
Abstract
Colorectal cancer (CRC) is a heterogeneous disease and a major contributor to world cancer mortality rates. Molecular subtypes of CRC have become standards for CRC classification and have established prognostic potential. Here, we attempt to corroborate and provide further insight pertinent to the fragile histidine triad (FHIT) gene in microsatellite instable (MSI), microsatellite stable (MSS), and CpG island methylator phenotype (CIMP) CRC subtypes. We employed array comparative genomic hybridization and multiplex ligation-dependent probe amplification (MLPA) techniques to survey genomic aberrations in FHIT gene and their effects on FHIT protein expression using immunohistochemistry (IHC) in a CRC cohort. We further studied FHIT protein expression by IHC in a larger CRC cohort defined for its mismatch repair (MMR) protein expression and genomic methylation profiles. Our results show FHIT genomic deletions centered in exons 4 and 5 in most of MSI-CRC samples. Moreover, we confirmed the significant association of FHIT protein expression diminution (p=0.035) with MSI-CRC. In the larger cohort, reduced FHIT protein expression was significantly associated with CIMP-high subtype of CRC (p=0.009) and loss of PMS2 protein expression (p=0.017). We conclude that FHIT expression may be a valuable marker for CRC subtyping, and its diagnostic, prognostic, and therapeutic potential should be perused.
Keywords
Introduction
Colorectal cancer (CRC) is the third most common cancer in both sexes and the fourth most frequent cause of cancer mortality in the world (Ferlay et al. 2010). There are three distinguishable molecular subtypes in CRC: microsatellite stable/chromosomal instable (MSS/CIN), microsatellite instable (MSI), and the CpG island methylator phenotype (CIMP) subtypes (Boland and Goel 2010). MSS-CRC makes up 75% of hereditary cases and 85% of sporadic cases (Lengauer, Kinzler, and Vogelstein 1998). MSI is associated with 20% to 25% of hereditary CRC but is reported in only 15% of sporadic CRC (Ionov et al. 1993; Lengauer, Kinzler, and Vogelstein 1998). CIMP-CRC overlaps with some MSI cases as a result of epigenetic silencing of the mismatch repair gene hMLH1 (Esteller et al. 1998). There are several distinct genetic markers that distinguish these CRC subtypes and are significantly associated with their pathogenesis and progression. The fragile histidine triad (FHIT) gene encodes a tumor suppressor protein that belongs to a nucleotide binding HIT protein family that is known to have enzymatic functions. HIT proteins are characterized by the hydrophobic histidine triad motif that is critical for substrate binding and cleavage. FHIT is a ubiquitously expressed dinucleoside 5′,5″′-P1,P3-triphosphate (Ap3A) hydrolase (Barnes et al. 1996). The FHIT gene is located on chromosome 3p14.2 and is composed of 10 exons that contribute differently to at least 8 known splice variants. Coding exons of the gene span exons 5 to 9 and express a 16.8 kDa small protein. Chromosomal location of FHIT overlaps with common fragile site FRAB3 that is subject to genotoxic and replicative stress (Ohta et al. 1996; Durkin et al. 2008). FHIT genetic aberrations and abnormal expression have been reported in different types of cancers (Wali 2010). FHIT aberrations in CRC have been extensively confirmed and are thought to associate more with MSI-CRCs’ loss of differentiation and escape from apoptotic control (Elnatan et al. 1999; Mimori et al. 2006; Cao et al. 2007). The exact role that FHIT plays in CRC remains inconclusive as reports speculate different roles for the protein in CRC initiation and progression (Chen et al. 1997; Elnatan et al. 1999; Hao et al. 2000; Mori et al. 2001; Dong et al. 2005). Here, we set out to interrogate a panel of cohorts comprising the three molecular subtypes of CRC for FHIT genetic aberrations and expression. Our data show that FHIT gene deletion is a common event in CRC. However, FHIT expression diminution or loss appears to be influenced by the extensive promoter methylation program manifested in CIMP-high CRC cases.
