
Abstract
Valve dystrophic calcification is a common disorder affecting normophosphatemic subjects. Here, cultured aortic valve interstitial cells (AVICs) were treated 3 to 28 days with phosphate (Pi) concentrations spanning the normal range in humans (0.8, 1.3, and 2.0 mM) alone or supplemented with proinflammatory stimuli to assess possible priming of dystrophic-like calcification. Compared with controls, spectrophotometric analyses revealed marked increases in calcium amounts and alkaline phosphatase activity for 2.0-mM-Pi-containing cultures, with enhancing by proinflammatory mediators. Ultrastructurally, AVICs treated with low/middle Pi concentrations showed an enormous endoplasmic reticulum (ER) enclosing organelle debris, so apparently executing a survival-related atypical macroautophagocytosis, consistently with ultracytochemical demonstration of ER-associated acid phosphatase activity and decreases in autophagosomes and immunodetectable MAP1LC3. In contrast, AVICs cultured at 2.0-mM Pi underwent mineralization due to intracellular release and peripheral layering of phospholipid-rich material acting as hydroxyapatite nucleator, as revealed by Cuprolinic Blue and von Kossa ultracytochemical reactions. Lack of immunoblotted caspase-3 cleaved form indicated apoptosis absence for all cultures. In conclusion, fates of cultured AVICs were crucially driven by Pi concentration, suggesting that serum Pi levels just below the upper limit of normophosphatemia in humans may represent a critical watershed between macroautophagy-associated cell restoring and procalcific cell death.
Keywords
Introduction
In developed countries, heart valve dystrophic calcification is a common disorder affecting normophosphatemic subjects. 1 Normal serum phosphate (Pi) levels conventionally range from a minimum of 0.8 to 1.45 mM2,3 to a maximum of 2.0 to 2.1 mM,4,5 which represent threshold values for proper functioning of cellular and systemic processes. Several cohort studies indicated high serum Pi levels within the normophosphatemic range to be associated with increased risk of cardiovascular diseases, including ectopic calcification.2,3,6–8 Actually, elevated Pi concentrations act as critical biomineralization determinants,4,9,10 triggering the precipitation of hydroxyapatite (HA) crystals in diseased tissues. Besides depending on Pi concentration, it is widely accepted that cardiovascular tissue calcification also correlates with active processes, including accumulation and/or alteration of lipids, inflammation, loss of anticalcific proteins, cell osteogenic differentiation, and cell death.11–16 In particular, apoptosis- and macroautophagocytosis-derived bodies are reported as HA-nucleating structures, playing a procalcific role as matrix vesicles do in mineralizing cartilage and bone.17–21 Using original in vitro models simulating either severe dystrophic calcification 22 or metastatic calcification, 23 aortic valve interstitial cell (AVIC) mineralization resulted to be triggered by elevated Pi alone and enhanced after superstimulation with lipopolysaccharide (LPS) and proinflammatory mediators derived from cultures of LPS-stimulated macrophages. The induced calcific process depended on a distinct cell degeneration involving membrane colliquation, lipid release, and formation of peripheral phthalocyanine-positive layers (PPLs) at the level of dying cells and derived cell debris, acting as major HA nucleators. Comparable features were previously reported for both in vivo experimentally induced aortic valve mineralization24–27 and actual ectopic calcification.11,28–30 In the present study, similar in vitro models were used to simulate different normophosphatemic-like conditions by treating AVICs with Pi concentrations spanning the normal range in humans and including the borderline ones, with or without superstimulation with proinflammatory mediators. AVICs treated with low/middle Pi concentrations were found to undergo atypical autophagy preserving cells from death and subsequent mineralization. In contrast, AVICs exposed to the highest Pi concentration underwent prominent non-apoptotic, lipid-release-associated procalcific cell degeneration and death, supporting the concept that concentration-dependent procalcific effects in organisms may be exerted by Pi at the most elevated levels even within the normal range.
Materials and Methods
AVIC Isolation
Tricuspid aortic roots were excised from hearts (n=3) of healthy bovines slaughtered in a local abattoir (Salumificio Pitaccolo G. Srl - Macellazione & sezionamento carni; Castions di Strada, Udine, Italy), in which the rules of Reg CE 1099/2009, September 24, 2009, concerning the protection of animals at the time of killing were respected. Cows were not killed specifically for the purpose of the present study, and no experiments were performed on animals before slaughtering. Aortic roots were placed in cooled DMEM (Sigma, St. Louis, MO) supplemented with 1% penicillin/streptomycin and 1 µg/ml amphotericin B. Once isolated from aortic roots and subjected to removal of samples destined to microscopic examination, all valve leaflets underwent AVIC extraction procedure. Cell extraction and subsequent in vitro treatments were performed separately for each aortic root to avoid pooling of AVICs from different animals. For AVIC extraction, aortic leaflets were (1) depleted of endothelial cells by gentle scraping, (2) minced into about 2- to 3-mm
3
pieces, and (3) enzymatically digested with type I collagenase (125 U/ml; Sigma), elastase (8 U/ml; Sigma), and soybean trypsin inhibitor (0.375 mg/ml; Sigma) for 30 min at 37C in a CO2 incubator. Then, digested pieces were transferred into 100-mm tissue culture Petri dishes (Greiner Bio-One, Frickenhausen, Germany) and cultured in DMEM supplemented with 20% FBS (Gibco–Invitrogen, Carlsbad, CA), 1%
Conditioned Medium (CM) From Bovine Macrophages
Peripheral blood was drawn from healthy bovines (n=2) during routine care. Blood was collected by the veterinarian in the whole respect of animal normal behavior and wellness, according to both Italian legislation (D.L. No. 116, January 27, 1992), in line with European Economic Community (EEC) regulation No. 86/609, November 24, 1986, and the professional ethics of Italian veterinarians (Federazione Nazionale Ordini Veterinari Italiani [FNOVI], June 12, 2011). Blood cell separation was performed separately for each animal. Namely, blood was diluted 1:2 with PBS solution and subjected to Ficoll (Sigma) density gradient centrifugation. Collected lymphocytes and monocytes were plated on tissue culture flasks and maintained in DMEM supplemented with 10% FBS, 1%
Cell Culture Treatments
At preconfluence, cultured AVICs were treated for 3, 9, 15, 21, 25, and 28 days with (1) DMEM supplemented with 10% FBS, 1%
Spectrophotometric Quantification of Calcium Content
After culture medium recovering, AVICs were scraped, centrifuged, and treated with a lysis buffer containing 50-mM TRIS-HCl, 150-mM NaCl, 5-mM EDTA, and 1% Triton X-100, pH 7.4, for 1 hr at 4C. After microcentrifugation at 2000 rpm for 5 min, part of supernatants (500 µL) was recovered for protein assay, whereas the remaining volumes were added to their original culture media and transferred into distinct Teflon vessels containing 1 ml of 65% suprapure grade nitric acid (Merck Millipore, Darmstadt, Germany) and 500 µL of 30% suprapure hydrogen peroxide (Merck Millipore). Then, samples were irradiated at 250 W for 2 min, 0 W for 2 min, 300 W for 5 min, 450 W for 5 min, and 650 W for 6 min using the Milestone High Performance Microwave Digestion Unit MLS 1200 Mega (Shelton, CT). After dilution with ultrapure water up to 100 ml of total solution, calcium quantification was assessed using the o-cresolphthalein complexone method (Chema Diagnostica, Monsano, Italy) reading the absorbance at 575 nm with the Varian Cary 50 Bio Spectrophotometer (Midland, Ontario, Canada). The estimated values were the mean of 10 readings of each three-times repeated culture (n=3), which were normalized on the basis of total protein content.
