
Abstract
Heparan sulfate proteoglycans (HSPGs) are implicated as inflammatory mediators in a variety of settings, including chemokine activation, which is required to recruit circulating leukocytes to infection sites. Heparan sulfate (HS) polysaccharide chains are highly interactive and serve co-receptor roles in multiple ligand:receptor interactions. HS may also serve as a storage depot, sequestering ligands such as cytokines and restricting their access to binding partners. Heparanase, through its ability to fragment HS chains, is a key regulator of HS function and has featured prominently in studies of HS’s involvement in inflammatory processes. This review focuses on recent discoveries regarding the role of HSPGs, HS, and heparanase during inflammation, with particular focus on the brain. HS chains emerge as critical go-betweens in multiple aspects of the inflammatory response—relaying signals between receptors and cells. The molecular interactions proposed to occur between HSPGs and the pathogen receptor toll-like receptor 4 (TLR4) are discussed, and we summarize some of the contrasting roles that HS and heparanase have been assigned in diseases associated with chronic inflammatory states, including Alzheimer’s disease (AD). We conclude by briefly discussing how current knowledge could potentially be applied to augment HS-mediated events during sustained neuroinflammation, which contributes to neurodegeneration in AD.
Keywords
Heparan Sulfate Proteoglycans and Heparanase
Heparan sulfate proteoglycans (HSPGs) are macromolecules composed of polysaccharide chains of heparan sulfate (HS) that are covalently attached to core proteins. The HSPGs are classified according to the identity of the core protein. The cell-surface HSPGs include the transmembrane family of syndecans (SDCs) and the glycophosphatidylinositol (GPI)-anchored glypicans (GPCs), while agrin and perlecan belong to the extracellular matrix (ECM) HSPGs. These molecules are abundantly expressed, and the HS side chains interact mainly through their negative charges with a multitude of proteins (e.g., growth factors and cytokines); accordingly controlling diverse biological and pathophysiological processes.1,2 HS biosynthesis involves a series of enzymatic reactions, generating heterogeneous molecular structures of HS that display cell-type specific patterns of expression. As many as 11 different enzymes are responsible for the biosynthesis of an HS chain; some of the enzymes are encoded by single genes, while others have multiple isoforms. This biosynthesis process is tightly regulated under normal conditions, 3 but is altered under various pathological states. 4 As certain HS structural motifs have been associated with specific biological activities, alterations to the polysaccharide structure are predicted to impact on the capacity of a given HS chain to perform its function.
Heparanase is an endoglucuronidase that specifically cleaves HS chains; it is expressed as a pro-enzyme with little enzymatic activity, but undergoes proteolysis by cathepsin-
HSPG Modulation of Cytokine and Chemokine Activity
HS and HSPGs have been assigned a variety of roles in inflammation, particularly with respect to cytokine and chemokine binding in the context of immune cell activation and recruitment of leukocytes to sites of infection. These functions have been reviewed extensively elsewhere,10,11 but a brief summary relevant to the focus of this review is presented here. Chemokines are inflammatory effectors secreted by innate immune cells to recruit additional arms of the immune system to sites of infection. HS facilitates the activation of chemokines by acting as a template on which they can oligomerize, a prerequisite for their signaling activity. 12 The importance of this chemokine-binding capacity of HS is epitomized in the example of leukocyte tethering, where HSPGs on the luminal surface of the endothelium present the chemokine CCL2 to circulating leukocytes. HS-bound CCL2 interacts with rolling leukocytes via its receptor CCR2, tethering the leukocyte to the endothelium and decelerating its rolling behavior, which ultimately permits the leukocyte to migrate across the endothelium to the injury site. 13 Our study observed similar results for the macrophage inflammatory chemokine CXCL2, where a chemokine gradient on endothelial HSPGs was required for efficient emigration of neutrophils from circulation. 14 Furthermore, the shedding of SDC1 carrying HS-bound chemokines from the surface of neutrophils attenuates the inflammatory response to the bacterial endotoxin LPS. 15 HSPGs also bind cytokines including APRIL, a member of the tumor necrosis factor α (TNFα) family, which induces B-cell activation. 16 Furthermore, in models of brain injury, the HSPGs GPC1, SDC1, and SDC3 are upregulated in glial cells to participate in neurite outgrowth regulation,17–19 but may well participate in binding chemokines and cytokines secreted as part of the associated inflammatory response. HSPGs may also act to sequester cytokines in an inactive state; subsequent heparanase-mediated degradation of the HS chains increases their availability,20,21 which represents one key example of the pro-inflammatory activity of heparanase.
The reactive nature of the negatively charged sulfate groups along HS chains permits reactions with a diverse range of protein ligands, and HSPGs are often found to fulfill a co-receptor function for ligand:receptor interactions. A degree of consensus exists among HS-binding motifs for various HS:protein interactions and in a recent report by Davis et al., a bioinformatics search of immune protein sequences yielded 235 candidates that carry potential HS-binding sites. 11 Given this vast array of possible HS interactions with modulators of the immune response, it is clear that specific HS chain types and HSPGs may participate in amplifying a specific inflammatory event, while others may serve to suppress the same event. In the following sections, these diverse, even contradictory, functions of HS in specific aspects of the inflammatory response in the brain will be discussed.
