
Abstract
Transglutaminases (TGs) are a family of widely distributed enzymes that catalyze protein crosslinking by forming a covalent isopeptide bond between the substrate proteins. We have shown that MC3T3-E1 osteoblasts express Factor XIII-A (FXIII-A), and that the extracellular crosslinking activity of FXIII-A is involved in regulating matrix secretion and deposition. In this study, we have investigated the localization and potential role of intracellular FXIII-A. Conventional immunofluorescence microscopy and TIRF microscopy analyses showed that FXIII-A co-localizes with caveolin-1 in specialized membrane structures, caveolae, in differentiating osteoblasts. The caveolae-disrupting agent methyl-β-cyclodextrin abolished FXIII-A staining and co-localization with caveolin-1 from the osteoblast plasma membrane. The presence of FXIII-A in caveolae was confirmed by preparing caveolae-enriched cellular fractions using sucrose density gradient ultracentrifugation followed by western blotting. Despite this association of FXIII-A with caveolae, there was no detectable transglutaminase activity in caveolae, as measured by monodansylcadaverine incorporation. TG inhibitor NC9—which can alter TG enzyme conformation—localized to caveolae and displaced FXIII-A from these structures when added to the osteoblast cultures. The decreased FXIII-A levels in caveolae after NC9 treatment increased c-Src activation, which resulted in caveolin-1 phosphorylation, homo-oligomerization and Akt phosphorylation, suggesting cellular FXIII-A has a role in regulating c-Src signaling in osteoblasts.
Introduction
Transglutaminases (TGs) are a family of widely distributed, Ca2+-dependent enzymes with diverse functions ranging from intracellular signaling to extracellular matrix stabilization. The TG family includes nine enzymes: TG1–7, Factor XIII-A (FXIII-A), and band 4.2. TG enzymes can catalyze several, two-step reactions that result in posttranslational modifications of glutamine residues; however, several TGs have been reported to also have noncatalytic functions (Beninati et al. 2009; Eckert et al. 2014; Iismaa et al. 2009; Lorand and Graham 2003; Muszbek et al. 2011). The best known and most studied catalytic function of TG enzymes is the protein transamidation reaction that occurs between a protein-bound glutamine residue and a protein-bound lysine or primary amine, resulting in the formation of a γ-glutamyl-ε-lysyl crosslink, also referred to as an isopeptide bond. This crosslinking in certain extracellular matrix substrate proteins has been reported to stabilize both collagenous and noncollagenous extracellular matrices (Al-Jallad et al. 2006; Akagi et al. 2002; Cui et al. 2014; Esposito and Caputo 2005; Hohenadl et al. 1995; Kaartinen et al. 2002). The crosslinking is also essential for cornified cell envelope formation in terminal keratinocyte differentiation, where covalent crosslinking occurs in proteins such as involucrin, loricrin, and small proline-rich protein (Eckert et al. 2005; Hitomi 2005). Crosslinking by TGs can also be involved in integrin-growth factor coupling, where FXIII-A-catalyzed extracellular crosslinking of β3-integrin to VEGFR-2 results in VEGFR-2 activation and the promotion of angiogenesis (Dardik et al. 2005). In addition to protein crosslinking, several noncatalytic function for TGs have been reported, and these include the function of TG2 as a fibronectin-binding co-receptor for β1-integrin in human fibroblasts and erythroleukemia cells (Akimov et al. 2000), and the cytoskeletal scaffolding function of band 4.2, which maintains membrane integrity in erythrocytes (Das et al. 1994; Satchwell et al. 2009).
FXIII-A is best known as a circulating, plasma TG enzyme responsible for the fibrin crosslinking that occurs as the last step of the blood coagulation cascade (Ariëns et al. 2000; Muszbek et al. 2011); however, circulating FXIII-A also contributes to wound healing and tissue repair (Iismaa et al. 2009; Muszbek et al. 2011; Verderio et al. 2004). FXIII-A is part of a heterotetrameric FXIII complex consisting of two FXIII-A units and two inhibitory FXIII-B units. The B units are released from the complex via thrombin cleavage of FXIII-A, which induces the TG activity of FXIII-A. The circulating FXIII-A is produced by cells of a bone marrow origin (Poon et al. 1989; Wölpl et al. 1987). FXIII-A (which is not bound to FXIII-B) is also found in many tissues and is expressed by monocytes, macrophages, megakaryocytes, chondrocytes, osteoblasts and adipocytes; in these cell types, FXIII-A is found in various cellular and extracellular locations: in the cytosol, at the plasma membrane/cell surface, and secreted into the extracellular matrix (Johnson et al. 2008; Malara et al. 2011; Muszbek et al. 1985; Myneni et al. 2014; Nakano et al. 2007; Nurminskaya and Kaartinen 2006; Piercy-Kotb, 2011). A number of reports show that FXIII-A and its crosslinking activity outside the cell (being either cell surface-bound or soluble), can regulate fibroblast adhesion, induce chondrocyte hypertrophy, regulate extracellular matrix secretion and stabilization, and promote angiogenesis (Al-Jallad et al. 2006; Cui et al. 2014; Dardik et al. 2005; Johnson et al. 2008; Nurminskaya and Kaartinen 2006; Ueki et al. 1996). FXIII-A is also found inside cells but whether it has a function the intracellular compartment, is not known.
Our recent work has focused on understanding the role of FXIII-A in osteoblasts during osteogenesis. We have shown previously that FXIII-A is produced by osteoblasts and is externalized to the extracellular matrix where its function has been linked to the stabilization of plasma fibronectin matrix (Al-Jallad et al. 2006, 2011; Cui et al. 2014; Piercy-Kotb et al. 2011). Cell-associated FXIII-A resides on the extracellular side of the osteoblast plasma membrane where it appears to facilitate the formation of high-molecular weight detyrosinated Glu-tubulin, which is linked to the secretion and elaboration of collagenous extracellular matrix in these cells (Wang et al. 2014). Both these extracellular functions involve transamidation activity. We also previously reported by immunofluorescence microscopy that FXIII-A in osteoblasts appears as small patches at the plasma membrane, not always co-localizing with Glu-tubulin (Al-Jallad et al. 2011; Wang et al. 2014). In the present study, we hypothesized that those patches represent FXIII-A, which is associated with the lipid structures, caveolae. Caveolae are specialized, omega-shaped lipid raft plasma membrane invaginations that are involved in vesicular transport processes such as transcytosis, endocytosis and potocytosis, as well as in signal transduction (Baker and Tuan 2013; Cohen et al. 2004; Razani et al. 2002). The aim of this study was to examine if intracellular FXIII-A is associated with caveolae and if it has an intracellular function in osteoblasts. We report here that intracellular FXIII-A does indeed localize to caveolae in differentiating osteoblasts, where it regulates caveolin-1 phosphorylation and its homo-oligomerization, and c-Src kinase activation in a manner that does not appear to involve its transamidation activity, thus suggesting that FXIII-A also has a noncatalytic function, as has been similarly reported for other TG enzymes.
