. 100 These data raise the intriguing possibility that MDSCs may respond to tenascin-C and that this interaction could serve as a natural means of turning off tenascin-C-driven inflammation. Whether this is the case, and whether this mechanism goes awry in chronic or pathological inflammation, remains to be seen.
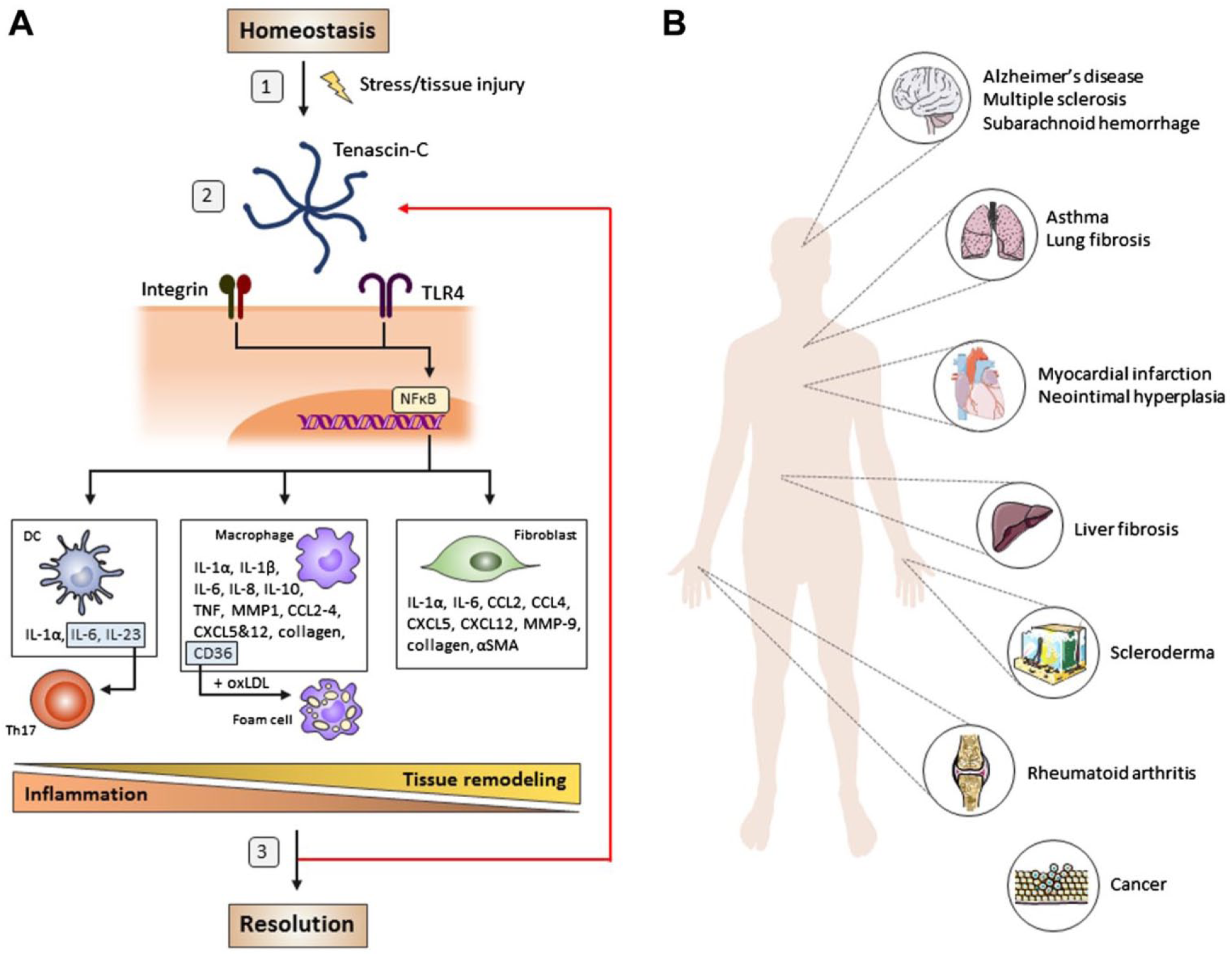
Tenascin-C modulates physiological and pathological inflammation. (A) Tenascin-C, upregulated upon cellular stress or tissue injury, is capable of binding to TLR4 and integrins α9β1 and αVβ3 on the cell surface and inducing NFκB-mediated gene expression. In macrophages and DCs, tenascin-C stimulates the secretion of a wide array of proinflammatory cytokines and chemokines, provoking an inflammatory response. Among these soluble mediators, IL-1β, IL-6, and IL-23 promote the differentiation of Th17 cells. Tenascin-C also polarizes myeloid cell phenotypes in a context-specific manner, for example, generating reparative macrophages or facilitating lipid uptake, promoting macrophages to form foam cells. In fibroblasts, tenascin-C stimulates expression of a different subset of mediators; this includes the cytokine IL-6 but also molecules such as αSMA, TGF-β and high levels of collagen that promote tissue remodeling. Tenascin-C is also capable of inhibiting naive T-cell activation. Typically, tenascin-C expression is downregulated, allowing for the resolution of inflammation and completion of tissue repair. However, a number of questions regarding the mode of action of tenascin-C still need answering: (1) What are the exact mechanisms leading to tenascin-C upregulation? (2) Does the form of tenascin-C (hexabrachion/monomer/domains cleaved by proteolytic enzymes) influence receptor binding and activation? (3) How exactly, and why, is tenascin-C expression downregulated and protein cleared from the tissue? Answering each of these questions will be crucial to understanding how and why persistent expression of tenascin-C leads to a chronic inflammation that can trigger the development of a number of inflammatory diseases, summarized in (B). Abbreviations: TLR4, toll-like receptor 4; NFκB, nuclear factor kappa-light-chain-enhancer of activated B cells; DC, dendritic cell; IL = interleukin; αSMA, alpha-smooth muscle actin; TGF-β, transforming growth factor beta; TNF, tumor necrosis factor; MMP, matrix metalloproteinase; CCL, C-C motif chemokine ligand; CXCL, chemokine receptor; oxLDL, oxidized low-density lipoprotein.
Due to its unique properties and characteristic expression pattern, tenascin-C has long been regarded as an attractive therapeutic candidate for targeting in clinics. A substantial part of ongoing work focuses on utilizing tenascin-C as a “postcode” for delivery of therapeutics directly to the tumor microenvironment with the means of aptamer or antibody technologies (reviewed in Spenle et al. 101 ). However, we are yet to explore applications targeting the proinflammatory potential of tenascin-C. To regain control over detrimental inflammation fueled by tenascin-C, two main strategies could be implemented. The first approach might involve regulation of tenascin-C production at the level