Spectrophotometric Quantification of Protein Content and Alkaline Phosphatase Activity
Supernatants obtained from lysed samples as described above were used to quantify both total protein content and alkaline phosphatase (ALP) activity. Total protein content was estimated using the Pierce Coomassie (Bradford) Protein Assay kit (Thermo Fisher Scientific, Waltham, MA) reading the absorbance at 595 nm using the aforementioned Varian spectrophotometer. ALP activity was assayed using a kinetic method based on measurement of 4-nitrophenol production (Chema Diagnostica) with absorbance reading at 405 nm, at 37C, within the first 5 min of enzymatic activity using the Varian spectrophotometer as above. Values were obtained by calculating the trend line gradients resulting from the mean of three readings of each three-times repeated culture (n=3), which were normalized on the basis of total protein content.
Alizarin Red S Calcium Staining
After culture medium removal, AVIC monolayers were rinsed with Pi buffer and fixed with neutral-buffered 5% formalin for 10 min. After further rinsing, AVIC monolayers were incubated in an aqueous solution of 2% alizarin red S (CARLO ERBA Reagents, Milano, Italy), pH 4.2, for 5 min; rinsed again to remove exceeding staining solution; and covered with distilled water. As negative controls, parallel AVIC monolayers were treated with a decalcifying solution containing 0.05-M sodium acetate buffer and 0.05-M magnesium chloride, pH 4.8, for 1 hr before alizarin red S staining. Observations and photographic records were made using the Olympus IX70 inverted microscope (Hicksville, NY).
Preembedding Cuprolinic Blue Reaction for Ultrastructural Polyanion Evidentiation
After culture medium removal, AVIC monolayers were incubated in 25-mM sodium acetate buffer, pH 4.8, containing 0.05% phthalocyanine Cuprolinic Blue (CB; Electron Microscopy Sciences, Hatfield, PA), 0.05-M magnesium chloride, and 2.5% glutaraldehyde overnight at room temperature keeping culture plates under continuous agitation. After washing, AVIC monolayers were postfixed with phosphate-buffered 2% osmium tetraoxide (Agar Scientific, Stansted, UK) for 1 hr at 4C, washed again, dehydrated in graded ethanols, and embedded in Epon 812 resin. Ultrathin sections were collected on formvar-coated 2 × 1-mm-slot copper grids and contrasted with uranyl acetate and lead citrate. Observations and photographic recordings were made using the Philips CM12 STEM electron microscope (Eindhoven, The Netherlands).
Postembedding von Kossa Silver Staining for Ultrastructural Calcium-Binding Site Evidentiation
Semithin sections of CB-reacted AVIC monolayers were mounted on glass slides, covered with a drop of 1% silver nitrate solution, and placed on an 80C warm plate for 15 min under direct sunlight. After washing with distilled water and drying, semithin sections were covered with a drop of 5% sodium thiosulfate reducing solution and warmed at 80C for 5 min. After further washing and drying, reacted semithin sections were reembedded by gluing onto the slides top-less conic BEEM capsules (Agar Scientific, Stansted, UK), so encircling each semithin section, which were filled with Epon-Araldite fluid. After resin polymerization, reembedded sections were detached from slides and cut to obtain ultrathin sections which were collected, contrasted, observed, and recorded as above.
Ultrastructural Localization of Acid Phosphatase Activity
After culture medium removal, AVIC monolayers were fixed with 3% paraformaldehyde for 30 min, washed with 0.05-M sodium acetate buffer, pH 5.0, and incubated in the same buffer supplemented with 0.01-M beta-glycerophosphate (Sigma) and 0.004-M lead nitrate for 45 min at 37C. As negative controls, incubating solutions lacking in beta-glycerophosphate were used. After washing with cooled 0.05-M sodium acetate buffer to block enzymatic activity, AVIC monolayers were (1) fixed with 2% glutaraldehyde for 15 min, (2) postfixed with 2% osmium tetraoxide for 1 hr at 4C, (3) dehydrated in graded ethanols, and (4) embedded in Epon 812 resin. Ultrathin sections were collected, contrasted, observed, and recorded as above.
Immunocytochemistry
Monolayers of AVICs seeded on 24 × 24-mm coverslips placed at the bottom of 35-mm culture plates were incubated in (1) 0.1% Triton X-100 solution for 10 min, (2) 3% hydrogen peroxide solution for 5 min, (3) 3% normal serum solution for 30 min, (4) 1:600 rabbit anti-MAP1LC3 polyclonal antibody (Merck Millipore) or 1:25 goat anti-annexin-V polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA) for 2 hr at room temperature in a humidified chamber, and (5) 1:600 anti-rabbit peroxidase-conjugated secondary antibody (Jackson ImmunoResearch, West Grove, PA) or 1:400 anti-goat peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology) for 30 min at room temperature. As negative controls, primary antibodies were replaced by normal serum. Peroxidase activity was revealed incubating AVIC monolayers in a solution containing diaminobenzidine tetrahydrochloride (DAB) and hydrogen peroxide (BioGenex, San Ramon, CA) for 3 to 6 min. After rinsing with distilled water to block enzymatic activity, cells were weakly counterstained with hematoxylin. Coverslips were then mounted on glass slides using an aqueous mounting medium, observed, and recorded using a Zeiss Axio Imager photomicroscope (Jena, Germany). Percentage values of immunopositive cells were estimated using the ImageJ software, averaging the number of reactive cells in five distinct areas of each three-times repeated AVIC culture (n=3).
Western Blotting
AVIC cultures were subjected to cell lysis and protein quantification as described above. Supernatants were then separated by SDS-PAGE and transferred on nitrocellulose membranes by electroblotting using the Bio-Rad Mini Trans-Blot system (Hercules, CA). After blocking with 3% skim milk (Sigma) solution, blotted membranes were incubated with 1:500 rabbit anti-cleaved-caspase-3 polyclonal antibody (Cell Signaling Technology, Danvers, MA) overnight at 4C and then with 1:15,000 anti-rabbit peroxidase-conjugated secondary antibody (Sigma) for 1 hr at room temperature. Immunoreactive bands were revealed using the Pierce enhanced chemiluminescence assay (Thermo Fisher Scientific) with a maximum exposure time of 4 hr. For accurate estimation of protein molecular weight, Precision Plus Protein Standards (Bio-Rad) were used. As positive controls, lysates of AVICs treated with 50-µM etoposide (Sigma) for 18 hr were used.