Materials & Methods
Antibodies and Reagents
Methyl-β-cyclodextrin, monodansylcadaverine and 2-(N-morpholino)ethanesulfonic acid (MES) were from Sigma-Aldrich (St. Louis, MO). Src family kinase inhibitor PP2, c-Src and p-c-Src (Tyr424) (antibody 9A6) antibodies were purchased from Santa Cruz Biotechnology (Dallas, TX). Anti-dansyl antibody was purchased from Molecular Probes (Eugene, OR). Polyclonal antibody against mouse FXIII-A was designed and generated by GenScript (Piscataway, NJ) (Al-Jallad et al. 2011). Mouse anti-p-caveolin-1(Tyr14) was from BD Biosciences (San Jose, CA). Rabbit anti-caveolin-1 antibody was from Abcam (Cambridge, MA). Anti-p-Akt (Ser473), anti-pan-Akt, anti-β1-integrin, horseradish peroxidase-linked anti-rabbit IgG and anti-mouse IgG were from Cell Signaling Technology (Whitby, ON, Canada) and GE Healthcare Life Sciences (Baie d’Urfe, QC, Canada), respectively. TG2 mouse monoclonal antibody (CUB7402/TG100), the Alexa Fluor secondary antibodies, DAPI, EZ-link Sulfo-NHS-SS-biotin, High-Capacity NeutrAvidin Agarose and protein BCA assay kit were from Thermo Scientific (Waltham, MA). All other reagents, if not otherwise specified, were purchased from Sigma-Aldrich or Fisher Scientific. Horseradish peroxidase linked anti-mouse IgG was from GE Healthcare Life Sciences (Baied’Urfe, QC, Canada).
Cell Culture and Treatments
Mouse calvarial preosteoblast (MC3T3-E1) subclone 14 cells were originally a generous gift from Dr. Renny T. Franceschi, University of Michigan, School of Dentistry, Ann Arbor, MI (Wang et al. 1999). Cells were cultured in a humidified 37°C incubator at 5% CO2 in modified alpha minimum essential medium (α-MEM) minus
Preparation of Protein Extracts
Total cellular protein preparations were obtained using buffer containing 150 mM NaCl, 10 mM Tris-HCl (pH 7.2), 5 mM EDTA, 0.1% sodium dodecyl sulphate (SDS), 1% Triton X-100 and 1% Na-deoxycholate. Cell extracts were homogenized by ultrasonication directly after extraction and were centrifuged at 14000 ×g for 15 min at 4°C. The supernatant was collected and hereafter is referred to as total protein extract. Orthovanadate and sodium fluoride were added into the above buffer for studying protein phosphorylation.
Sucrose Density Gradient Ultracentrifugation
MC3T3-E1 cells were cultured on 150-mm plates and cells were detached from the plates by gentle scraping in the presence of 1% Triton-X 100/MES-buffered saline (MBS). Cell preparations were transferred to a pre-chilled Dounce (glass-glass) homogenizer and incubated on ice for 20 min. Cells were then homogenized on ice and mixed with 90% sucrose/MBS and transferred into an ultracentrifuge tube. MBS/Triton X-100 containing 35% sucrose was applied on top of the sample/90%sucrose/MBS layer. A visible interface remained evident between the two density layers. Finally, MBS/Triton X-100 containing 5% sucrose was layered on top of the 35% sucrose layer. The preparation was centrifuged for 16–20 hr at 39,000 RPM (4°C) in a SW41Ti rotor (Beckman); this provided a maximum force equivalent (bottom of tube) to approximately 260,000 × g and an average force (middle of the tube) of approximately 188,000 × g. At centrifugation, a faint light-scattering band, which consisted of the buoyant lipid raft/caveolar material, became visible at the 35% sucrose-5% sucrose interface. Samples were collected from the gradient from the top down as 1 ml fractions. Fractions 4 and 5 are defined as bona fide caveolae-enriched fractions (Parolini et al. 1999). Fractions were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and western blotting.
SDS-PAGE and Western Blotting
Twenty five (25) µg of each sample was boiled in SDS sample buffer for 5 min, followed by electrophoretic separation of proteins by SDS-PAGE. For visualization of caveolin-1 oligomers, samples were not boiled before loading. Following electrophoresis, proteins from the gels were transferred to PVDF membranes (Bio-Rad) and blocked for 30 min in 5% non-fat dry milk in TBS-Tween (TBS-T), and then incubated at 4°C overnight in primary antibodies diluted in TBS-T. Following the incubation with primary antibodies, membranes were rinsed in TBS-T, and then incubated with appropriate secondary antibodies conjugated to horseradish peroxidase at room temperature. Secondary antibodies used were sheep anti-mouse (Invitrogen, Carlsbad, CA) and goat anti-rabbit (Cell Signaling Technology). Bands were visualized using the ECL Plus kit (GE Healthcare) and chemiluminescence was detected using Kodak Biomax film (Kodak, Rochester, NY).
Immunofluorescence Microscopy
Cells were seeded on Nunc Lab-Tek chamber slides (Fisher Scientific) and cultured as described above. On day 5, cells were fixed with 3.7% formaldehyde in PBS for 10 min at room temperature. Slides were washed three times with PBS and blocked with 2% bovine serum albumin (BSA) in PBS. This was followed by primary antibody incubation for 2 hr at room temperature, and a washing step with 0.1% BSA in PBS and incubation with Alexa Fluor secondary antibody conjugates. Nuclei were stained with DAPI (4’, 6-diamidino-2-phenylindole). Samples were mounted with Prolong Gold Anti-Fade medium (Invitrogen) and dried overnight at room temperature. Cells were imaged using an Axioskop2 upright fluorescence microscope equipped with an Axio CamMRm camera and AxioVision v4.8 imaging software from Zeiss (Oberkochen, Germany).
Total Internal Reflection Fluorescence Microscopy
TIRF microscopy was done as described by us previously (Wang et al. 2014). Briefly, MC3T3-E1 cells were plated on FluoroDish TM plates (World Precision Instruments, Inc, Sarasota, FL) that were coated with 1 μg/ml fibronectin (Sigma-Aldrich) and cultured as above. On day 5, cells were fixed with 3.7% formaldehyde in PBS for 10 min at room temperature. Immunofluorescence staining of the cells was done by following the same protocol as for conventional immunofluorescence microscopy as above. After the final wash after DAPI staining, samples were covered with PBS and kept in the dark at 4°C prior to analysis. Samples were observed and visualized with an incident angle of 68° using a ZeissAxio ObserverZ1 microscope in the multi-color TIRF illumination mode. Antibody controls for the TIRF analysis are presented in Supplemental Fig. 1.
Results
FXIII-A Associates with Caveolae in Differentiating Osteoblasts
To test our hypothesis that intracellular FXIII-A is associated with caveolae, we examined FXIII-A co-localization with caveolin-1 (Cav-1) in nondifferentiating (preosteoblasts) and differentiating osteoblasts. Cav-1 is a marker for caveolae, and a necessary protein for both the structure and function of caveolae (Razani et al. 2002). Conventional immunofluorescence microscopy showed only very weak Cav-1 and FXIII-A staining in medium only (M)-treated preosteoblasts, whereas differentiating osteoblasts (treated with differentiating media, DM) showed strong staining as well as clear co-localization of FXIII-A and Cav-1 in small patches (Fig. 1A). Color correlation distribution analysis that reflects the extent of co-localization is presented in Fig. 1B; the data shows that not all stainable FXIII-A is found to co-localize with Cav-1. The induction of FXIII-A upon differentiation treatment (DM media) was consistent with that observed in our previous studies (Al-Jallad et al. 2011; Piercy-Kotb et al. 2012; Wang et al. 2014).