Methods
Clinical CRC Samples
One hundred and sixteen sporadic CRC formalin-fixed paraffin-embedded (FFPE) samples were used for the array CGH part of this study. The clinicopathological characteristics of this cohort are shown in Table 1. For methylation analysis, we utilized another cohort composed of 131 CRC cases. Genomic DNA was isolated from macrodissected FFPE tumor tissues according to an established protocol (Bosso and Al-Mulla 2011). Briefly, hematoxylin- and eosin-stained sections were used to determine regions with the highest number of tumor cells for each case. The slides were deparaffinized in xylene and rehydrated in 100% and 95% alcohol. Areas with greater than 80% tumor were macrodissected from 5 to 7 sections of 10-μm thickness using a sterile needle. DNA was extracted using the Gentra Puregene Tissue Kit (Qiagen; Hilden, Germany). All steps were carried out according to the manufacturer’s protocol. Isolated genomic DNA was assessed for concentration and quality using spectrophotometry and 1.5% agarose gel electrophoresis.
Demographic and Clinicopathological Characteristics of Colorectal Cancer Cohort Included in Array-Comparative Genomic Hybridization Analysis.

Abbreviations: MSI, microsatellite instable; MSS, microsatellite stable.
Microsatellite Stability Profiling
Microsatellite fragment analysis was performed on FFPE-extracted DNA using MSI Analysis System Version 1.2 kit (Promega; Madison, WI). Spectral calibration on the Applied Biosystems 3130 genetic analyzer was carried out using the Powerplex Matrix Standards 3100/3130 kit (Promega). The MSI Analysis System includes fluorescently labeled primers for co-amplification of seven markers: five mono-nucleotide repeat markers (Bat-25, BAT-26, NR-21, NR-24, and MONO-27), and two penta-nucleotide repeat markers (Penta C and Penta D). The mono-nucleotide markers are used to determine the MSI status, whereas penta-nucleotide markers are used to detect potential sample mix-up by confirming that tumor and matching normal samples are from the same individual. DNA concentrations of 10 to 20 ng from normal and tumor samples were subjected to a fluorescent PCR-based assay. The allelic profiles of microsatellite markers generated by amplification of normal and tumor DNA were compared to determine microsatellite instability. Internal lane size standard ILS600 was added to amplified samples to assure accurate sizing of alleles. A loading cocktail was prepared by mixing the ILS600-PCR product with highly deionized formamide and was denatured prior to loading onto the 3130 Genetic Analyzer for capillary electrophoresis. The input sample’s fragment separation output data were analyzed using GeneMapper software version 4.0. CRC samples were classified as MSS if no marker showed any length variation compared with its matching normal mucosa. When two or more of the markers showed length mutation in CRC compared with its matching normal mucosa, the CRC sample was labeled as MSI-high.
Array CGH Hybridization and Analysis
Array-comparative genomic hybridization (aCGH) was carried out on 116 CRC samples following our standard published protocol (Al-Mulla 2011). To summarize, 2 µg of tumor DNA and pooled sex-matched reference DNA (Promega) were fragmented using sonication in a water bath. The universal linkage system (ULS) Cy3 and Cy5 (Agilent Technologies; Santa Clara, CA) dyes were used, according to the manufacturer’s protocol, to label the DNA. Differentially labeled DNA were purified by Agilent-KREApure columns (Agilent Technologies). The purified labeled tumor and matching reference samples were hybridized onto Human Genome CGH array 244 A slides (Agilent Technologies) in SureHyb Chambers (Agilent Technologies) for 40 hr at 60C. Washing and scanning were performed according to the manufacturer’s protocol. The slides were scanned immediately on an Agilent microarray scanner, at 5-μm resolution, to minimize the impact of environmental factors on signal intensities. Data were extracted from microarray image files using Feature Extraction Software (version 9.5) and analyzed using Nexus Copy Number software (BioDiscovery; Hawthorne, CA). Quality values ranged between 0.05 and 0.40, and to minimize false positive calls and random copy number variations, a Fast Adaptive State Segmentation Technique (FASST2) with a stringent significance threshold of 5.0 × 10−6 was used to determine copy number aberrations. Moreover, a systematic method termed Genomic Identification of Significant Targets in Cancer (GISTIC) was used to identify biologically significant copy number aberrations in these samples (Beroukhim et al. 2007).