Statistical Analysis
Continuous variables were summarized as mean ± standard deviation. Data were tested for normal distribution using the Shapiro–Wilk test. Equality of variance was assessed using the Levene test. The variation of calcium amounts or ALP activity between groups (control, Pi, and Pi-LPS-CM) throughout incubation times was explored using the linear mixed models for longitudinal data. Comparisons between the three groups for each incubation time were performed using the ANOVA test or Kruskal–Wallis test, as appropriate. Comparisons between two groups were performed using the t-test or Mann–Whitney test, as appropriate. Comparisons between incubation times within each group were achieved using the paired t-test or Wilcoxon signed-rank test. Bonferroni correction for multiple comparisons was applied.
Results
Spectrophotometric Quantifications
Spectrophotometric analyses indicated that calcium amounts and ALP activity associate with Pi concentrations and incubation times, with both parameters being enhanced by additional proinflammatory stimuli to various degrees. In more detail, there was slight time-dependent increase in both calcium amounts (Fig. 1A) and ALP activity (Fig. 1B) for 0.8-Pi-cultures and 0.8-Pi-LPS-CM-cultures, with the same trend resulting for control-cultures. Additional slight calcium increases resulted for 1.3-Pi-cultures and 1.3-Pi-LPS-CM-cultures, with these latter showing more significant increase starting from day 21 (Fig. 1C). Concerning ALP activity, evident increase was restricted to 1.3-Pi-LPS-CM-cultures (Fig. 1D). Also, 2.0-Pi-cultures showed slight time-dependent mineral increases, which were more marked starting from day 25. Conversely, sudden increases in calcium amounts resulted for 2.0-Pi-LPS-CM-cultures, which underwent further striking increase starting from day 21 (Fig. 1E). Sudden, even more marked increases in ALP activity characterized 2.0-Pi-LPS-CM-cultures again, whereas enzyme activity remained at stable levels in 2.0-Pi-cultures, except for its dramatic surge starting from day 25 (Fig. 1F).
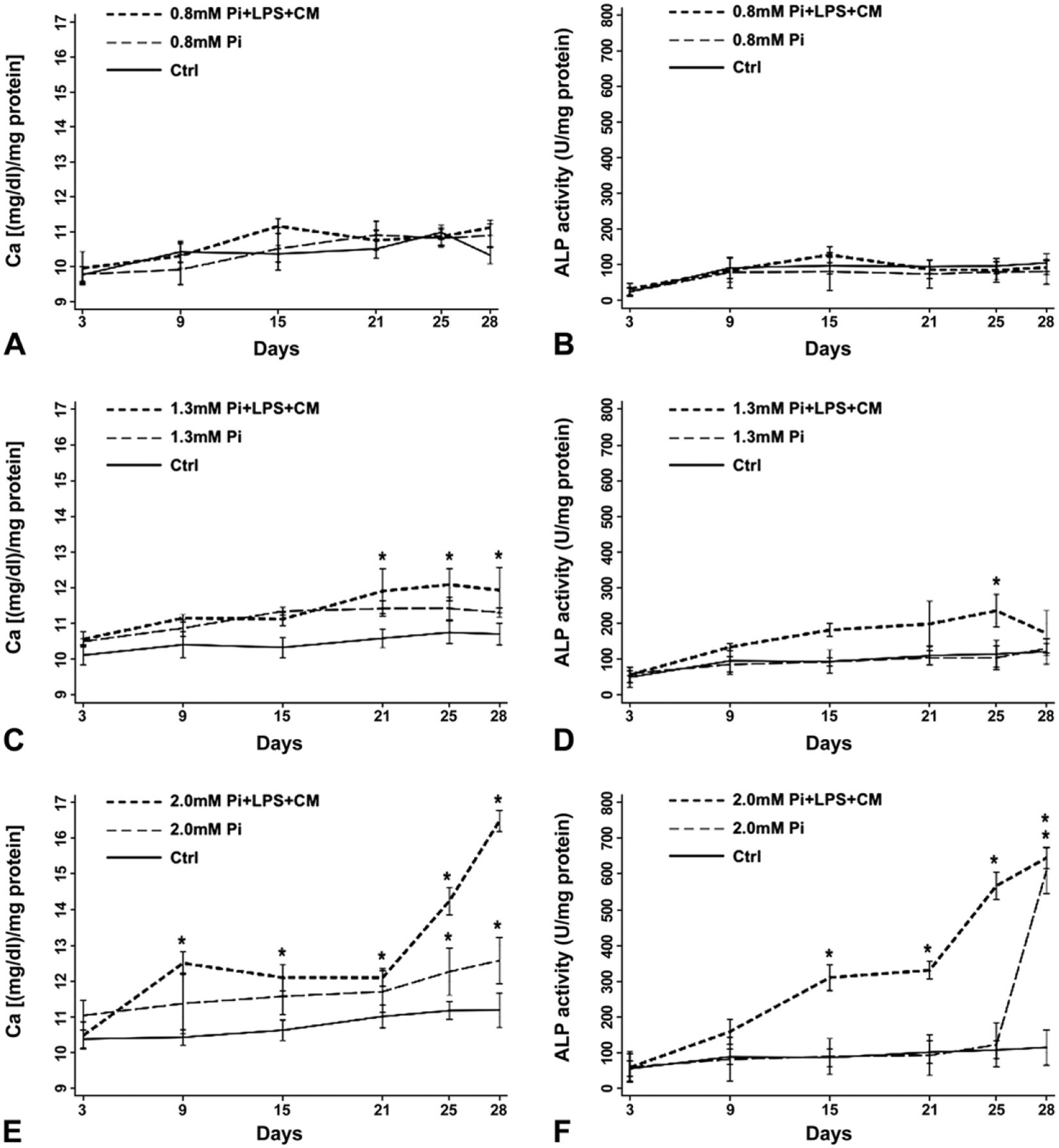
Spectrophotometric quantification of calcium amounts and alkaline phosphatase activity. Calcium content (left column) and enzymatic activity (right column) in 3- to 28-day-long (A–F) control AVICs and AVICs treated with (A, B) 0.8-mM Pi, (C, D) 1.3-mM Pi, and (E, F) 2.0-mM Pi solely or superstimulated with LPS and CM derived from LPS-stimulated macrophages. Values are reported as mean ± SD. Statistically significant values are indicated with asterisks. Abbreviations: AVIC, aortic valve interstitial cell; Pi, phosphate; LPS, lipopolysaccharide; CM, conditioned medium.