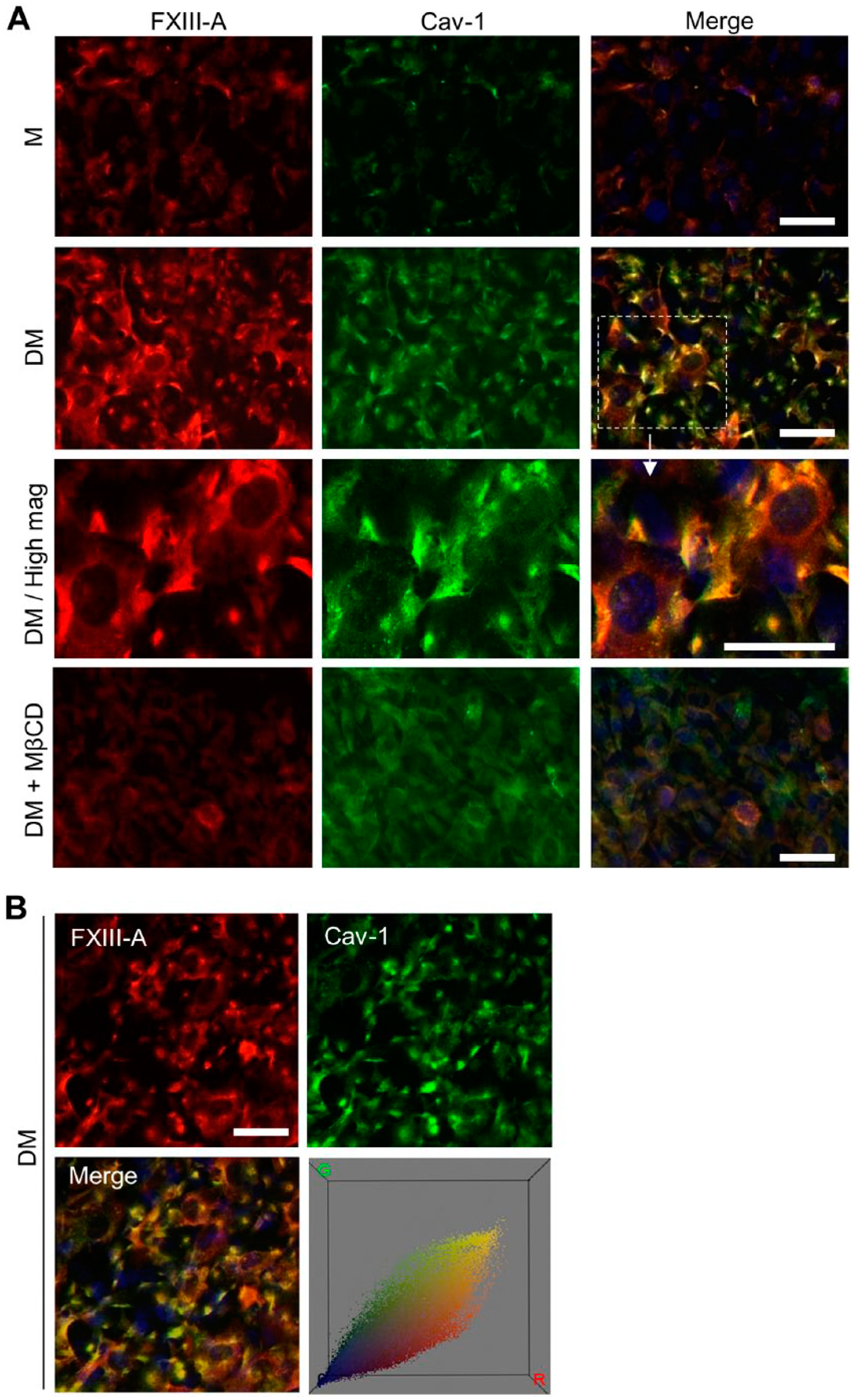
Cellular FXIII-A co-localizes with caveolin-1 in differentiating osteoblasts. (A) Immunofluorescence microscopy of FXIII-A (red) and caveolin-1 (Cav-1) (green) in osteoblasts on day 5. Medium-only treated, nondifferentiating preosteoblasts (M) showed very weak FXIII-A and Cav-1 staining, whereas differentiating osteoblasts (DM) showed strong staining and co-localization (yellow) of the two proteins in a patchy pattern (DM/High mag). Incubation of the cells with a caveolae-disrupting agent methyl-β-cyclodextrin (MβCD) for 30 min at 37°C abolished both FXIII-A and Cav-1 staining. (B) Color correlation distribution constructed from co-localization images using the Color Inspector 3D plug-in of Image J (NIH, Bethesda, MD). Scale, 20 μm.
To further investigate whether the presence of FXIII-A in the patches is dependent on the integrity of caveolae, we used a caveolae-disrupting agent, methyl-β-cyclodextrin (MβCD), which depletes cellular cholesterol and thus eliminates caveolae (Razani et al. 2002). As expected, a 30-min treatment of the cells with MβCD abolished Cav-1 staining and simultaneously eliminated FXIII-A staining in the patches (Fig. 1). To further confirm the association of FXIII-A with Cav-1, we used TIRF microscopy, which is a high-resolution, specialized technique to study cell-surface- and plasma membrane-associated molecules (Axelrod 2001; Jaiswal and Simon 2007; Mattheyses et al. 2010). As seen in Fig. 2, FXIII-A co-localized with Cav-1 in differentiating osteoblasts at day 5, as analyzed by TIRF microscopy. The higher magnification image revealed punctate and patchy caveolae staining and the clear presence of FXIII-A in these specialized, small membranous structures. Although secondary antibodies gave a slightly dotted background in TIRF microscopy (Supplemental Fig. 1), the rounded patchy staining was only observed when the primary antibody was present (Supplemental Fig. 1B). Also, to confirm that the fixation and permeabilization method used allows for staining of an integral membrane protein, we stained the cells for insulin receptor β-subunit (IR-β), which is located in the inner leaflet of the plasma membrane and known to associate with caveolae. As seen in Supplemental Fig. 2, IR-β is stained and is found in similar patches as Cav-1.
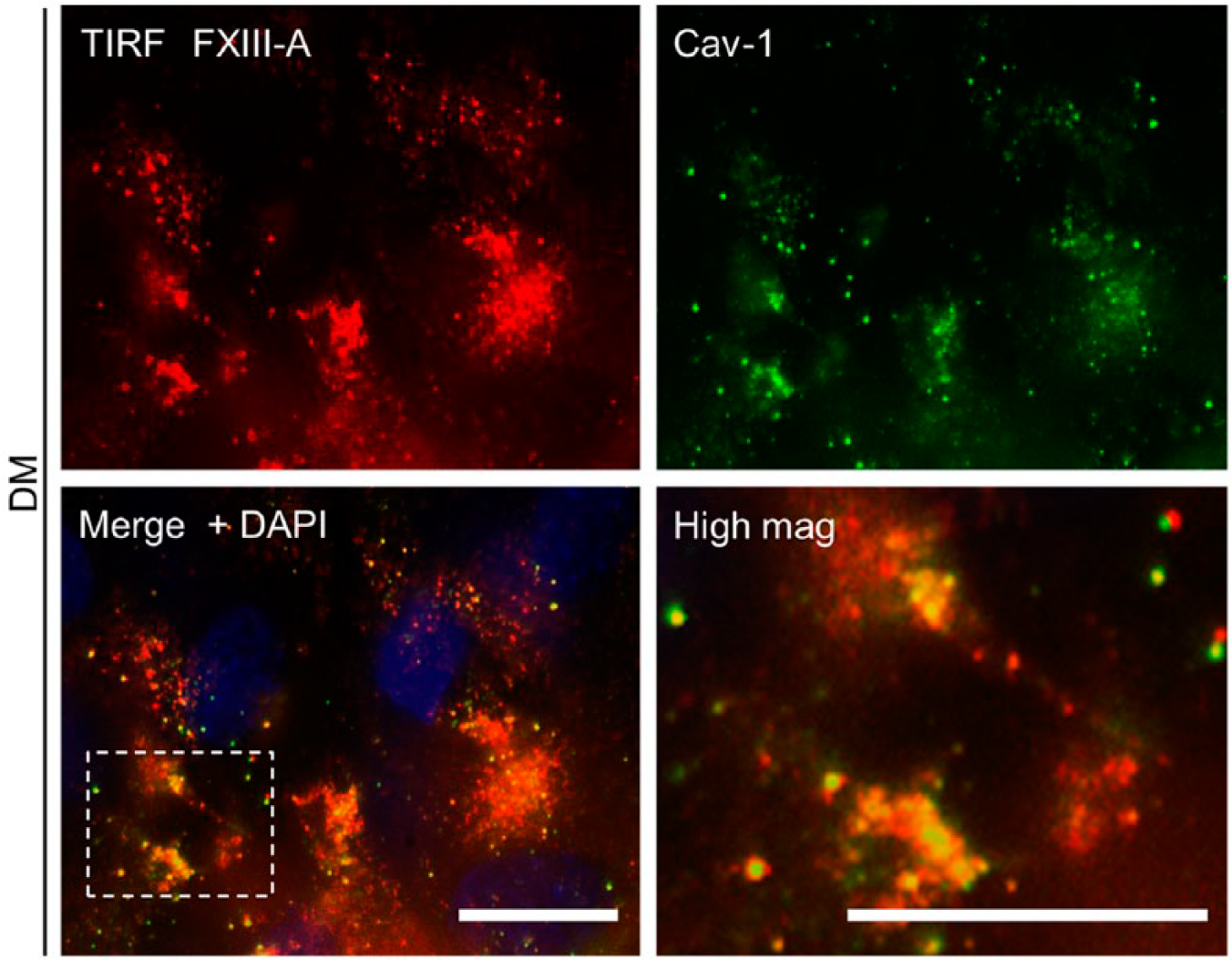
TIRF (Total Internal Reflection Fluorescence) microscopy of FXIII-A (red) and caveolin-1 (Cav-1; green) co-localization in differentiated osteoblasts on day 5. The analysis confirms the co-localization, and the higher magnification image (lower panel, right) reveals punctate caveolae staining and shows the clear presence of FXIII-A in these specialized membrane structures. Box in the lower left panel is magnified in the lower right panel. Scale, 20 μm.