A Note on Modeling HS Loss-of-Function
The essential biological roles of HS have been demonstrated in experimental animals by selective disruption of the genes involved in HS biosynthesis (see recent reviews22–24). Complete elimination of some of the genes resulted in severe alterations to HS molecular structures leading to lethality in mouse models,25–27 which prevented further investigation of the pathological implications of HS in diseases. Thus, the critical roles of HS in disease states have been demonstrated using mice in which the genes encoding biosynthetic enzymes are targeted by conditional knockout strategies.13,28,29 Alternatively, manipulating the expression of heparanase also leads to altered molecular structures of HS,30,31 providing mouse models for studying HS and heparanase under pathological conditions. Heparanase overexpression results in a substantial shortening of HS chains but is also accompanied by an increased degree of HS chain sulfation and an increased rate of HSPG turnover. 4 In most cases, the phenotype derived from heparanase overexpression can be ascribed to loss of HS function due to increased chain fragmentation; however, a number of non-enzymatic functions of heparanase have also been described. For example, heparanase induces migration of endothelial cells by facilitating Akt phosphorylation 32 ; furthermore, this function of heparanase reveals a dependency on integrins. 33 Such non-enzymatic roles of heparanase are suggested to rely on a heparanase receptor(s), the identity of which has yet to be confirmed. However, in the context of inflammation, Blich et al. propose that heparanase may potentiate an inflammatory response by mediating the activation of toll-like receptors. 34 Elimination of the heparanase gene yields extended HS chain length, but does not affect the sulfation pattern. 31 Many important functions of HS and heparanase have been revealed using these heparanase transgenic models, especially with regard to the role of HS during inflammatory states.14,35–37
Context-Specific Functions of HSPGs and Heparanase During Inflammatory Events
The classical signs of inflammation, calor (heat), dolor (pain), rubor (redness), and tumor (swelling), were recorded by Celsus in the first century. While today we have a greater understanding of the mechanisms that permit inflammatory responses, we have yet to fully elucidate the complete complement of molecular interactions that cause specific inflammatory stimuli to elicit responses from specific cell types. Cells of the innate immune system are responsible for the initial inflammatory response to pathogens and pathological molecules, and among these, macrophages play important roles. Many tissue types host a population of resident macrophages that serve as first responders to infectious agents, including Kupffer cells in the liver, alveolar macrophages in the lungs, and microglia in the brain. In their resting state, macrophages may be considered as tissue sentinels, continually monitoring their environment for signs of intruders or tissue damage. The activation of macrophages, including microglia, has long been discussed in terms of the polarized M1 or M2 states, also termed classical and alternative activation states. However, macrophages have proven unwilling to neatly accommodate such a binary definition and, instead, reveal a complex spectrum of often overlapping behavior, ranging from seek-and-destroy to repair-and-remove.38,39 The activation program initiated by a macrophage is specialized for the specific inflammatory insult but is also influenced by its environment. For example, LPS dissociated from bacteria induces macrophages to release a pro-inflammatory storm of cytokines creating a hostile environment for the invading pathogen. However, this response is also associated with tissue damage as the immune system attacks the infection ferociously, and in the brain, sustained attacks are associated with neurotoxicity.40,41 The resolution of these inflammatory episodes is also dependent on macrophages, which can adopt a phagocytic phenotype, clearing cell and pathogen debris and secreting cytokines associated with repair and recovery. Heppner et al. suggest that given the dynamic functionality that can be attributed to macrophages, they should be viewed as having “near-infinite plasticity.” 42