MLPA Assay and Analysis
MLPA assay was performed according to the manufacturer’s standard protocol (MRC-Holland; Amsterdam, The Netherlands) using Tumor loss P294-A1 MLPA probemix (lot no. 1008). FFPE-extracted DNA was processed for hybridization, ligation, and amplification steps according to standard protocols without any modification (Monticone et al. 2012). The amplified PCR product was mixed with formamide, CEQ-600 marker, and the MLPA PCR sample was added to each well of a 96-well plate and topped with a drop of mineral oil. Fragment separation was carried out on a CEQ8000 Genetic Analysis System (Beckman Coulter; Brea, CA) according to the manufacturer’s protocol. CSV files generated from runs were imported into the Coffalyser.NET software, which is free MLPA data analysis software created by MRC-Holland. CRC samples’ MLPA data were normalized and quality checked against the MLPA data of several reference controls. Normalization was based on tumor analysis methods custom designed by the manufacturer to accommodate variation levels associated with tumor samples. MLPA result reports, including descriptive statistics, ratios, 95% confidence intervals, and predictions, were exported to Microsoft Excel software data sheets (Microsoft; Redmond, WA).
Methylation Analysis
One hundred and thirty-one CRC cases were subjected to promoter methylation assessment of the samples’ DNA according to our previously established protocol (Dallol et al. 2011). In short, 500 ng of tumor DNA was treated with sodium bisulfite using the Epitect Bisulfite Kit (Qiagen). SssI-treated human genomic DNA (methylated control) and unmethylated DNA were used as controls. DNA methylation was quantified in seven CIMP-specific promoters—IGF2, CDKN2A, SOCS1, CRAPB1, NEUROG1, CACNA1G, and RUNX3—via MethyLight real-time PCR using an ABI 7500 instrument (Applied Biosystems; Foster City, CA). PCR amplifications were performed for COL2A1 in triplicate and a standard curve was generated for bisulfite-converted human genomic DNA at four different concentrations (in a 5-fold dilution series). The percentage of methylated reference DNA (referred to as methylation index) at a specific locus was calculated in each case. Cases where ≥4 methylated markers were detected were assigned as CIMP-high, whereas the presence of 1 to 3 methylated markers was designated as CIMP-low. CIMP-0 was given to cases where none of the markers were detected.
Immunohistochemistry
FFPE CRC sections of 4-μm thickness were used for immunodetection. Sections were deparaffinized and dehydrated in xylene and graded alcohol series, respectively. Antigen retrieval was achieved by microwaving in 10 mM sodium citrate buffer at pH 6 for 12 min. Peroxidase was blocked with 3% hydrogen peroxide, and 5% normal goat serum was used to block nonspecific protein binding. Incubation of mouse monoclonal anti-MLH1 (clone ZM001; Zymed, San Francisco, CA; working dilution: 1/80), mouse monoclonal anti-MSH2 (clone FE11; Zymed; working dilution: 1/100), mouse monoclonal anti-PMS2 (clone A16-4; BD Pharmingen, San Diego, CA; working dilution: 1/100), mouse monoclonal anti-MSH6 (clone 44/MSH6; BD Transduction Laboratories, San Jose, CA; working dilution: 1/150), and rabbit polyclonal anti-FHIT (clone ZP54, cat. no. 71-9000; Invitrogen, Carlsbad, CA; working dilution: 1/500) primary antibodies was performed overnight at 4C. Sections were immunostained with anti-rabbit biotinylated secondary antibody (IgG; Vector Laboratories, Burlingame, CA) for FHIT antibody, and biotinylated anti-mouse for MLH1, MSH2, PMS2, and MSH6 antibodies. The VECTASTAIN Elite ABC system (Vector Laboratories) was used for detection according to the manufacturer’s instructions. Visualization was performed using DAB chromogen (Dako; Carpinteria, CA). Negative control sections were incubated with species-specific secondary antibody with omission of the primary antibody. Sections were counterstained with hematoxylin, dehydrated, and mounted with Dpex media.