To further confirm that FXIII-A is found in caveolae, we prepared caveolae-enriched cell fractions using sucrose density gradient centrifugation of osteoblast homogenates. Caveolae membranes are highly enriched in glycol-sphingo lipids and cholesterol, making them extremely light; therefore, they have a low buoyant density in sucrose (Cohen et al. 2004; Razani et al. 2002). They are also resistant to solubilization by nonionic detergents (Triton X-100, NP-40). Triton X-100 solubilizes most of the cellular proteins, which remain in the 45% sucrose layer. This leaves the caveolae and lipid rafts in the insoluble interface between the 35% and 5% sucrose layers. Further separation of the sucrose gradient into 12 analyzable 1-ml fractions showed that caveolae are found in fractions 4 and 5, as analyzed by Cav-1 detection by western blotting (Fig. 3). These two fractions have been identified as the bone fide caveolae fractions (Parolini et al. 1999). The rest of the material includes non-lipid raft-associated proteins, and Cav-1 was also found in this material. As demonstrated in Fig. 3A and 3B, FXIII-A, with a molecular weight of between 50 and 75 kDa (as we have published before for mouse tissues) (Al-Jallad et al. 2011; Myneni et al. 2014) co-purified with caveolae-enriched fractions 4 and 5 in differentiating osteoblasts. FXIII-A cannot be detected in fractions 4 and 5 of preosteoblasts, which is consistent with our immunofluorescence data (Fig. 3A). Disrupting caveolae with MβCD reduced Cav-1 levels in fractions 4 and 5 (Fig. 3C) but did not completely eliminate the detection of FXIII-A. It is possible that even the low Cav-1 levels in caveolae maintain an association with FXIII-A. Attempts to abolish caveolae completely were unsuccessful, as higher concentrations of MβCD were toxic to the cells. The fairly weak detection of FXIII-A in western blots merely reflects the quality of the antibody in this application and does not likely reflect the actual quantity of FXIII-A in these structures. The strong detection of FXIII-A in caveolae by immunocytochemistry with the same antibody strongly supports our conclusion and its presence in these structures. In summary, intracellular FXIII-A in osteoblasts associates with specialized membrane structures, caveolae, which are increased in differentiating osteoblasts. The co-localization of FXIII-A with Cav-1 (which resides on the inner membrane leaflet) is dependent on the integrity of the caveolae.
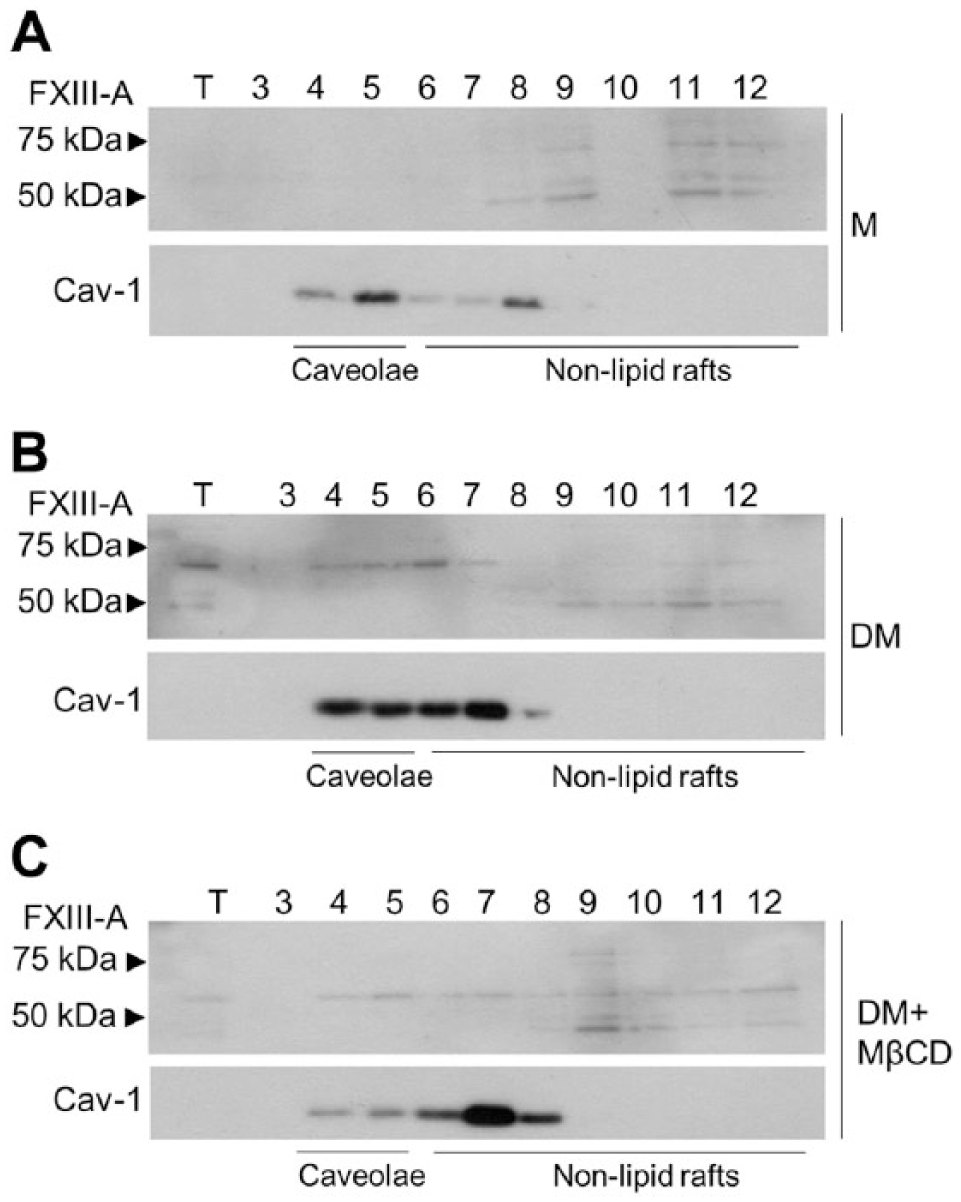
Western blotting of FXIII-A in the caveolae-enriched membrane fractions of osteoblasts. Caveolae-enriched fractions were prepared using sucrose density gradient centrifugation of osteoblast homogenates that were fractionated into 1-ml fractions and of which fractions 3–12 were analyzed by western blotting. Fractions 4 and 5 are bone fide caveolae-containing fractions, and fractions 6–12 contain non-lipid raft membranes. Caveolin-1 (Cav-1) is used as a caveolae marker; however, it can also be found in non-raft fractions. Upper blots were detected with anti-mouse FXIII-A antibody. Arrowheads point to molecular weight markers. (A) Nondifferentiating (medium only treated, M) preosteoblasts show no FXIII-A detection in fractions 4 and 5. (B) FXIII-A is detected in caveolin-enriched fractions 4 and 5 in differentiating osteoblasts (differentiating media, DM). (C) Treatment of differentiating osteoblast cultures with the caveolae-disrupting agent methyl-β-cyclodextrin (MβCD) decreased Cav-1 levels in fractions 4 and 5, and also resulted in a reduction in FXIII-A; however, the treatment did not abolish either completely.