Light and Electron Microscopy
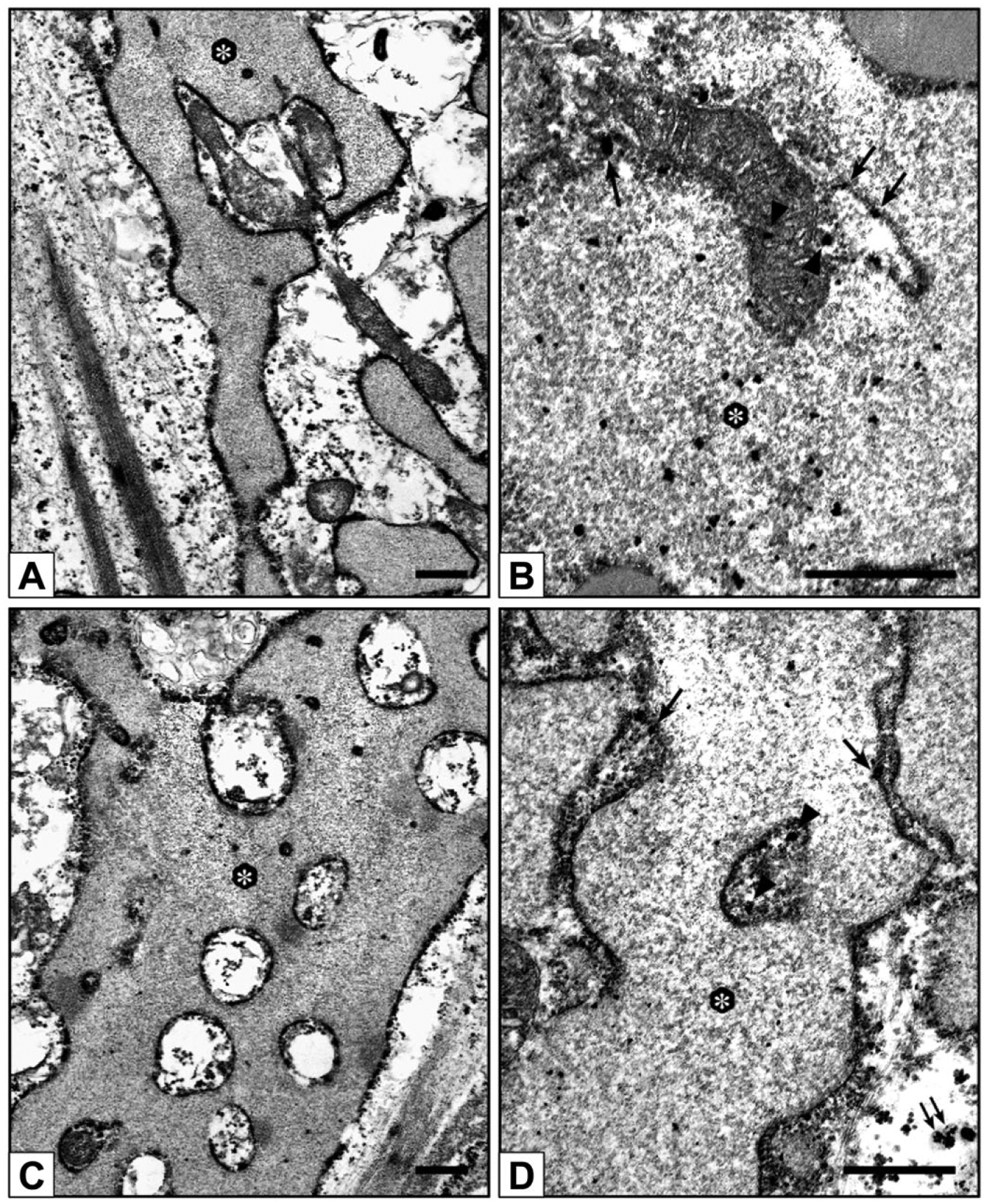
Under the inverted microscope, in contrast with control-cultures (not shown), 0.8-Pi-cultures, 0.8-Pi-LPS-CM-cultures, 1.3-Pi-cultures, and 1.3-Pi-LPS-CM-cultures (Fig. 2A–D), calcific nodules appeared for 2.0-Pi-cultures and, at greater extent, 2.0-Pi-LPS-CM-cultures starting from day 21 (Fig. 2E and F). Ultrastructurally, control AVICs exhibited well-preserved organelles including a lot of autophagic vacuoles as usually shown by cultured cells (Fig. 3A and B). Compared with controls, AVICs from 0.8-Pi-cultures and 1.3-Pi-cultures showed mild time-dependent changes, whereas those from 0.8-Pi-LPS-CM-cultures and 1.3-Pi-LPS-CM-cultures underwent an evident appearance of hypertrophic rough endoplasmic reticulum (RER) enclosing distinct cytoplasm regions containing mitochondria and preexistent autophagosomes (Fig. 3C). Further time-dependent RER swelling resulted in a lot of narrow cytoplasm compartments enveloping degenerating mitochondria, disrupting autophagosomes, and remnants of other cytoplasmic components (Figs. 3D and 4A, C). Positivity for acid phosphatase activity resulted at the level of lumens of enlarging RER, its membranes, and enveloped mitochondria (Fig. 4B and D). In contrast, AVICs from 2.0-Pi-cultures and above all those from 2.0-Pi-LPS-CM-cultures appeared to have undergone more and more severe alterations consisting in (1) marked decrease in autophagic vacuoles in the absence of apparent RER hypertrophy; (2) swelling of intracytoplasmic organelles with dissolution of their membranes paralleled by appearance of lipid droplets showing acidification, as revealed by increasing reactivity to preembedding CB reaction (Fig. 5A1), and calcium binding capacity, as revealed by postembedding von Kossa silver staining (Fig. 5A2); (3) progressive lipid droplet melting with concurrent intracytoplasmic accumulation of phthalocyanine-positive and silver-reactive amorphous material (PPM; Fig. 5A2); (4) PPM layering at cell edges so forming PPLs (Fig. 5B1), showing enhanced affinity for metallic silver deposition after von Kossa reaction (Fig. 5B2) and representing initial HA crystal nucleation foci (Fig. 5C); and (5) AVIC fragmentation into irregularly shaped and sized products lined by concentric PPLs in their turn stemming paracrystalline PPL-lined calcospherulae with persistent reactivity to CB and HA nucleation capacity (Fig. 5D, E1, and E2). Of interest, early degenerative patterns characterizing most AVICs from 2.0-Pi-LPS-CM-cultures, such as lipid droplet accumulation and PPM formation, were occasionally found also in AVICs from 1.3-Pi-LPS-CM-cultures (not shown).