We used the Hpa-tg mouse model to study the role of HSPGs in the neuroinflammatory response to two distinct inflammatory stimuli. To study the functional requirement for HS in immune cell-dependent clearance of the AD associated Aβ peptide, Aβ fibrils were injected into the brain of wild-type (Ctrl) and heparanase-overexpressing (Hpa-tg) mice. Aβ was readily cleared from Ctrl mice, as evidenced by a robust immune cell response consisting of infiltrating macrophages, microglia, and astrocytes that surrounded the injection site, while this response was significantly impaired in Hpa-tg brains. In a second model, LPS was administered intraperitoneally, and the activation of macrophages was significantly suppressed in Hpa-tg brain compared with Ctrl. 9 We concluded that heparanase-mediated HS fragmentation in the endothelium reduced the presentation of chemokines and, thus, impaired the recruitment of circulating monocytes into the brain (Fig. 1A), similar to the role HS plays in the recruitment and transmigration of neutrophils.13,14 Importantly, glycosaminoglycans (GAGs) including HS and chondroitin sulfate (CS) have also been found to facilitate the entry of bacterial pathogens into the central nervous system (CNS) 43 ; consequently, the impaired macrophage activation in the LPS-exposed Hpa-tg mice may also relate to a reduced HS-dependent influx of LPS to the brain.
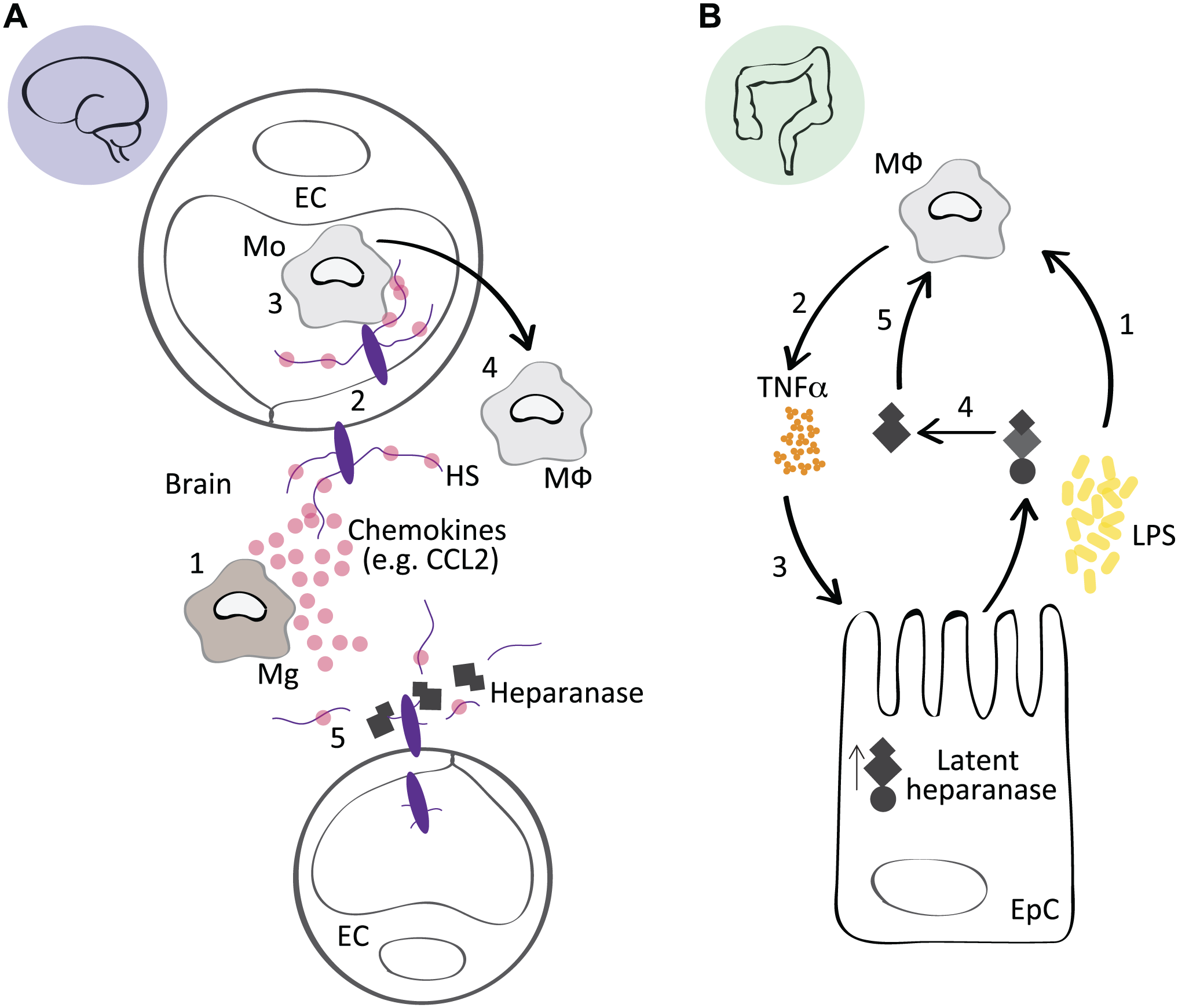
Context-specific effects of heparanase during inflammation. (A) In response to a systemic administration of LPS, or a local inflammatory stimuli, for example, Aβ, activated Mg in the brain secrete chemokines such as CCL2 (1). HSPGs on EC bind CCL2 and transcytose the chemokine to the luminal surface of the endothelium (2), where it participates in the recruitment of circulating Mo (3) that transmigrate across the blood brain barrier, where they differentiate into MΦ and participate in the inflammatory response (4). In Hpa-tg mice, heparanase-mediated fragmentation of endothelial HPSGs suppressed the presentation of chemokines on the endothelial lumen, reducing the recruitment of monocytes, thus attenuating the inflammatory state in the brain. This model is adapted from Zhang et al.
9
(B) The inflammatory condition ulcerative colitis is associated with impaired barrier function of epithelial cells in the colon (EpC), which leads to an increased exposure of MΦ to bacterial debris from the intestinal flora, including LPS (1). LPS induces TNFα and cathepsin-
Heparanase, however, is not readily assigned to the pro- or anti-inflammatory camp and has yielded context-specific effects. Lerner et al. used a variety of approaches, including the Hpa-tg mouse model to study colitis, an inflammatory bowel disease, and concluded that heparanase was responsible for eliciting a chronic inflammatory state in macrophages. Specifically, impaired epithelial barrier function in the colon results in macrophage exposure to bacterial flora (LPS), which induces TNFα. TNFα stimulates the colon epithelium to express latent heparanase, which is activated by macrophage-derived cathepsin-
Reconciling these apparently opposing effects of heparanase in these different inflammatory contexts is challenging. Given that heparanase can be viewed as a constant in all these conditions, HS chains are the most likely variables that may underpin the observed conflicting effects of heparanase. The specific disaccharide composition and degree of sulfation of the HS chains attached to different core proteins may permit HS to interact with as yet unknown inflammatory modulators specific to certain cell types. The distribution of inflammatory ligand-binding sites along HS chains (e.g., for chemokine activation) in different cell types may also have an impact on whether HS is acting to facilitate or suppress an inflammatory signaling event (Fig. 2A and B). Equally, the effect may also be determined by whether heparanase is releasing HS-bound chemokines from immune cells and, thus, attenuating the inflammatory response, or from non-immune cells and potentially increasing the pool of soluble HS-bound chemokines available to stimulate immune cells (Fig. 2C). As mentioned, HSPG shedding releases HS-bound chemokines and attenuates the inflammatory response, 15 and it is conceivable that there are cell-type specific variations in the dynamics of HSPG shedding during inflammation (Fig. 2D). HS may also sequester ligands from their receptor and prevent signaling events, as observed for interferon γ (IFNγ). The HS-binding site for IFNγ is similar to its receptor-binding domain; consequently, HS-bound IFNγ has a reduced affinity for its receptor, 44 in this setting, the activity of heparanase may serve to release this HS-inhibition on ligand:receptor binding (Fig. 2E and F).