NC9 Decreases FXIII-A Levels in Caveolae, which Increases Phosphorylation and Homo-oligomerization of Cav-1 in Caveolae-enriched Fractions and c-Src Activity
To continue to understand the function of FXIII-A in caveolae, we hypothesized that NC9—an irreversible TG inhibitor—may be able to interfere with/alter its function, as it has been shown to associate noncovalently with enzymes inside the cell (Al-Jallad et al. 2011) and it has been reported to change the conformation of TG enzymes from the closed to open conformations (Caron et al. 2012). As seen in Fig. 4A, detection of the dansyl group of NC9 by immunofluorescence microscopy showed its co-localization with Cav-1, suggesting that NC9 interacts with caveolae inside the cells. We then examined if NC9 had any effects on the levels of FXIII-A in caveolae fractions 4 and 5 on days 2, 4 and 6 of cell differentiation. Western blotting showed that NC9 decreased the presence of FXIII-A in caveolae (Fig. 4B). NC9 did not affect the total FXIII-A levels in the cells, as seen by immunoblotting of total cell extracts (Fig. 4B), nor did it change Cav-1 levels in caveolae (Fig. 4C). This strongly suggests that the association of NC9 with FXIII-A promotes the dissociation of FXIII-A from caveolae. To examine whether NC9 has effects on Cav-1 levels and/or function in caveolae, we considered that Cav-1 can be phosphorylated at Tyr14 by Src-kinases (Aoki et al. 1999), which promotes Cav-1 self-association to form noncovalent, high-molecular weight homo-oligomers that contain 14 to 16 individual Cav-1 molecules. These Cav-1 oligomers represent the functional assembly units of caveolae (Cohen et al. 2004; Parolini et al. 1999; Razani et al. 2002). Thus, we examined Cav-1 phosphorylation and homo-oligomerization in the caveolae fractions at the same time points. The analysis showed that the decreased FXIII-A levels in caveolae were associated with an increase in both homo-oligomerization and phosphorylation of Cav-1 (Fig. 4B). The Cav-1 levels in fractions 4 and 5, and in the total cell extracts, were not affected by NC9 treatment (Fig. 4B and 4C). TG2, which is also expressed by osteoblasts and co-localizes with FXIII-A (Al-Jallad et al. 2011), was not detected in the caveolae fractions (Fig. 4B); although it was clearly expressed by the cells and found in total cell extracts (Fig. 4B). TG2 also did not co-localize with Cav-1 (Supplemental Fig. 3). Taken together, these data show that noncovalent interaction of NC9 with FXIII-A interferes with its ability to associate with caveolae, and that the decreased levels of FXIII-A in caveolae affect Cav-1 phosphorylation and homo-oligomerization.
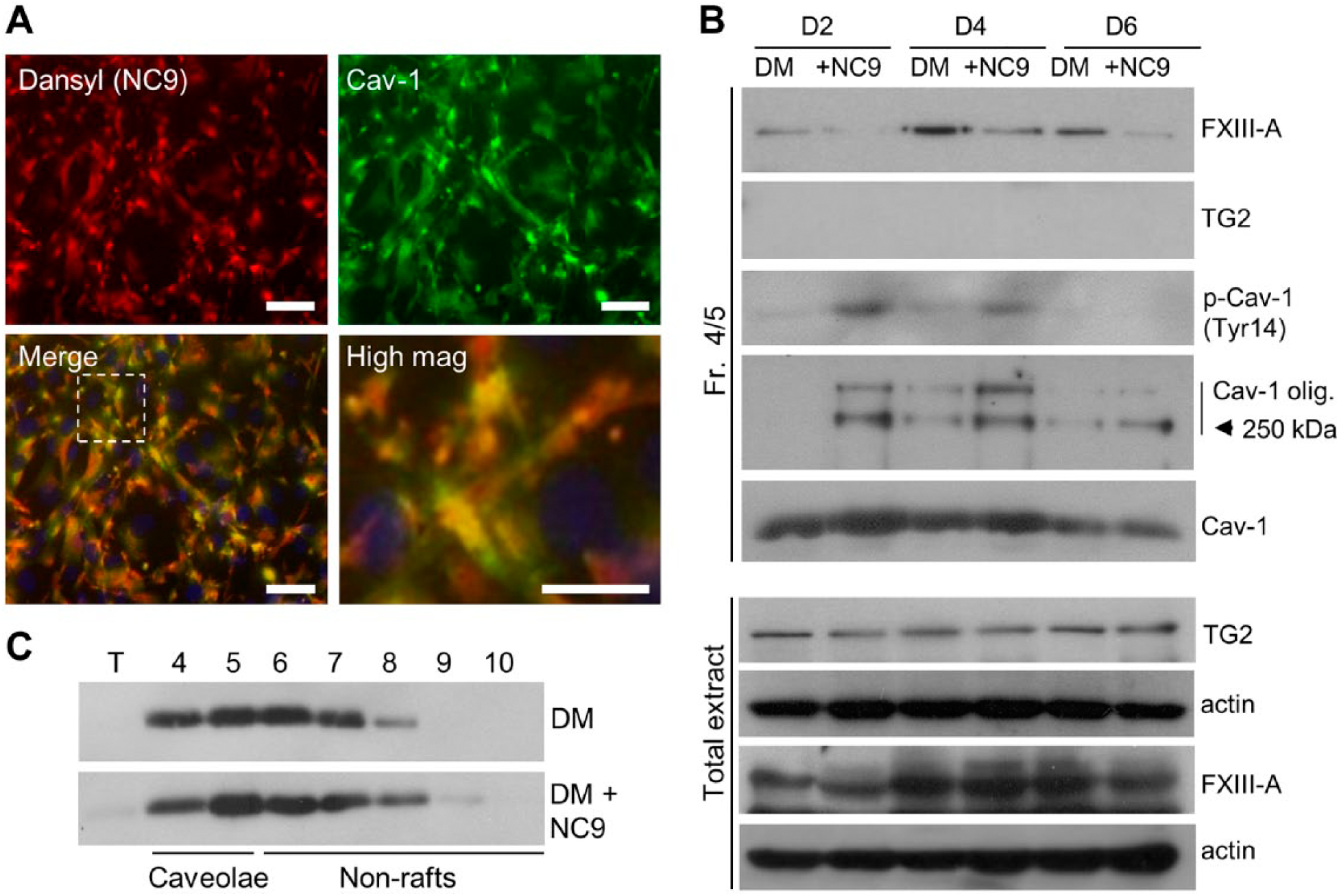
NC9 displaces FXIII-A from caveolae and affects the homo-oligomerization and phosphorylation status of Cav-1 in caveolae-enriched fractions. (A) Immunofluorescence microscopy of NC9 in differentiating osteoblasts. NC9 (dansyl, red), when administered to the osteoblast cultures, associates with caveolae (Cav-1, green) as seen by their co-localization (yellow). Scale, 20 μm. (B) Western blotting of FXIII-A levels and Cav-1 status in caveolin-enriched fractions (4/5) and total cell extracts of osteoblasts. NC9-treated cells have decreased FXIII-A levels whereas total extracts show no change in FXIII-A protein levels. The decreased FXIII-A levels in caveolae is associated with increased Cav-1 phosphorylation (p-Cav-1(Tyr14)) and increased homo-oligomerization. Total Cav-1 levels remain similar in NC9-treated and control cultures. TG2 is not detected in the caveolae fractions, but is abundant in all total cellular fractions. (C) Western blot analysis of Cav-1 from all fractions from the membrane component isolation confirmed that total levels and the distribution of Cav-1 remains the same with or without NC9 treatment.