Scoring Analysis
For all antibodies used, scoring accounted for both representation of the areas and intensities of the stains as previously described (Al-Mulla et al. 2006). Briefly, the score is the sum of the percentage of positive cells (1: less than 25% positive cells; 2: 26–50% positive cells; and 3: more than 50% positive cells) and the staining intensity (0: negative; 1: weak; 2: moderate; 3: strong). Sums between 0 and 2 were scored as negative, sums of 3 and 4 were scored as weakly positive, and sums of 5 and 6 were scored as positive.
Statistical Analysis
Two-sided Chi-square or Fisher’s exact tests were used for statistical associations. Detection of cancer recurrence in the forms of metastasis or local recurrence was used as an endpoint measurement for disease-free survival. Log-Rank test was used for the measurement of significance for Kaplan-Meier Survival curves. All statistical analyses were performed using SPSS statistical package v17 (SPSS Inc.; Chicago, IL).
Results
Genomic Aberrations of FHIT Using aCGH and MLPA
Patients’ demographics and CRC profiles are shown in Table 1. Microsatellite stability was successfully assayed for 108 CRC cases (93%), 8 cases having non-discriminatory results. The Agilent aCGH platform incorporated 188 different probes dispersed throughout the full length of the FHIT gene. FHIT-related aberrations on the chromosome 3p arm were found in 42 (36.2%) of 116 assayed CRC samples (Fig. 1b). These aberrations, which were largely deletions, involved most of the FHIT gene with a minimal common region of deletion involving exon 5 (Fig. 1a). To confirm these data, 44 selected CRC cases were used in MLPA analysis, which is an assay