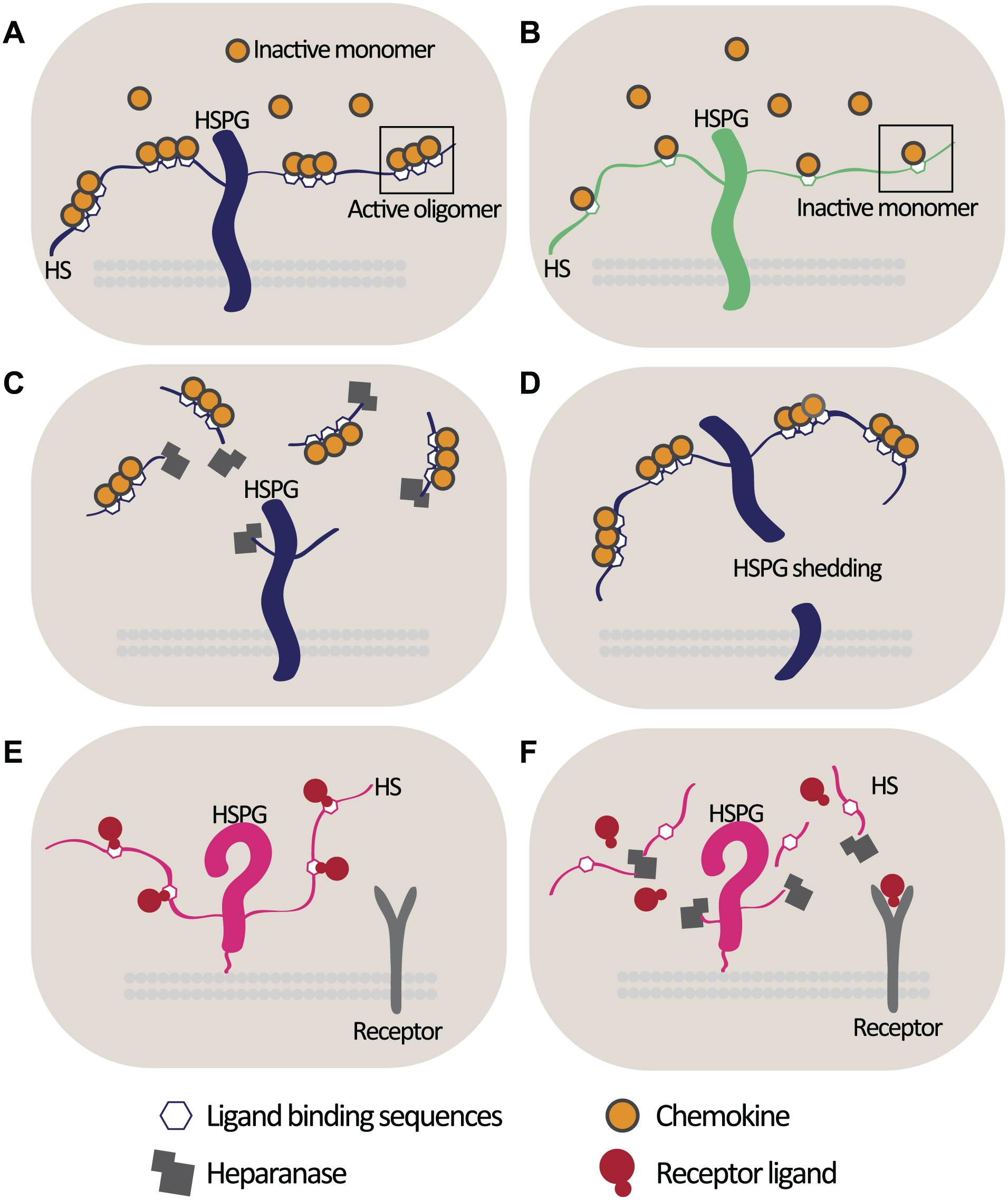
Cell-specific HS chain effects during inflammation. (A) The spatial distribution of ligand-binding sites along the HS chain will determine the oligomerization and thus activation state of certain chemokines. 12 (B) Sparse distribution of chemokine-binding sites on an HS chain may result in sequestration of inactive chemokine monomers, accompanied by an attenuated inflammatory response. (C) Heparanase release of HS-bound chemokines (or other inflammatory effector ligands) may facilitate the attenuation of the inflammatory response in an immune cell; however, heparanase release of HS-activated chemokine oligomers from non-immune cells may increase the soluble pool of activated chemokines capable of paracrine activation of immune cells. (D) HSPG shedding releases chemokines from the cell surface and suppresses immune cell activation. 15 The dynamics of HSPG shedding may differ between different cell types, and as in C, the outcome of releasing HS-bound inflammatory ligands from the cell surface will differ depending on cell type. (E) HS may bind and compete with receptors for ligand-binding sites, as demonstrated for IFNγ and its receptor, 44 thus preventing receptor activation. (F) Heparanase degradation of HS may permit the dissociation of a ligand from its inhibitory interaction with HS, allowing it to stimulate signaling of its receptor. Abbreviations: HS, heparan sulfate; HSPGs, heparan sulfate proteoglycans; IFNγ, interferon γ.
HSPG Interactions With TLR4
TLR4 is one of the major pathogen pattern recognition receptors and features in several studies of HS in the innate inflammatory response. The LPS-binding protein (LBP) binds to gram-negative LPS, which together interact with TLR4 and promote its activation, and ultimately signaling via the NFκB pathway. This induces pro-inflammatory cytokine expression, including interleukin-1β (IL1β) and TNFα. 45 However, while LPS can signal directly via TLR4, 46 in most cases, a robust response requires that LPS is initially bound at the cell surface by the TLR4 co-receptor CD14, a GPI-anchored protein for which the structure of the LPS-binding site has been studied in detail. 47 CD14 is itself upregulated as part of the pro-inflammatory response, and its expression is induced by TNFα signaling via its receptors, 48 suggesting that LPS-induced TNFα can subsequently induce CD14 as part of an autocrine signaling loop. While the GPI-bound form of CD14 is presented at the membrane (mCD14), a soluble form of this LPS-receptor (sCD14) can be derived from the same precursor.49,50 TLR4-competent cells that lack mCD14 can activate their TLR4 signaling pathway in response to LPS bound to sCD14. This may serve to instate a hierarchy to the inflammatory response such that cells expressing both mCD14 and TLR4, which includes cells of the innate immune system, will be particularly sensitive to bacterial insults and will engage the pathogen as the first responders at the infection site. However, if the infection persists, the inflammatory response may be expanded to include the activation of TLR4 on cells that are dependent on a paracrine source of sCD14. An additional critical surface component required for TLR4 functionality is lymphocyte antigen 96 (MD2), a secreted soluble protein that physically interacts with TLR4. 51
Kodaira et al. provided one of the earliest demonstrations that soluble HS chains were capable of promoting the maturation of dendritic cells (derived from bone marrow) and of eliciting an inflammatory cytokine response, including IL1β and TNFα release 52 (reviewed in Saadi et al. 53 ). Johnson et al. proposed a model in which released soluble fragments of HS were detected via TLR4 as part of a system to monitor tissue well-being. 54 Brunn et al. described ECM HS as a limiting factor for TLR4 activation, possibly through the action of HS-bound molecules capable of inhibiting TLR4, and proposed that the activity of proteases and heparanase releases this HS-mediated inhibition of TLR4 (Fig. 3A). 55 Importantly, once released from the ECM, the soluble HSPG and HS fragments, previously impeding TLR4 activation, can then assume a TLR4 agonist role, which was substantially increased in CD14-competent cells. However, the ability of HS to induce activation in an NFκB reporter system was reduced by pre-treatment with heparanase. Equally, heparanase pre-treatment of HSPG fragments, which were obtained following a protease digestion of an endothelial-derived ECM, also revealed impaired NFκB activation. This attenuation effect was lost if heparanase was first heat inactivated. 