It is known that Cav-1 is phosphorylated at Tyr14 by Src kinases (Aoki et al. 1999), which affects caveolae function and regulates osteoblast maturation (Del Pozo et al. 2005; Peruzzi et al. 2012). It has also been shown that the internalization of certain integrin-regulated membrane domains requires the same phosphorylation of Cav-1 on Tyr14 (Del Pozo et al. 2005). Cav-1 phosphorylation is generally orchestrated by c-Src kinases that are activated by autophosphorylation at Tyr424 (in mouse) (Roskoski 2005); this is promoted by c-Src association with Cav-1. To examine whether c-Src kinase activity and signaling are increased by NC9 treatment, we analyzed c-Src activation/phosphorylation at Tyr424. The data presented in Fig. 5A show a clear increase in p-c-Src in total cell extracts upon NC9 treatment levels, thus implying activation. To examine if c-Src is responsible for the observed NC9-induced increased phosphorylation and homo-oligomerization of Cav-1, we applied a Src kinase inhibitor PP2 to the cells and examined its effects on the NC9-mediated increase of p-Cav-1 and Cav-1 oligomers on day 2 of cell differentiation. As seen in Fig. 5B, PP2 treatment markedly decreased the NC9-induced phosphorylation and homo-oligomerization of Cav-1 down to the levels of control cultures, without affecting total Cav-1 levels. Analysis of c-Src and p-c-Src levels in the caveolae-enriched fractions on days 2, 4 and 6 of cell differentiation showed that only nonphosphorylated c-Src was associated with caveolae and that NC9 increased this association (Fig. 5C). This supports the concept that FXIII-A acts as a blocking molecule between the Cav-1 and c-Src interaction. The observation that no p-c-Src was seen in caveolae fractions also suggests that once autophosphorylated, p-c-Src dissociates from caveolae and may continue its cytosolic signaling function (Fig. 5C). Indeed, the increased cytosolic p-c-Src induced by NC9 was associated with increased phosphorylation of the c-Src downstream target, p-Akt (Ser473). The p-Akt levels were decreased with c-Src inhibitor (PP2) treatment (Fig. 5D). These data strongly suggest that NC9-mediated displacement of FXIII-A from caveolae stimulates c-Src activation and the induction of related signaling pathways. c-Src was found as two bands on day 6. This may be linked to its fragmentation and perhaps inactivation when preosteoblasts mature into osteoblasts around that time point (Miura et al. 2004; Park et al. 2004).
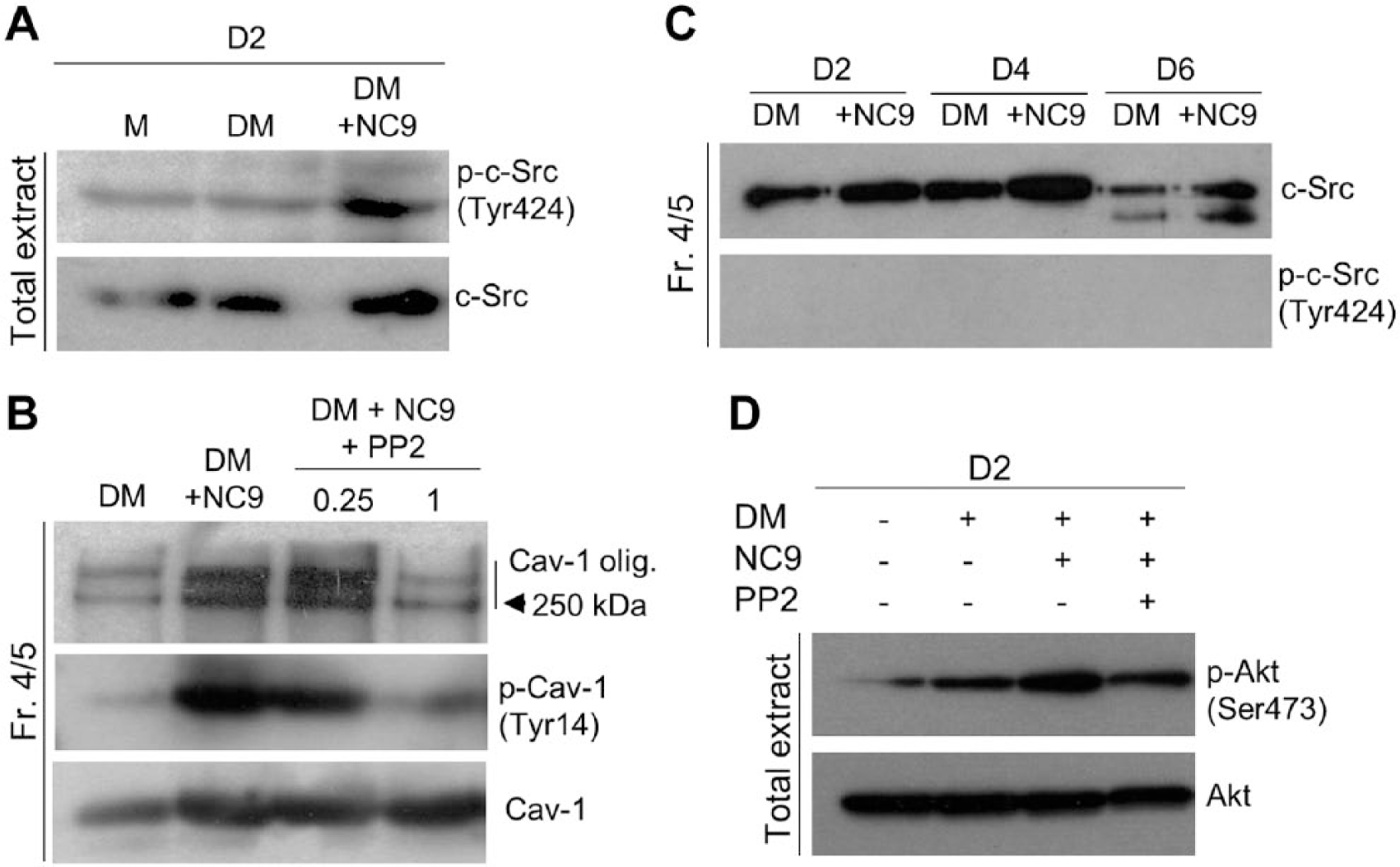
NC9 promotes Src-kinase phosphorylation and activation. (A) Analysis of phosphorylation levels of c-Src (p-c-Src (Tyr424)) in total extracts of differentiating (DM) osteoblasts show increased phosphorylation in NC9-treated cells as compared with controls. (B) Inhibition of Src kinase activity in NC9-treated cultures with PP2 markedly decreased both phosphorylation and homo-oligomerization of Cav-1 in caveolae-enriched fractions (Fr. 4/5) to the levels of control cultures, without affecting total Cav-1 levels, thus showing the involvement of c-Src in these modifications of Cav-1. (C) c-Src is associated with caveolae-enriched fractions (Fr. 4/5) and its presence there is increased in NC9-treated, differentiated osteoblasts. p-c-Src(Tyr424) does not remain associated with caveolae and is mostly found in the cytosol, as seen in the above analysis of total osteoblast extracts. (D) Analysis of phosphorylation of a c-Src downstream signaling target, Akt, in differentiating osteoblasts (DM) treated with NC9 show an increase in p-Akt compared with that in controls. Src kinase inhibitor PP2 reduces the NC9-induced increase in Akt phosphorylation.