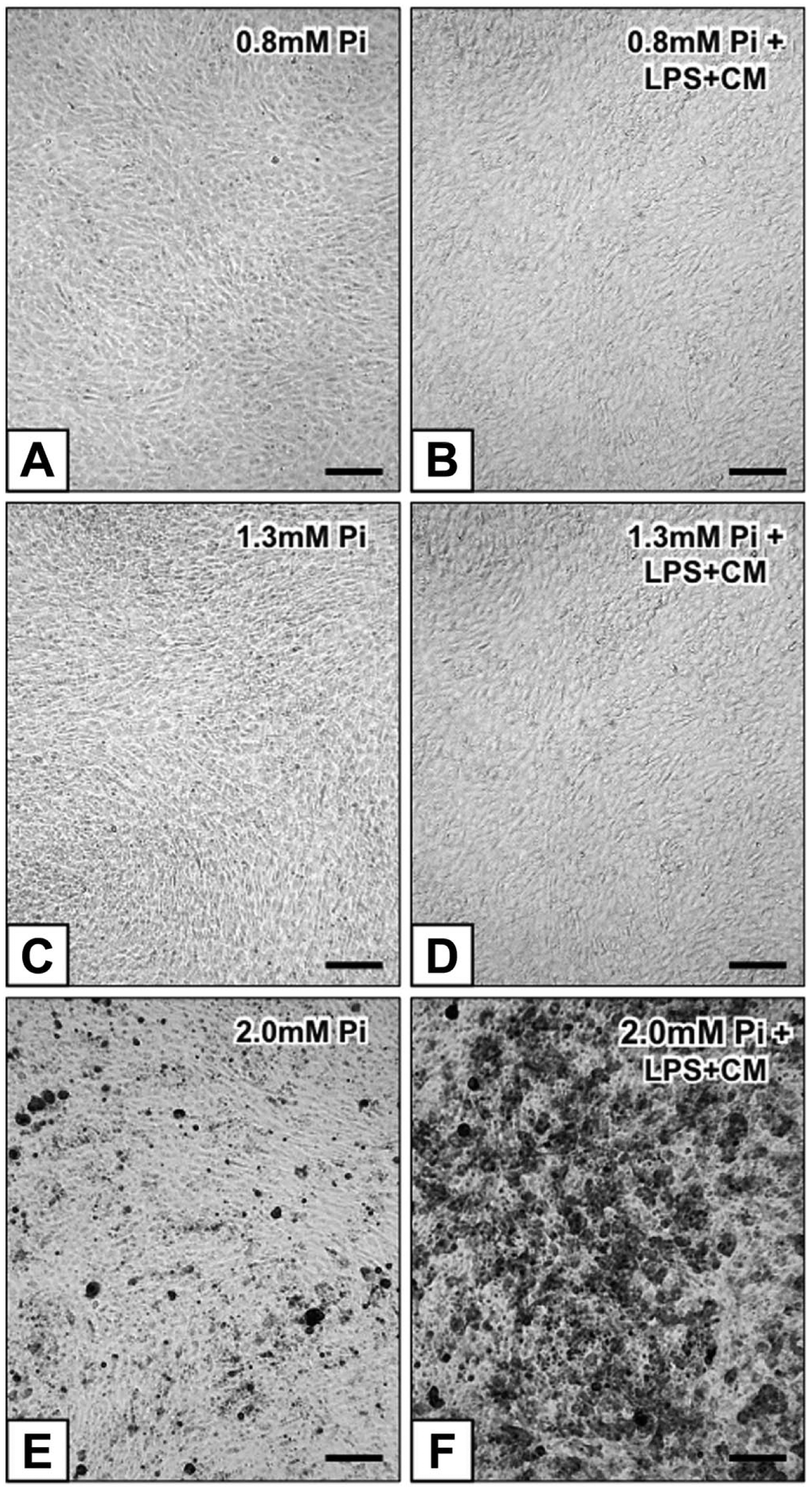
Inverted microscope micrographs of 21-day-long AVIC cultures subjected to alizarin red S calcium staining: (A) unreacted 0.8-Pi-cultures, (B) 0.8-Pi-LPS-CM-cultures, (C) 1.3-Pi-cultures, (D) 1.3-Pi-LPS-CM-cultures, (E) calcium precipitation in 2.0-Pi-cultures, and (F) its increase in 2.0-Pi-LPS-CM-cultures. Abbreviations: AVIC, aortic valve interstitial cell; Pi, phosphate; LPS, lipopolysaccharide; CM, conditioned medium. Scale bars: A–F = 100 µm.
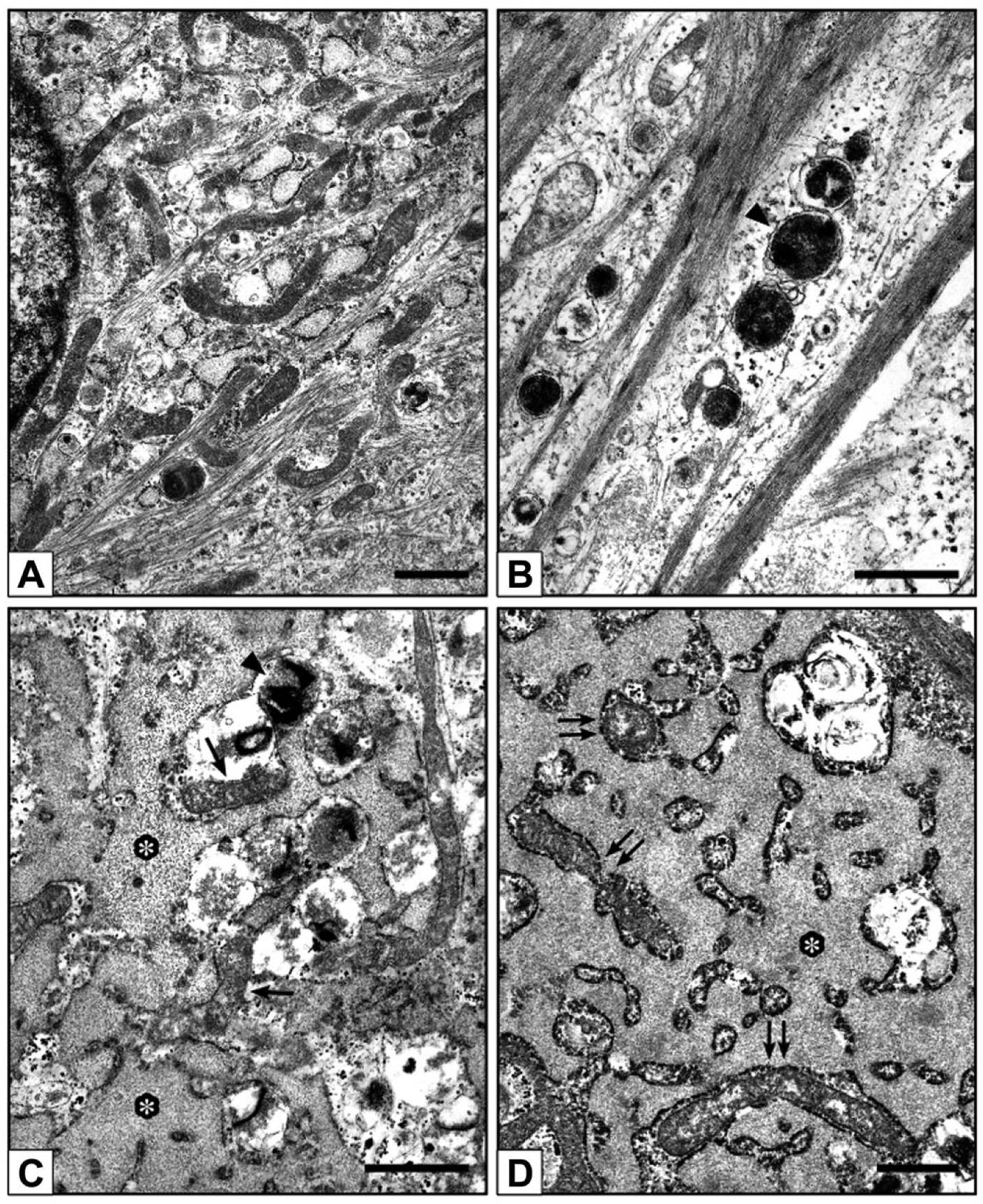
Electron microscope micrographs of 21-day-long AVIC cultures. (A, B) Control AVICs showing normal features including abundant autophagic vacuoles (arrowhead in B). (C, D) AVICs from 0.8-Pi-LPS-CM-cultures showing an abnormal enlargement of the rough endoplasmic reticulum (asterisks), enveloping mitochondria (arrows in C and double arrows in D), autophagosomes (arrowhead in C), and other degrading cytoplasmic components. Abbreviations: AVIC, aortic valve interstitial cell; Pi, phosphate; LPS, lipopolysaccharide; CM, conditioned medium. Scale bars: A–D = 1 µm.