55 This suggests that a certain minimum HS chain length may be required for its ability to activate TLR4, and that heparanase-mediated HS fragmentation impairs this effect. Notably, a number of subsequent studies support the ability of soluble HS to function as a TLR4 agonist.56,57 Lerner et al. found that heparanase increased cytokine induction in peritoneal macrophages exposed to LPS (Fig. 3B); importantly, this effect was also dependent on the enzymatic function of heparanase. 35 This result was discussed in terms of the model proposed by Brunn et al. in which the added heparanase would relieve an HS-induced inhibition of TLR4, and in doing so increase its sensitivity to LPS. It would be reasonable to assume that in these macrophage cultures, to which no artificial ECM was added, the major contribution of HS would be derived from cell-surface HSPGs, such as SDCs and GPCs. Therefore, such a macrophage HSPG would interact directly or via an HS-bound TLR4 inhibitor to hinder TLR4 activation by LPS, and heparanase would act to relieve this constraint (Fig. 3B). This conclusion contrasts with our study of microglial HSPGs and their function in LPS activation of the CD14-dependent TLR4 pathway. We reported that the LPS-induced IL1β and TNFα response was substantially impaired in Hpa-tg microglia, but little effect was observed in astrocytes. We proposed that the CD14-positive status of microglia underpinned the differences between the cell types, such that a microglial HSPG likely acts as a co-receptor for CD14 permitting maximal activation of TLR4 (Fig. 3D). In contrast, LPS-induction of inflammatory cytokines in CD14-lacking astrocytes would be limited to the response available from TLR4 alone, and therefore would be independent of the cell’s HSPG status. 58 Heparanase through its ability to fragment microglial HS could attenuate this pro-inflammatory effect. To some extent, this finding is in line with the heparanase effect observed by Brunn et al., where it also attenuated the ability of HS to activate TLR4. However, a number of important caveats remain, as the HS in that context was soluble and served as the sole TLR4 agonist (i.e., in the absence of LPS). Importantly, Lerner et al. studied the effect of LPS in combination with exogenous heparanase added to wild-type macrophages, while we added LPS to heparanase-overexpressing microglia. It is difficult to predict the full effect that the continual pruning of cell-surface HSPGs in the Hpa-tg model may have on the microglial surface. CD14, TLR4, and MD2 were equally expressed in unstimulated wild-type and Hpa-tg microglia, but it is possible that the ongoing remodeling of the cell surface has as yet unexplored effects on the LPS activation pathway. In addition, a recent study utilizing macrophages derived from a heparanase knockout mouse revealed that their basal level of cytokine expression was suppressed relative to wild-type controls. 59 It is, of course, tempting to consider that the conflicting effects may reflect tissue and cell-type specific differences in HS chain length and structure, and/or the specific complement of HSPGs on bone marrow–derived macrophages versus brain-derived microglia. Future studies utilizing specific HSPG knockout models would help clarify whether different HSPG types exert different effects on TLR4 activation. To complicate matters further, heparanase has also been assigned functional roles independent of its HS-degrading activity. In the context of pro-inflammatory signaling, the work of Blich et al. is particularly relevant, where heparanase was found to promote NFκB signaling (Fig. 3C), which demonstrated a dependence on TLR4. 34 In this respect, alternative model systems in which specific elements of the HS biosynthesis machinery have been depleted would help to elucidate to what extent the functions attributed to HS in the context of TLR4 activation can be reproduced in a model that is not reliant on heparanase for HS loss-of-function.
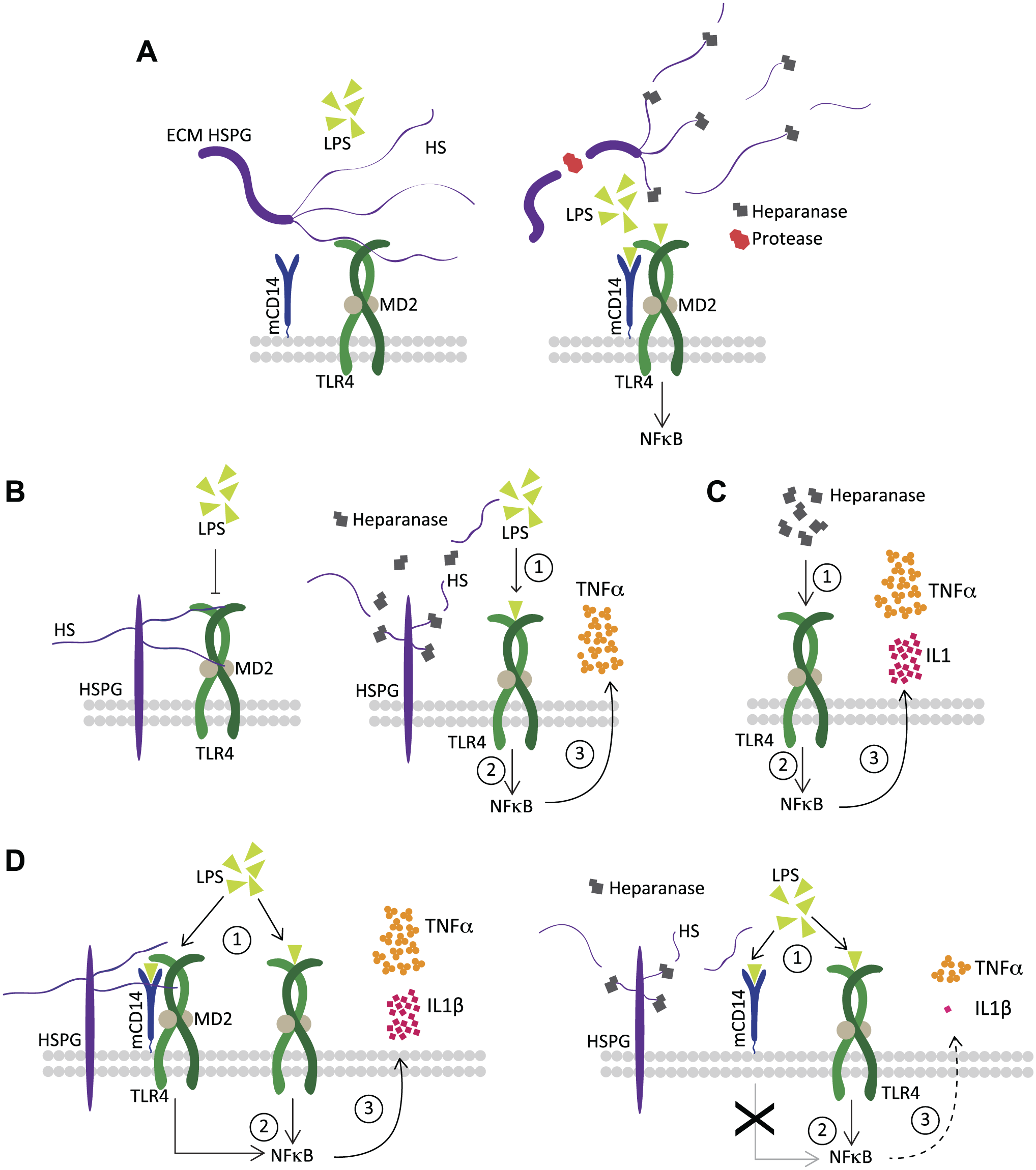
Proposed functional roles of HSPGs and heparanase during LPS-mediated TLR4 activation. (A) HS chains connected to core proteins in the ECM exert an inhibitory effect on TLR4 activation by LPS, potentially through direct contact with the receptor or alternatively by binding TLR4 inhibitors (left panel). In combination with an inflammatory insult, protease and heparanase expression increases, and remodels the ECM such that both the HSPG core protein and its HS chains are fragmented, which releases its inhibition on TLR4 and permits its activation by LPS (right panel). 55 (B) HSPGs on the surface of murine peritoneal macrophages impair the TLR4 response to LPS (left panel), heparanase treatment releases this HS-dependent inhibition and increases sensitization to LPS as evidenced by an increase in TNFα release (right panel). 