of higher resolution when compared to aCGH. The MLPA test panel had two different FHIT probes, one for exon 4 and another for exon 5 (Fig. 1c). Thirty-four (77%) of the 44 cases had deletions involving either exon 4 or 5. Among those 34 cases, 14 (41%) cases had deletions in the FHIT gene, as detected by aCGH (p=0.01). In 18 (53%) of the 34 cases with MLPA-detected FHIT deletions, the deletions centered on exon 5. Among these 18 cases, only 10 (56%) had corresponding FHIT deletion data generated by the aCGH analysis. These data illuminate a significant correlation (p<0.0001) for the FHIT gene deletion obtained by two independent techniques. Given its higher sensitivity, MLPA detected more FHIT deletions involving exons 4 and 5 compared to aCGH.
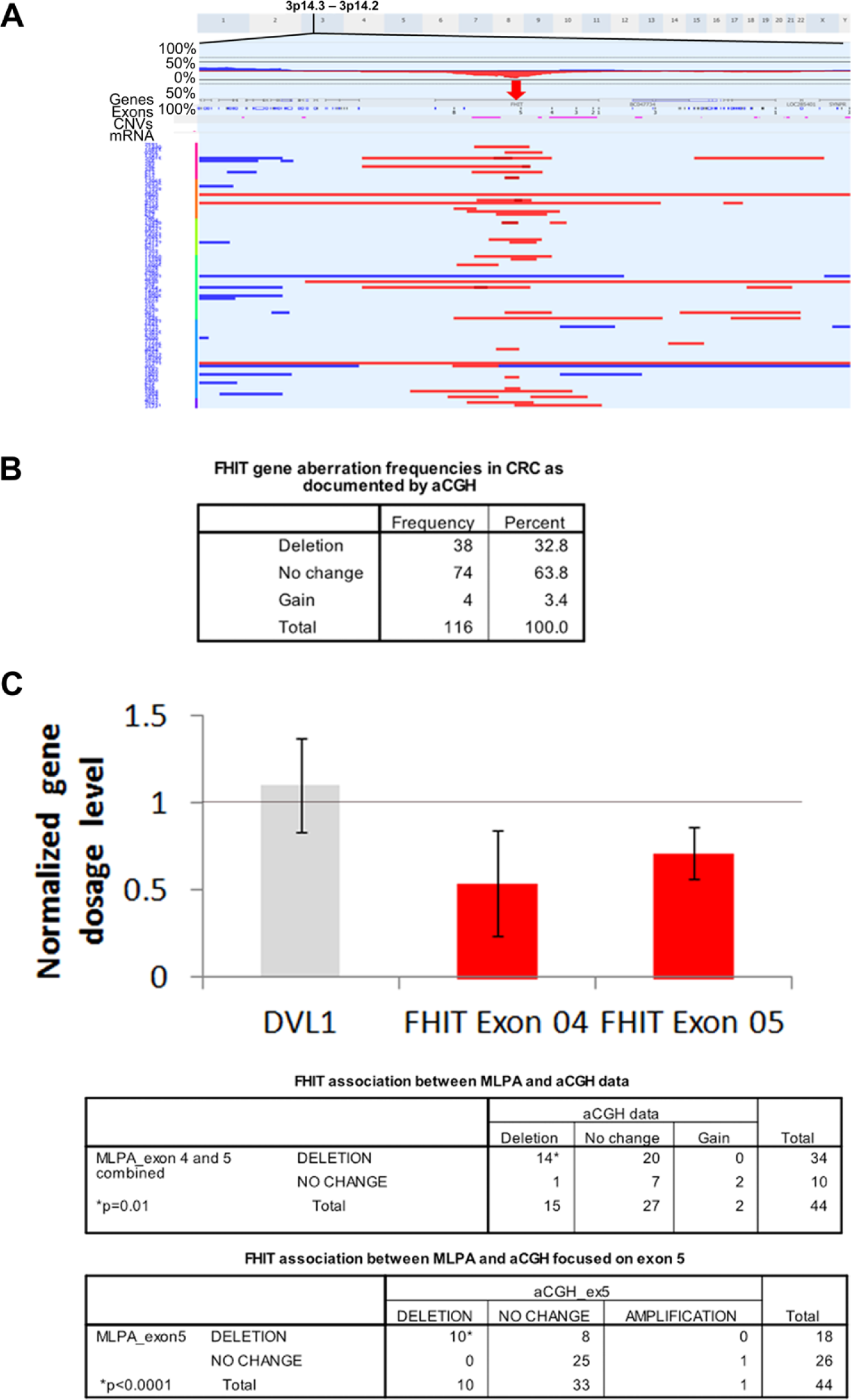
Fragile histidine triad (FHIT) gene deletions in colorectal cancer (CRC) as documented by array-comparative genomic hybridization (aCGH) and multiplex ligation-dependent probe amplification (MLPA). (A) aCGH focused on the FHIT gene (marked with red arrow). FHIT exons are numbered. Red horizontal lines indicate the deleted regions with each row representing a patient. Dark red lines demarcate homozygous deletion events. Blue horizontal lines indicate gains. (B) Frequencies of FHIT gene aberrations in 116 cases of CRC as identified by aCGH. (C) Normalized FHIT exons 4 and 5 dosage using MLPA with DVL1 gene as an example of an unaltered gene dosage (crossing 1-line mark). The MLPA data generated bar chart and tables confirm the deletions of the FHIT gene found using aCGH and the highly significant correlation between MLPA and aCGH data sets. *p values were calculated using Fisher’s exact test.