Electron microscope micrographs of 21-day-long 1.3-Pi-LPS-CM-cultures. (A, C) AVICs showing prominent segregation of cytoplasm components by hypertrophic rough endoplasmic reticulum (asterisks). (B, D) AVICs from parallel 1.3-Pi-LPS-CM-cultures showing positivity to acid phosphatase activity as revealed by lead precipitation at the level of reticulum lumen (asterisks) and membrane (arrows in B and D) as well as entrapped organelles (arrowheads in B and D) and organelle remnants (double arrows in D). Abbreviations: Pi, phosphate; LPS, lipopolysaccharide; CM, conditioned medium; AVIC, aortic valve interstitial cell. Scale bars: A–D = 0.5 µm.
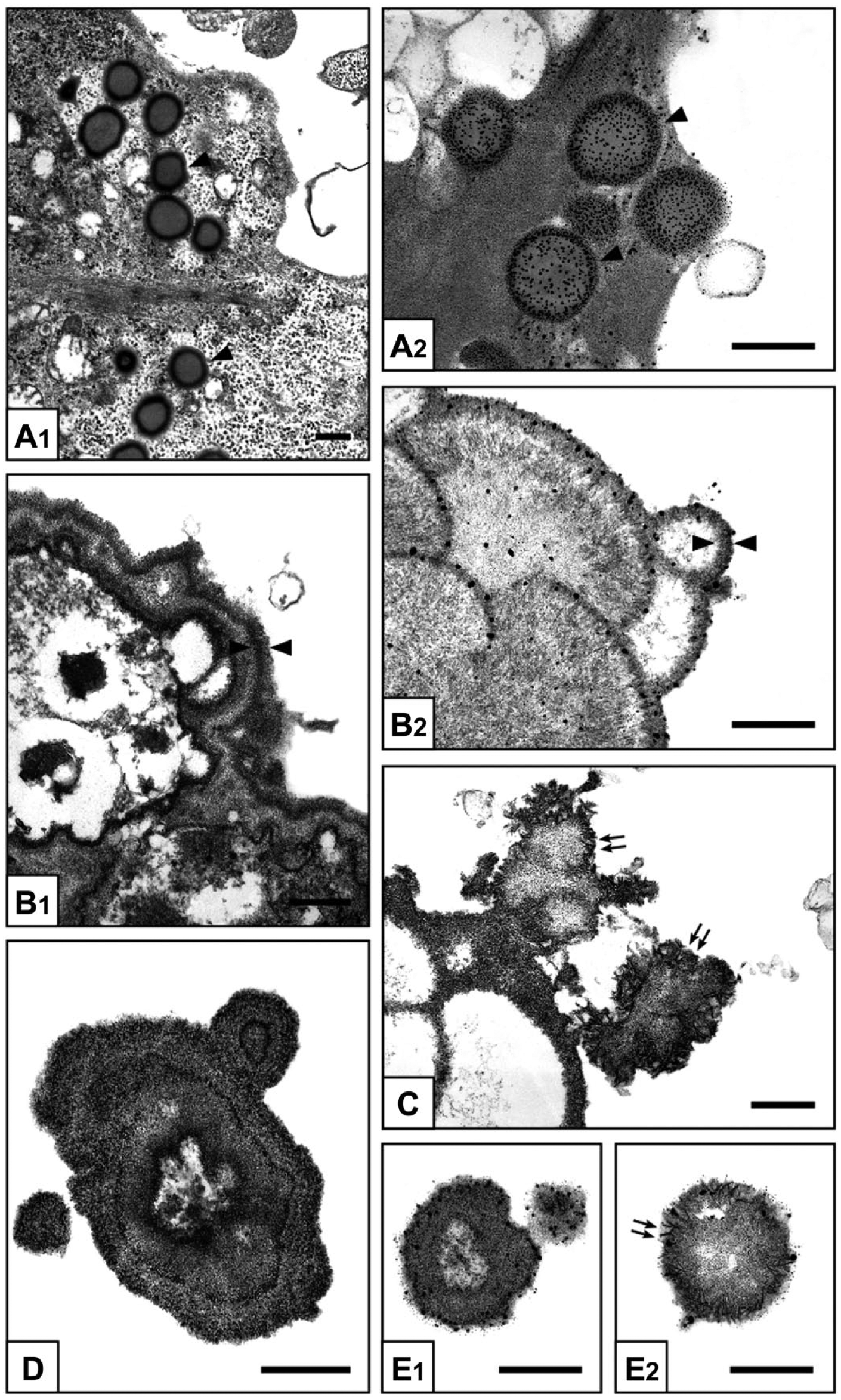
Electron microscope micrographs of 3- to 28-day-long 2.0-Pi-LPS-CM-cultures. AVICs show progressive procalcific degenerative patterns consisting in (A1) intracellular accumulation of lipid droplets (arrowheads), (A2) which are positive for von Kossa silver staining (arrowheads); (B1) margination of released lipids forming CB-positive layers at cell edges (counterposed arrowheads), (B2) which are sites of silver particle deposition after von Kossa reaction (counterposed arrowheads) and (C) HA crystal nucleation (double arrows); and (D) cell disruption into irregularly shaped cell-derived products, stemming (E1) small calcospherulae preserving positivity to von Kossa reaction and (E2) HA nucleation capacity (double arrows). Abbreviations: Pi, phosphate; LPS, lipopolysaccharide; CM, conditioned medium; AVIC, aortic valve interstitial cell; CB, Cuprolinic Blue; HA, hydroxyapatite. Scale bars: A, B1,2, C, D, E1,2 = 0.5 µm.