35 (C). Heparanase, acting independently of its ability to fragment HS, can activate the NFκB pathway in a manner that is likely dependent on TLR4 at the surface of macrophages. 34 (D). Microglial HSPGs facilitate the CD14-dependent activation of TLR4 in response to LPS, promoting the release of IL1β and TNFα (left panel), transgenic heparanase overexpression fragments microglial HS and impairs the inflammatory response to LPS (right panel). This model is adapted from O’Callaghan et al. 58 Abbreviations: HSPGs, heparan sulfate proteoglycans; LPS, lipopolysaccharide; TLR4, toll-like receptor 4; HS, heparan sulfate; ECM, extracellular matrix; TNFα, tumor necrosis factor α; NFκB, nuclear factor kappa-light-chain-enhancer of activated B cells; IL1β, interleukin-1β.
While there are many aspects of HS/HSPG interactions with TLR4 and its counterparts that have yet to be fully understood, it is clear that HS chains can play important regulatory functions in restricting, facilitating, or even amplifying the TLR4-dependent inflammatory response. Given that dysregulation of the TLR4 activation pathway has been associated with diverse disease states including autoimmune conditions and several neurodegenerative diseases, our understanding of these conditions will be increased when we further elucidate what role HSPGs and heparanase may play in these settings.
HSPGs in the Glial Response to AD
HS is an established co-deposit of the majority of amyloidoses described to date. The Aβ deposits that accumulate in the parenchyma of individuals with AD are one of the major pathological hallmarks of the disease and are typically positive for HS. 60 Studies of the functional relevance of HS co-deposition with amyloid proteins have focused on its well-established in vitro capacity to accelerate amyloid fibril formation (reviewed in 61 ). HS chains act as linear scaffolds with neatly distributed binding sites for amyloid monomers or small amyloid aggregates. This elevates the local concentration of the amyloid precursors and can vastly reduce the lag phase of Aβ fibrillization. Thus, HS can provide an infrastructure upon which amyloid fibril formation and deposition can be focused. In addition, there are a number of reports which indicate that HS interactions with amyloid proteins, particularly Aβ, are mutually protective such that protein and GAG shield each other from degrading enzymes. 62
A question that we have attempted to address is what specific cell type(s) is responsible for contributing the HS found accumulated with Aβ deposits in AD. Lesions in the brain, including amyloid deposits, result in local neuronal damage and death, and a number of mechanisms are in place to prevent aberrant regrowth of neurons. One characteristic feature of such lesions is the surrounding glial scar, a cellular barrier formed by recruited microglia and astrocytes that collaborate to isolate the injury site, and in doing so, impede interactions between the lesion and surrounding healthy tissue.63,64 In AD, many Aβ deposits are surrounded by a reactive glial population; however, certain diffuse Aβ deposit types do not appear to elicit this glial response. Early studies regarding the HSPG source for the HS found associated with Aβ deposits implicated members of the ECM HSPGs such as perlecan and agrin,65,66 and given their presence throughout the basement membranes of the highly vascularized brain, they undoubtedly contribute to the formation of Aβ deposits. Nonetheless, we observed that the degree of HS associated with diffuse Aβ deposits was significantly lower than that observed with so-called dense-core deposits. Diffuse deposits also revealed a much less pronounced glial response. We established that in AD brain microglia and astrocytes in close proximity to dense-core Aβ deposits expressed the HSPGs syndecan 3 (SDC3) and glypican 1 (GPC1), neither of which was readily detected in diffuse deposits. Using primary microglia and astrocytes isolated from mice, we determined that exposure to Aβ elevated the levels of SDC3 and GPC1 in both cell types. 67 The ability of Aβ to upregulate cellular expression of HSPGs, including GPC1, has also been observed by others. 68 Bruinsma et al. have also addressed the presence and the nature of HS associated with different Aβ deposit types using phage display antibodies that specifically detect different HS chain modifications. They concluded that a greater variety of HS modifications were associated with fibrillar deposits, while HS in diffuse deposits was limited to highly sulfated forms. 69 These differences in HS chain types associated with different Aβ deposit types may reflect differences in both the cell and proteoglycan core-protein source for these HS chains.