FHIT Expression in CRC Subtypes
To examine whether FHIT gene deletions influenced its protein expression, we examined FHIT protein expression by immunohistochemistry. Loss of FHIT protein expression has been reported frequently in CRC. Moreover, a loss of expression event has been shown to be significantly associated with MSI-CRC. Using this cohort, FHIT staining was successfully performed on 73 (63%) of the 116 cases that were part of the aCGH cohort (Fig. 2). FHIT expression was positive in 35 (48%) of 73 cases and weak or negative in 38 (52%) of 73 cases. Interestingly, there was no significant correlation observed between FHIT gene deletion and its protein expression both at aCGH or MLPA levels (Supplementary Tables 1 and 2). Most CRC tissues that lost FHIT protein expression (23/38, 60.5%) had no deletion events recorded using aCGH. For MLPA, this proportion was less (7/18, 39%). Our data indicate that other events, in addition to genetic deletions, are responsible for silencing the FHIT gene. FHIT gene promoter methylation is a frequent event in cancer and this is especially interesting given the fact that FHIT protein loss was associated with MSI-CRC, which is itself associated with the CIMP-high subtype. In this cohort, of the 116 CRC samples, only 68 (59%) had interpretable results for microsatellite stability and FHIT IHC. Among these, 78.5% of MSI cases were negative or stained weakly for FHIT, confirming the significant association between the diminution/loss of this protein and MSI reported previously (Table 2).
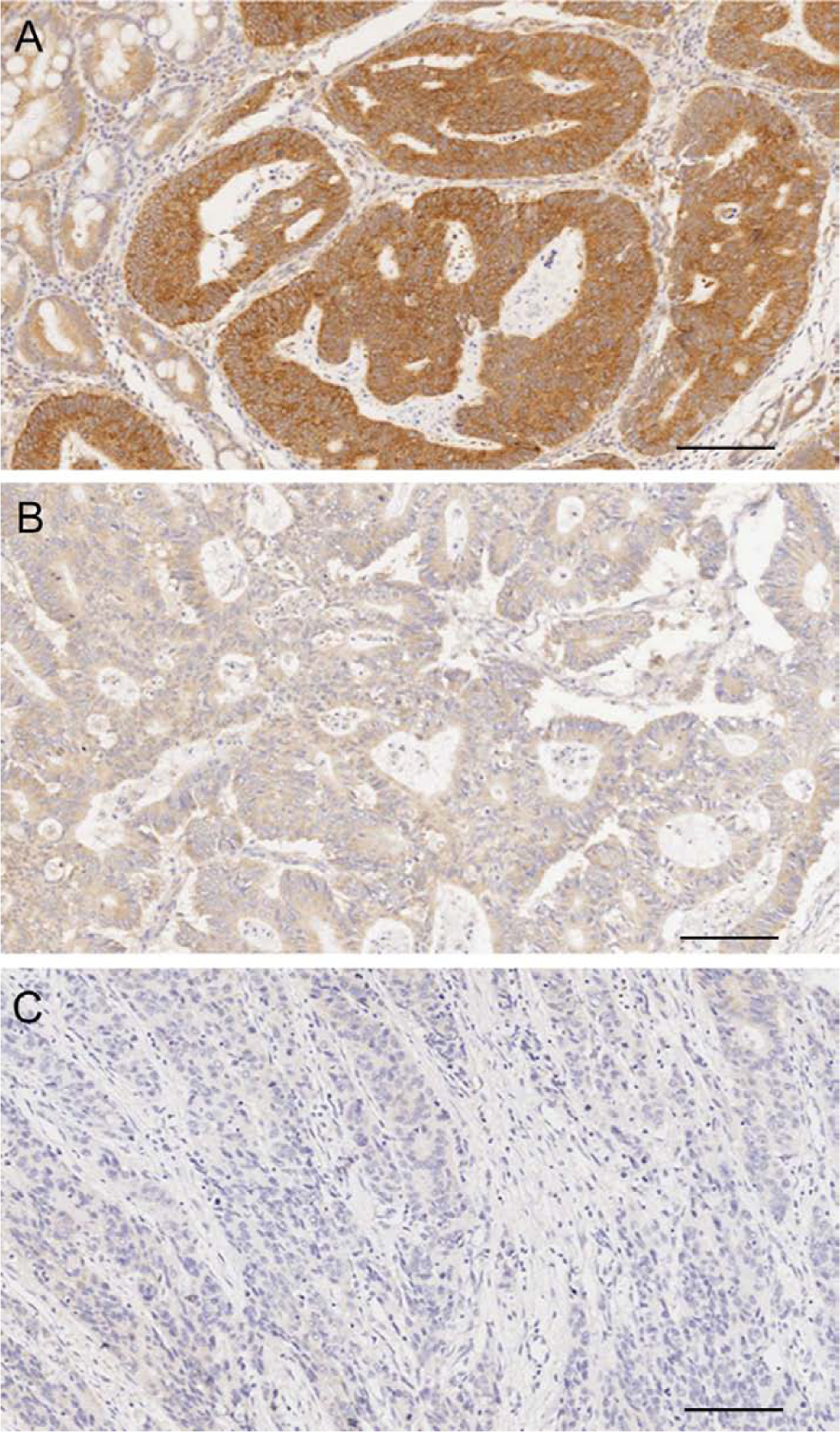
Examples of colorectal cancer (CRC) tissues immunostained for fragile histidine triad (FHIT) protein expression representing variable staining intensities used in scoring analysis. (A) Positive cytoplasmic staining of invasive CRC surrounded by normal mucosa. (B) Weakly positive staining in colorectal carcinoma. (C) Negative staining in a poorly differentiated colorectal cancer. Bars = 100 µm.