Caveolae Show No Transamidase Activity
Finally, we examined whether FXIII-A has transamidase activity in caveolae. For this, we used monodansylcadaverine (MDC), a TG activity probe that is capable of penetrating the cell membrane and incorporating into substrate proteins in the presence of an active enzyme. As seen in Fig. 6A, the dansyl group of MDC did not co-localize with Cav-1, as assessed by immunofluorescence microscopy. MDC also was not incorporated covalently into any substrates in caveolae-enriched fractions 4 and 5, as seen by the lack of dansyl detection in western blots of these fractions yet strong labeling in the total cell extracts (Fig. 6B); the isolation of caveolae does not involve harsher extraction conditions that would render all labeling undetectable. Furthermore, cells treated with MDC throughout differentiation showed no changes in Cav-1 homo-oligomerization as compared with NC9-treated cells (Fig. 6C), suggesting that transamidation activity is not required to regulate Cav-1 homo-oligomerization. This suggests that FXIII-A in caveolae is likely inactive as a transglutaminase.
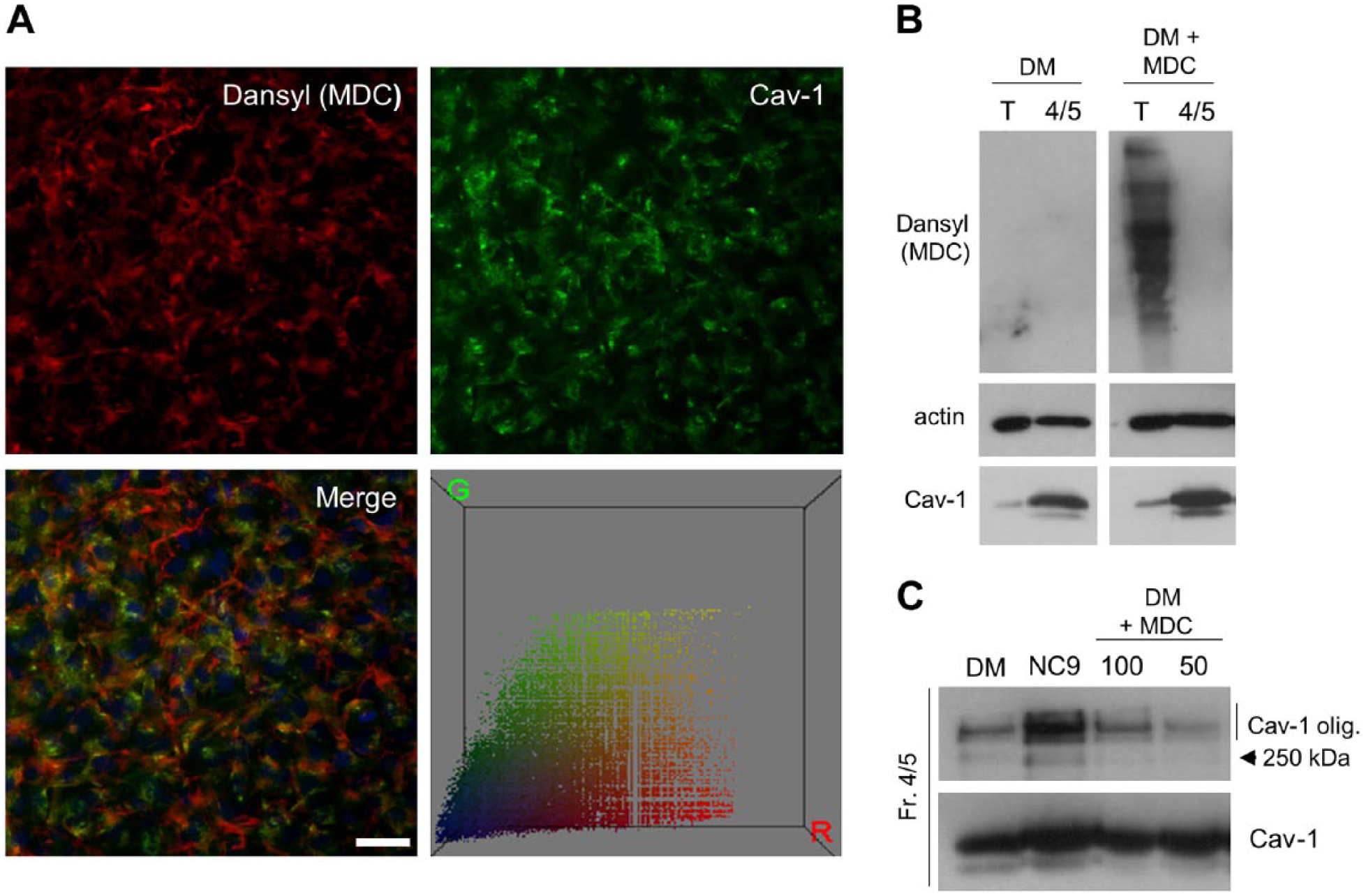
No transamidase activity is detected in caveolae. (A) In situ transglutaminase (TG) activity was analyzed in differentiating osteoblast cultures by growing the cells in the presence of monodansylcadaverine (MDC), as described in the Materials & Methods. Covalent incorporation of MDC into TG substrates to reveal enzyme activity was visualized by detecting the dansyl-group of MDC by immunofluorescence microscopy. No co-localization of the MDC/dansyl-group (red) with caveolin-1 (Cav-1) (green) was observed, although MDC was incorporated into cell layers, mostly to extracellular matrix fibrils. Color correlation distribution was constructed using the Color Inspector 3D plug-in of Image J and shows no correlation. Scale, 20 μm. (B) Western blotting of MDC in total osteoblast extracts (T) versus caveolae fractions (4/5) of differentiating osteoblasts (DM) showing no detection of covalently linked MDC in caveolae, even though it was clearly detected in the total extracts. Actin and Cav-1 were used as loading controls for total cell extracts and caveolae-enriched fractions, respectively. (C) Cells treated with MDC from day 0 to day 5 with two concentrations (50 μM and 100 μM) showed no changes in Cav-1 homo-oligomerization as compared with NC9-treated cells, which showed an increase in the formation of homo-oligomers.