Immunocytochemical and Immunoblot Assays
To explore whether the AVIC responses described above may correlate with macroautophagocytosis occurrence, immunocytochemical assays of marker MAP1LC3 were performed. Marked reactivity was restricted to 3-day-long control-cultures and 1.3-Pi-LPS-CM-cultures, with a drastic decrease after longer incubation times (not shown). Percentages of immunopositive cells are displayed in Fig. 6A. To assess possible involvement of apoptosis in driving AVIC responses, immunocytochemical detection of late marker annexin-V was carried out. Negligible reactivity resulted for all cultures irrespective of incubation times (not shown). Percentages of immunopositive cells are displayed in Fig. 6B. In addition, immunoblot assays revealed a complete absence of the 17-kDa caspase-3 cleaved form for all cultures (data not shown). Moreover, ultrastructural examination of 2.0-Pi-LPS-CM-cultures treated with pancaspase inhibitor Boc-D-FMK showed AVICs to undergo degenerative patterns as those observed for AVICs from 2.0-Pi-LPS-CM-cultures not subjected to caspase inhibition (not shown).
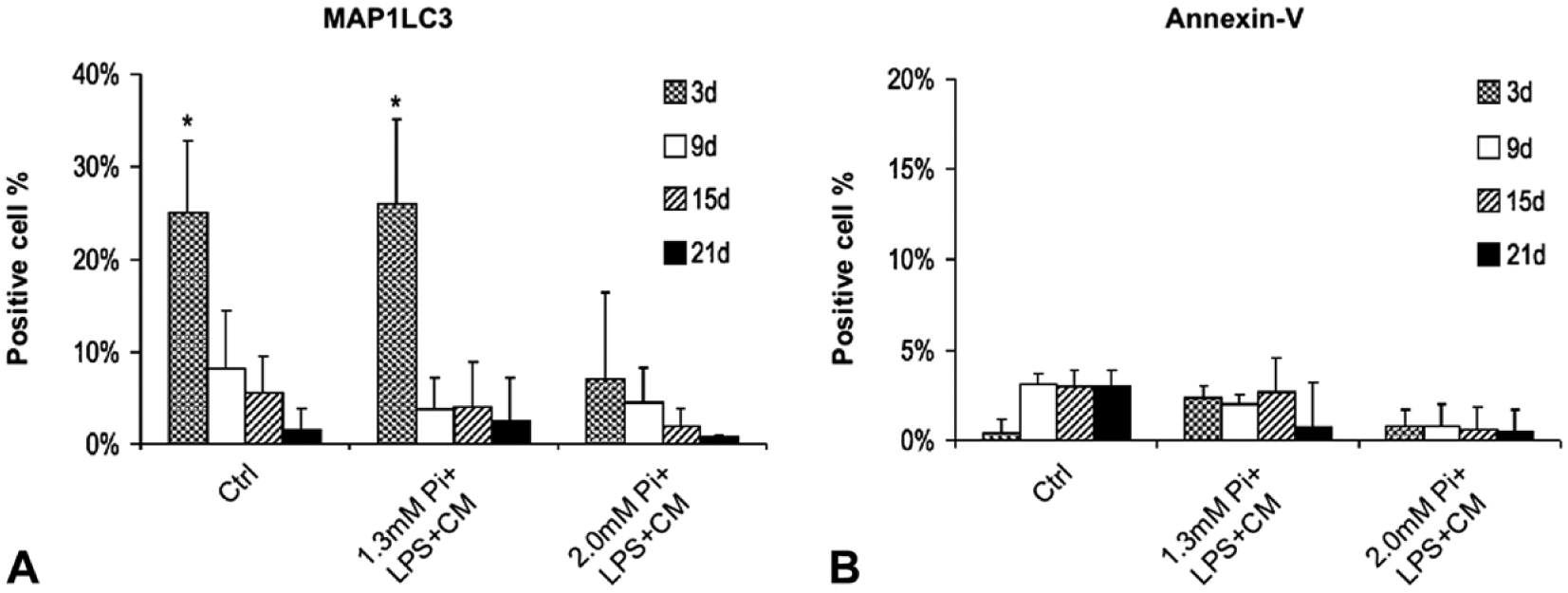
Percentage value histograms. (A) Percentage of AVICs immunopositive for mature autophagosome marker MAP1LC3 and (B) late apoptosis marker annexin-V in 3- to 21-day-long control-cultures, 1.3-Pi-LPS-CM-cultures, and 2.0-Pi-LPS-CM-cultures. Values are reported as mean ± SD. Statistically significant values are indicated with asterisks. Abbreviations: AVIC, aortic valve interstitial cell; Pi, phosphate; LPS, lipopolysaccharide; CM, conditioned medium.
Discussion
In the present investigation, possibility of different Pi-concentration-dependent cell responses to be induced was tested in vitro treating AVICs with three Pi concentrations within the normal range in organisms which included maximum and minimum threshold values. Two distinct cell fates, consisting in autophagy-related survival versus procalcific death, resulted to depend on Pi concentration, with possible enhancement caused by additional proinflammatory mediators. Namely, an atypical autophagic activity characterized by an RER-dependent organelle segregation and digestion took place in AVICs cultured at 0.8- and 1.3-mM Pi. Conversely, a peculiar lipid-release-associated procalcific death occurred in AVICs cultured at 2.0-mM Pi, according to the cell degenerative patterns previously described for bovine AVICs cultured under severe dystrophic- and metastatic-like conditions22,23 as well as porcine AVICs populating aortic valve leaflets subjected to xenogeneic subdermal implantation.24–27 Consistently, observational studies reported an increased risk of cardiovascular diseases including calcific events in individuals with serum Pi levels just below the upper limit of the conventional normophosphatemic range.2,3,6–8 Moreover, 1.3-mM Pi concentration representing a critical threshold for augmented valve disease risk2,6,7 was here supported by the finding that some AVICs from 1.3-Pi-LPS-CM-cultures underwent initial procalcific degeneration, suggesting that such a Pi level can actually be permissive of cell mineralization to some extent. However, notable cell procalcific degeneration was restricted to AVIC cultures containing 2.0-mM Pi for which spectrophotometric analyses revealed linear increases in calcium amounts starting from day 21, consistently with appearance of alizarin-red-S-positive calcific nodules and von-Kossa-positive cell-derived products acting as major HA nucleators, with these latter mirroring those reported for actual aortic valve calcification28–32 as well as in vivo and in vitro procalcific conditions.22–27 Additional increases in calcium amounts and ALP activity were observed for AVICs superstimulated with CM from LPS-stimulated macrophages plus LPS in the presence of 2.0-mM Pi. This effect may depend on simultaneous AVIC stimulation by proinflammatory mediators contained in the CM 33 and LPS, with this latter possibly causing ALP activity enhancement and concurrent removal of mineralization inhibitor pyrophosphate as reported for cultured human AVICs.34–36 Because such inflammation-dependent mineralization enhancement was restricted to AVIC cultures containing 2.0-mM Pi, proinflammatory mediators seem to act as stimuli exacerbating the Pi-dependent calcific effects rather than procalcific factors per se. Indeed, as we previously reported, 23 cell mineralization does not occur in AVIC cultures free from elevated Pi supplementation. Accordingly, the drastic increase in ALP activity occurring at day 25 also for LPS-free 2.0-Pi-cultures supports the idea that Pi concentration is the crucial factor involved in cell mineralization.4,9,10 Even if ALP activity increase was often reported to be associated with the expression of an osteoblast phenotype by mineralizing AVICs,13,37,38 in the present study, no cell osteoblastic transdifferentiation was ultrastructurally found but only the peculiar AVIC procalcific degeneration and death described. These degenerative features are superimposable to those already reported for cultured AVICs in experimental dystrophic- and metastatic-like calcific conditions,22,23 as well as in subdermally implanted aortic valve leaflets,24–27 according to a process which is more consistent with the concept that ectopic mineralization occurs independently of AVIC osteoblastic differentiation and subsequent bone neogenesis. This result does not call into question whether ossification can take place in calcifying valves,12,13,16 but the concept is supported that AVIC mineralization is not necessarily related to this histogenetic process. 