of mRNA transcription and protein expression. This would in principle resolve the key issue underlying tenascin-C pathology—its misregulated expression. Global inhibition of tenascin-C expression, however, would ablate all the effects exerted by this large multimodular molecule in all tissues, which would include undesirable, prolonged engagement of proinflammatory signaling, but which also might prevent desirable functions, for example, those supporting physiological tissue remodeling. The restricted pattern of prolonged expression of tenascin-C in the adult to pathological sites may make this a viable option. Moreover, the phenotype of the tenascin-C knockout mouse implies a level of redundancy for this molecule during development, and its absence does not have a profound effect in reported cases of acute tissue repair, suggesting an association with immune persistence that may provide an avenue to treat chronic diseases without significantly affecting underlying biology. Alternatively, elucidating mechanisms that control the induction of tenascin-C expression at the transcriptional level, or tissue removal at the protein level, would provide information that might enable manipulation in a tissue- or disease-specific manner, adding a further level of refinement to this approach.
The second approach would involve targeting tenascin-C signaling by specifically preventing its activation of receptors that mediate undesirable inflammatory signaling. However, this strategy also comes with its own challenges. Little is known about how different domains in tenascin-C interact with each other at the functional level; for example the extent of synergy between domain signaling is unclear, nor do we know how individual interactions are influenced by the structural context of the molecule as a whole. We also have limited knowledge of the different ligand binding modes of this matrix molecule; this includes understanding whether interactions are mediated as a result of ligand binding exclusively to a unique epitope within tenascin-C, or to more general motifs with multiple potential ligands, or whether a combination of both possibilities exists. For example, RGD sequences bind to more than one combination of integrin dimers, and integrins can also bind to non-RGD containing motifs in tenascin-C. In addition, one site among the tripartite TLR4-binding epitope within tenascin-C comprises an exposed cationic ridge, 22 which might conceivably interact with other charged molecules. Understanding more about how this multimodular molecule binds to its wide range of ligands therefore is key to understanding not only its biological complexity but also inhibitor development.
Footnotes
Competing Interests
KSM has filed patents around the use of tenascin-C in disease diagnosis and treatment, and is the founder of, and consultant to, Nascient Ltd.
Author Contributions
AMM and KSM contributed to the conception and preparation of the manuscript, and wrote the manuscript. Both authors reviewed and approved the final version of the manuscript.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Arthritis Research UK and the Kennedy Trust for Rheumatology Research.