Chronic inflammation is a contributing factor in the progression of AD, and microglia are implicated as major sources of neurotoxic cytokines. 40 Aβ interacts with microglial CD14 and TLR4, 70 and in the brain is thought to induce pro-inflammatory cytokines such as IL1β and TNFα, both with established neurotoxic effects.71,72 Cells of the innate immune system, including microglia, have also been identified as victims of cellular senescence, whereby aging cells begin to lose regulatory control over their activation states 73 ; this may also drive chronic pro-inflammatory activation at the expense of phagocytic abilities. It will be important to determine whether the HS biosynthesis machinery is perturbed in microglia in the aging and AD brains. Shao et al. performed a glycomic analysis of the HS and CS content in leukocytes and determined that while the abundance of CS was greater than that of HS, the HS chains carried by all leukocytes presented with a degree of sulfation more comparable to heparin than to that typically observed in tissue-derived HS. 74 Interestingly, the interaction of HSPGs with apolipoprotein E (ApoE) has also been established to suppress monocyte proliferation in the context of artherosclerosis. 75 Taking this and our finding that microglial HSPGs facilitate CD14-dependent TLR4 activation 58 into account, it is conceivable that age-related effects that result in an imbalance in cell-surface HS content and structure could well predispose cells toward adopting more sustained pro-inflammatory phenotypes. Similarly, the ability of Aβ to increase the levels of microglial and astrocyte HSPGs 67 (Fig. 4) could also contribute to a cell-surface landscape that was more sensitized toward adopting a pro-inflammatory state. Increased expression of heparanase has also been observed in AD brains, 76 and heparanase was readily detected with Aβ deposits, 9 which may also contribute to the release of HS chains from core proteins at the deposit site (Fig. 4).
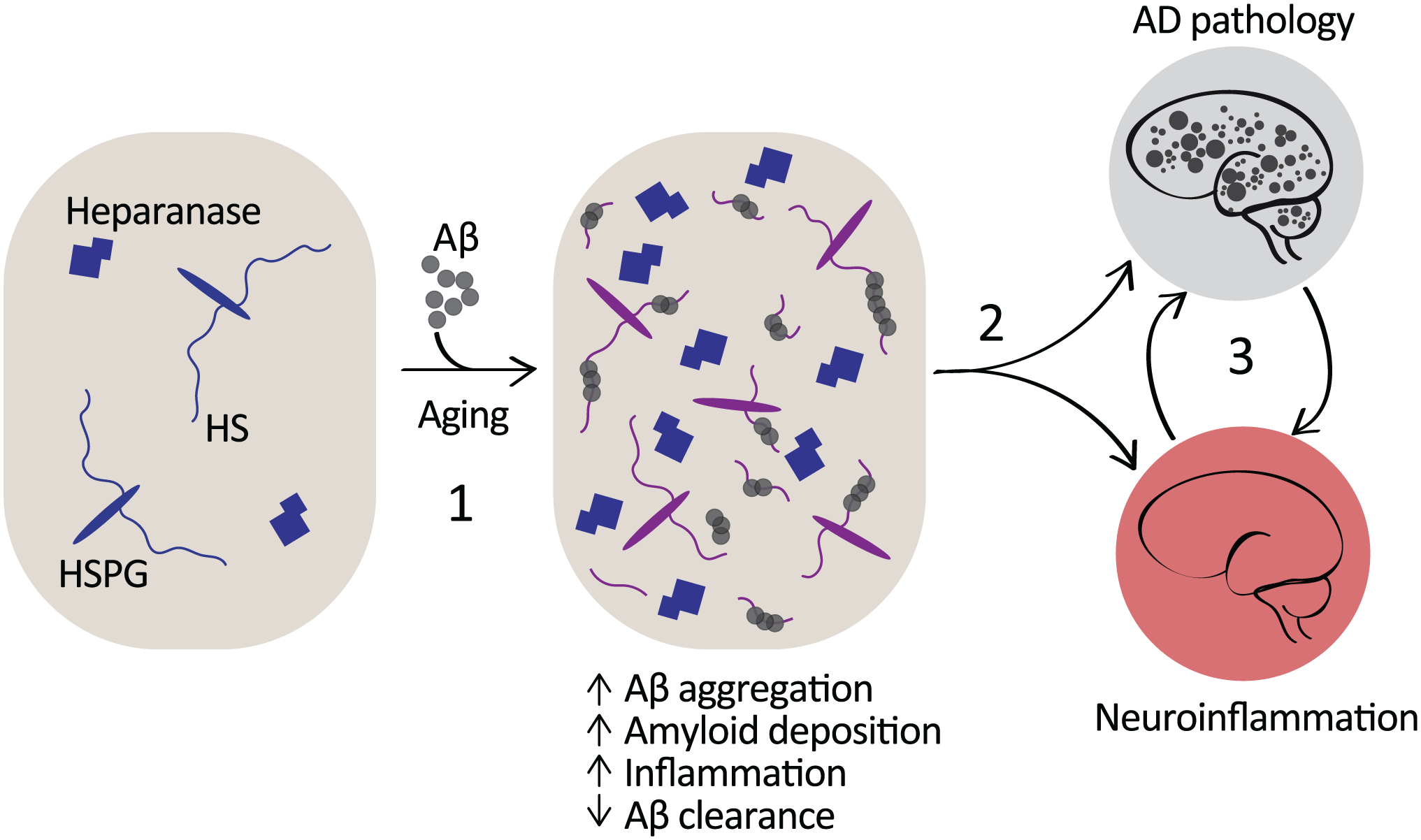
HSPGs and heparanase in the amyloidosis and inflammation of AD. HS serves as a template for Aβ aggregation, fibril stabilization, and deposition.77,78 HS-positive Aβ deposits recruit astrocytes, and both resident (microglia) and circulating macrophages. Exposure of microglia and astrocytes to Aβ elevates levels of associated cell-surface HSPGs, and increased heparanase expression is localized to the Aβ deposits.9,67 Aβ clearance is in part dependent on endothelial HSPGs via their role in facilitating the emigration of phagocytic macrophages into the brain. 9 In addition, neuronal HSPGs sequester Aβ, obstructing its clearance and promoting fibrillization (1). 78 Consequently, a combination of direct and indirect interactions between Aβ and HS contributes to the amyloid pathology and associated chronic state of neuroinflammation in AD (2), and each of these pathologies is likely to exacerbate and contribute to the persistence of the other (3). Abbreviations: HSPGs, heparan sulfate proteoglycans; AD, Alzheimer’s disease; HS, heparan sulfate; Aβ, amyloid-β.