11 As a matter of fact, bone metaplasia was reported to be histologically recognizable in less than 13% of hundreds of aortic valves affected by dystrophic calcification.13,39 Although AVIC mineralization is reported to be restricted to specific cell subtypes,40–42 the choice to use the complete AVIC population was made with the rationale to better reproduce actual in vivo conditions, including cell-to-cell interactions. In mineralizing AVICs, the CB reactivity and von Kossa positivity observed starting from the periphery of the early new-formed lipid droplets may correlate with initial outer lipid acidification,24,25 with this chemical change likely depending on oxidative effects exerted by endogenous reactive oxygen species (ROS) released by degenerating mitochondria, 43 possibly reinforced by additional macrophage-derived ROS contained in the CM. 44 Such oxidizing environment may also cause phospholipid disarray of lysosome membranes, with release of acid hydrolases, as well as mitochondrial and RER membranes, with calcium leakage and activation of intracellular calcium-dependent enzymes, enabling the occurrence of the autolytic colliquative process observed for mineralizing AVICs, besides causing cytoskeleton disintegration 45 with plasma membrane blebbing and pinching off of multiple procalcific cell by-products. In addition, cell exposure to oxidative stress was reported to contribute to RER stress, leading to the activation of the signaling pathway called unfolded protein response (UPR).46,47 Taking into account that UPR execution requires RER expansion, 48 the unusually RER oversizing observed for 0.8-Pi-LPS-CM-cultures and 1.3-Pi-LPS-CM-cultures might depend on toxic effects exerted by oxidative stress together with low Pi concentrations, with triggering of transient compensatory mechanisms by cells. Consistently, replacement of stimulating media with standard culture medium was associated with disappearance of RER hypertrophy for these AVIC cultures (unpresented data). As hypertrophic RER was found to segregate damaged mitochondria into more and more narrow compartments, it is likely that their sequestration may ensure prompt buffering of increasing intracytoplasmic calcium via its direct uptake into RER cisternae. This relationship is consistent with the occurrence of direct communication between mitochondrial intermembrane space and RER lumen at the level of topical contact sites, as reported.49,50 On the contrary, the seemingly unaltered mitochondria entrapped by the hypertrophic RER might uptake calcium from RER cisternae reinforcing their bioenergetic response and improving cell adaptation to stressing conditions. 51 Conversely, the occasional identification of RER hypertrophy in mineralizing AVICs suggests that elevated Pi concentrations may lead to cell death because of excessive intoxication compromising the activation of compensatory responses by the stressed cells. Of note, the ultracytochemical identification of acid phosphatase activity at the level of membranes and lumen of the hypertrophic RER strongly suggests its involvement in an alternative, non-lysosomal mechanism of organelle digestion taking place in AVICs cultured under low/middle Pi concentrations, as shown for other cells using the same approach.52–54 As autophagosomes themselves appeared to be removed by such RER-associated degrading activity, this atypical autophagocytosis may somehow represent a time-saving process providing faster recovery of homeostatic conditions by stressed cells than the activation of orthodox macroautophagocytosis machinery can do. Consistently, sudden decrease in autophagic vacuoles and immunoreactivity to mature autophagosome marker MAP1LC3 were observed, suggesting that macroautophagocytosis is just to be conceived as a constitutive house-keeping process providing adaptive cell responses to the culture environment 55 instead of a procalcific process as reported. 20 RER-associated autophagic activity possibly playing a cell survival role was further supported by the fact that negligible immunopositivity to late apoptosis marker annexin-V as well as a complete absence of the 17-kDa caspase-3 cleaved form resulted for all AVICs cultured under low/middle Pi concentrations. As these results were also shared by AVICs cultured at 2.0-mM Pi, AVIC mineralization can be assumed to take place independently from apoptosis occurrence, despite its widely accepted involvement in cardiovascular tissue mineralization.17–19,21 Moreover, apoptotic cascade to be not activated in mineralizing AVICs was supported by the occurrence of lipid-release-associated procalcific cell death also in AVICs from 2.0-Pi-LPS-CM-cultures treated with pancaspase inhibitor Boc-D-FMK, besides the fact that typical apoptosis features, such as cell shrinkage and chromatin condensation/fragmentation, were not ultrastructurally encountered. As main features associated with oncosis, such as chromatin clumping, cytoplasm blebbing, and cellular swelling, were not found too, occurrence of an alternative type of cell death in mineralizing AVICs could be assumed, depending on a distinct process resulting in a lipid-release-associated cell degeneration, as observed in the present procalcific conditions and other valve mineralization processes.22–27 In conclusion, the present findings suggest that Pi represents a specific cell stressor, the concentration of which is critical for AVIC fate, being elevated Pi concentration the major determinant of a distinct procalcific cell death other than apoptosis or oncosis whereas low/middle Pi concentrations trigger the activation of compensatory mechanisms preserving cell functionality via an unusual RER-dependent autophagic activity. In addition, the concept is supported that distinct serum Pi levels are predictive of dystrophic valve calcification incurrence although they are comprised within the conventional normophosphatemic range, suggesting that more meticulous statement of Pi threshold values in organisms is mandatory to properly evaluate the risk of this valve disorder.
Footnotes
Acknowledgements
The authors thank Dr. Emanuela Tesei, official veterinarian of Pitaccolo slaughterhouse, for her kind support in providing the animal tissues used in the present study.
Competing Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
AB: manuscript writing, contribution in designing this study, ultracytochemical and immunocytochemical reactions, and ultrastructural analyses; ADM: cell extraction, culturing, and treatments, and Western blot analyses; MC: image processing and panel preparation; GG: statistical analyses; FT: spectrophotometric analyses and data elaboration; MM: manuscript revision; and FO: design and supervision of this investigation, and manuscript revision.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.