Discussion
Osteoblasts express FXIII-A TG, which is found both in cells and as secreted to the extracellular matrix. In our previous work, we showed that the secreted, extracellular soluble FXIII-A promotes plasma fibronectin stabilization, crosslinking and fibrillogenesis into matrix, whereas the cell-associated FXIII-A is located on the outer plasma membrane of osteoblasts where its crosslinking action is linked to the regulation of microtubule stabilization, which, in turn, is associated with the secretion of type I collagen (Al-Jallad et al. 2006, 2011; Piercy-Kotb et al. 2011, Wang et al. 2014). Both these functions involve its crosslinking activity. In this study, we show that FXIII-A also co-localizes inside the cells with a major caveolae protein, Cav-1, which is known to associate with the inner leaflet of plasma membrane caveolae. Caveolae are specialized membrane structures that regulate endocytosis and signal transduction via clustering receptors and many cell signaling molecules into a confined space. FXIII-A is associated with caveolae specifically in differentiated MC3T3-E1 osteoblasts, and its presence there is dependent on the integrity of caveolae, as demonstrated by their co-reduction from the plasma membrane after treatment with the caveolae-disrupting agent, MβCD. Caveolae-associated FXIII-A does not appear to have transamidating activity; however, the TG inhibitor, NC9, interacts with the enzyme causing what appears to be displacement of FXIII-A from the caveolae. This displacement may be mediated by an NC9-induced conformational change and the opening of the FXIII-A structure, or by NC9 attacking the exposed FXIII-A active site that may also be used for its potential interaction with Cav-1. We further showed that the decreased FXIII-A levels in NC9-treated caveolae was associated with increased phosphorylation and subsequent homo-oligomerization of Cav-1, which was mediated by the increased activity of c-Src. It has been suggested Cav-1 binds to and maintains Src family kinases in an inactive configuration, and that Cav-1 homo-oligomer formation modifies the interaction of Cav-1 with Src kinases in a manner that may lead to the release of these kinases from caveolae (Li et al. 1996; Wei et al. 1999). Furthermore, it has also been shown that Src kinase-mediated phosphorylation of Cav-1 increases Cav-1 homo-oligomerization, suggesting that Cav-1 and Src kinases amplify each other’s activation (Del Pozo et al. 2005; Li et al. 1996; Wei et al. 1999). Our results above suggest that the presence of cellular FXIII-A in caveolae attenuates both Cav-1 homo-oligomerization and c-Src kinase activity, which indicates that FXIII-A may act as a suppressor (and a switch) of their functions. The precise mechanism of how FXIII-A regulates this remains unknown; however, it may involve a direct interaction between FXIII-A and Cav-1 or an interaction via a mediator protein. We hypothesize that this FXIII-A: i) inhibits cytosolic c-Src from associating with Cav-1, ii) inhibits Cav-1-mediated activation (autophosphorylation) of c-Src, and iii) inhibits p-c-Src-mediated Cav-1 phosphorylation. This may be viewed as FXIII-A in caveolae acting as a blocking molecule that inhibits c-Src and Cav-1 interaction to maintain c-Src in an inactive state. The action of FXIII-A in this process does not seem to require its TG activity.
Our previous studies have focused on the role of extracellular FXIII-A (both secreted and plasma membrane-associated) in matrix deposition and stabilization in osteoblast cultures. The present study extends the possible roles for FXIII-A in bone biology, describing for the first time in osteoblasts a potential function for intracellular FXIII-A. The observation that FXIII-A regulates c-Src activity is well-aligned with the results that we have obtained previously using NC9 as an inhibitor of osteoblast culture maturation, which results in an arrest of matrix deposition and osteoblast maturation. c-Src belongs to the SRC family of nonreceptor tyrosine kinases, which includes at least ten members (Lyn, Fyn, Lck, Hck, Fgr, Blk, Yrk, Gfr, Yes and c-Src) all sharing high homology in their domain structure (Brown and Cooper 1996). It has been reported that c-Src is involved in the regulation of osteoblast differentiation, where decreased c-Src expression enhances osteoblast differentiation and bone formation, and c-Src kinase activity is essential for osteoclast function and bone resorption (Marzia et al. 2000; Miyazaki et al. 2004); this makes c-Src a major negative regulator of bone mass. c-Src-deficient osteoblasts exhibit a cell autonomous alteration that leads to increased osteoblast numbers and increased bone formation in vivo, as well as accelerated osteoblast differentiation and associated increased matrix production (Marzia et al. 2000). Here, consistent with these previous findings, we have shown that Akt phosphorylation is increased as part of c-Src activation. p-Akt plays a critical role in controlling the survival and apoptosis of cells, as well as in insulin signaling, which integrates bone remodeling and energy metabolism in osteoblasts (Ferron et al. 2010; Hermann et al. 2000; Tonks et al. 2013). Interestingly, cell-surface TG2 has also been reported to inhibit c-Src activity via its role in promoting integrin clustering on the cell surface (Condello et al. 2013; Janiak et al. 2006). We have shown previously that TG2 and FXIII-A co-localize on the osteoblast surface (Al-Jallad et al. 2011) and a number of studies have shown tight functional links and synergy between the roles of TG2 and FXIII-A in bone and cartilage, as well as compensation in the case where one is absent (Deasey et al. 2012; Johnson et al. 2008; Nurminskaya and Kaartinen 2006; Tarantino et al. 2009). Thus, it is tempting to speculate that the potential synergistic role of the two enzymes in osteoblasts could be linked to the regulation of c-Src activity and subsequent signaling in a manner where TG2 would modulate its activity on the outer leaflet of the cell membrane, and FXIII-A would modulate its activity on the inner leaflet of the cell membrane.
Caveolae have been linked to cellular events that can both promote or inhibit osteoblast differentiation (Fulzele et al. 2010; Li et al. 1996; Marzia et al. 2000; Yamamoto et al. 1998). The role of caveolae and Cav-1 in the negative regulation of bone formation is based on solid animal studies (Rubin et al. 2007). Skeletal phenotyping of Cav-1-knockout mice has shown increased trabecular number and thickness. The authors speculated that Cav-1 deficiency leads to increased osteoblast differentiation, suggesting that Cav-1 helps to maintain osteoblast progenitors in a less-differentiated state. Links to the positive regulation of osteoblast function are based on the observations that caveolae sequester bone morphogenetic protein (BMP)-2 and vitamin D receptors (Baker and Tuan 2013; Bonor et al. 2012; Chen et al. 2012; Huhtakangas et al. 2004) on the osteoblast plasma membrane and regulate insulin signaling, at least in mesenchymal stem cells (Baker and Tuan 2013). Because of the potential dual role of caveolae in both the promotion and inhibition of osteoblast differentiation, it is plausible that additional factors, such as FXIII-A, are required to modulate these specific functions. In future studies, we will continue to address and refine the role of FXIII-A in the regulation of other caveolae-related functions.
In summary, our study suggests that intracellular FXIII-A is associated with caveolae in osteoblasts where its presence inhibits c-Src activation and c-Src-mediated Cav-1 phosphorylation, homo-oligomerization and function. This function for FXIII-A is novel, and appears not to involve its enzymatic transamidase activity, adding to the known noncatalytic function of TG enzymes in other cell types. The data from this paper, together with our previous work, adds a new modality as to how intracellular FXIII-A may contribute to the regulation of bone formation.
Footnotes
Acknowledgements
We would like to thank Dr. Erika Wee of the Advanced BioImaging Facility (ABIF) of McGill University’s Life Science Complex for excellent assistance in using TIRF microscopy, and Dr. Simon Tran (McGill University) for the use of his immunofluorescence microscope. This study was supported by a grant to MTK from the Canadian Institutes of Health Research (CIHR). SW received studentship stipends from the China Scholarship Council (CSC) and Faculty of Dentistry of McGill University.
Competing Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
SW designed the study and performed immunofluorescence microscopy, TIRF microscopy, western blotting, carried out all data analysis and wrote the manuscript. MTK designed the study and edited the manuscript. Both authors have read and approved the final manuscript.
Funding
This study was supported by grants to MTK from the Canadian Institutes of Health Research (CIHR) (MOP-119403) the CIHR Institute for Musculoskeletal Health and Arthritis (IMHA) (IMH-89827). SW received studentship stipends from the China Scholarship Council (CSC) and Faculty of Dentistry, McGill University.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.