Age-related changes present a particularly difficult challenge when attempting to model the functional relevance of HS in a given setting. Changes in HS chain structure would presumably have effects on the distribution of HS-binding molecules as well as receptors that rely on HSPGs as co-receptors. HS chains induce conformational changes in bound ligands that in the case of chemokines are important for their activation. 12 Furthermore, the HS-analogue heparin induces conformational changes in the ECM component fibronectin, exposing an otherwise cryptic-binding site for the angiogenesis effector vascular endothelial growth factor. 79 This function was dependent on the composition and chain length of the polysaccharide; hence, multiple direct and indirect effects of age- and/or disease-specific changes to HS structure can be envisaged. Consequently, it is complicated to assess whether HS in the setting of neuroinflammation, particularly in the aging brain in a condition like AD, contributes to the problem or the solution. Several lines of investigation suggest that the Aβ deposits in AD are relatively inert, while the soluble Aβ oligomeric precursors are viewed by many as the most neurotoxic species of the peptide. 80 In this respect, the formation of the deposit, which is promoted by HS, may be viewed as part of a protection mechanism. In addition, HSPGs in consort with the low-density related lipoprotein receptor 1 (LRP1) and ApoE are implicated in increasing the cell association and uptake of Aβ. Kanekiyo et al. revealed this mechanism as a contributing factor to Aβ-mediated neurotoxicity. 81 We proposed that under healthy conditions, these interactions may facilitate clearance of Aβ across the blood brain barrier, while during the pathogenesis of AD, the balance may tip in favor of Aβ deposition in the vasculature as cerebral amyloid angiopathy. 82 Jendresen et al. revealed that heparanase-mediated fragmentation of HS significantly reduces the Aβ deposit burden in a transgenic mouse model of AD, suggesting that HSPGs in the CNS are instrumental in promoting the pathogenesis of AD (Fig. 4). 77 Further support for this role was provided by a study using mice specifically lacking neuronal HSPGs, achieved by knockdown of the HS glycosyltransferase gene Ext1, in which Aβ was more efficiently cleared and less prone to aggregation due to the lack of HSPGs (Fig. 4). 78 Future work will be required to determine whether HS-modifying agents, such as heparanase, can be effectively engineered and deployed under specific disease states to essentially delete HS in settings where it has been determined to exacerbate a problem, such as neuroinflammation.
HS presents as both friend and foe in its relationship with Aβ, during the pathogenesis of AD. HSPGs appear to participate in the clearance of Aβ from the brain and through promoting Aβ fibril formation, and deposition may even serve a neuroprotective role. Microglial HSPGs facilitate a robust inflammatory response to bacterial insults, which is required for the resolution of infection. However, the ability of Aβ to elevate levels of microglial HSPGs may well contribute to sustained inflammatory states in the AD brain. Defining this tipping point at which the benefits of HS in protecting against AD become outweighed by its contribution to the disease state is a challenge for future research in this field.
In the brain, the ability of heparanase to fragment HS chains has been associated with the attenuation of inflammatory states, while in other organs subjected to autoimmune conditions such as colitis and rheumatoid arthritis, heparanase acted to potentiate the associated inflammation. Consequently, while heparanase has proven itself a powerful modulator of HS function in inflammatory states, more work will be required before a consensus can be reached with regard to what factors underlie these context-specific functions. Nonetheless, in AD, it would be interesting to explore the efficacy of HS mimetics or heparanase administration as a strategy to impair HS-mediated Aβ fibrillogenesis and cellular toxicity, and simultaneously attenuate the HS-dependent inflammatory response.
The ability of HS chains to act as molecular relays that are integral to the transmission of signals in a multitude of pathways and cellular interactions is in part dependent on chain length, disaccharide structures, and the identity of HSPG core proteins. Therefore, it seems reasonable to assume that tissue and cell-type specific populations of HS and HSPGs contribute to the diversity of functions that have been assigned in different inflammatory mechanisms and disease states. In amyloid disorders, knowledge of the specific composition of HS found to accumulate with amyloid pathologies has contributed to the development of diagnostic imaging tools. 83 In addition, with the current advances in techniques to analyze HS chains and determine their position and distribution on specific core proteins,84–87 it will soon be possible to gain a greater appreciation for what the interactive capacity of a given cell type’s HS “landscape” is. This will hopefully help predict the potential consequences of targeting these HSPGs during inflammatory events, both during the resolution of infection and in chronic disease states, such as AD.
Footnotes
Competing Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
POC wrote the manuscript and prepared the figures; XZ edited the manuscript and participated in discussions regarding content, layout, and figures; J-PL edited the manuscript and participated in discussions regarding content, layout, and figures. All authors agreed upon the final submitted draft of the manuscript.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors have received financial support from the Swedish Research Council (2015-02595), the Swedish Alzheimer Foundation (Alzheimerfonden grants AF-552581, AF-649251, and AF-554871), and Polysaccharide Foundation, Uppsala.