Fragile Histidine Triad (FHIT) Protein Expression’s Association with Microsatellite Instable Colorectal Cancer (CRC) Subtype.

Abbreviations: IHC, immunohistochemistry; MSI, microsatellite instable; MSS, microsatellite stable.
The data represent 68 of the 116 CRC cases that had successful FHIT IHC and microsatellite stability typing. *p=0.035.
We next used an independent and larger CRC cohort to confirm the association between FHIT and MSI with particular focus on mismatch repair protein expression. Moreover, given the link between FHIT methylation and cancer, we wished to examine the association between FHIT and CIMP in colorectal cancer, which has not been studied before.
FHIT Reduced or Lost Protein Expression Is Associated with MSI and CIMP-High in CRC
Table 3 shows the clinicopathological characteristics in a new cohort of 131 patients with CRC in relation to FHIT protein expression. In this cohort, MMR was assessed using allelotyping and immunohistochemistry against four MMR proteins, namely, hMLH1, MSH2, MSH6, and PMS2 (Fig. 3). As shown, FHIT protein expression loss was significantly associated with MSI, confirming previous data presented here by us and others (Hadaczek et al. 2001; Mimori et al. 2003; Sarli et al. 2004; Leopoldo et al. 2008). Interestingly, FHIT protein expression diminution was seen in 90% and 83% of CRC cases with hMLH1 and MSH2 deficiency, respectively, and was significantly associated with loss of PMS2 protein (Table 3). Moreover, FHIT protein reduction was documented in 71% of cases with a low level of global methylation and all 10 cases of CIMP-high (Table 3 and Supplementary Table 3). Our data indicate that global methylation and deletion mutation play an integral part in silencing the FHIT gene in CRC. It is well established that CIMP-high is usually associated with the MSI genotype in CRC. Therefore, it may be of interest to explore whether the FHIT protein loss in CIMP-high CRC also had MSI. Unfortunately, MSI data were available for only 5 of the 10 CIMP-high cases, and 4 of the 5 cases belonged to the MSI subtype whereas 1 case was MSS (Supplementary Table 4). It is worthy to note that FHIT protein loss was also documented in unmethylated MSI CRC cases (Supplementary Table 4), and it would be interesting to compare whether CIMP-high/MSI/FHIT-negative cases differ clinicopathologically from the CIMP-negative/MSI/FHIT-negative CRC subtype (Fig. 4). CRC with FHIT gene deletions had similar overall or disease-free survival compared to cases with maintained FHIT gene (data not shown). Equally, at the protein level, loss or diminished FHIT protein expression did not influence disease-free survival in CRC (Fig. 4a). Moreover, limiting the data to MSS genotypes confirms the limited role of FHIT protein loss in influencing disease progression in CRC (Fig. 4b). Future work may benefit from confirming these data in a larger cohort.
Association between Fragile Histidine Triad (FHIT) Protein Expression and Colorectal Cancer Clinicopathological Characteristics.
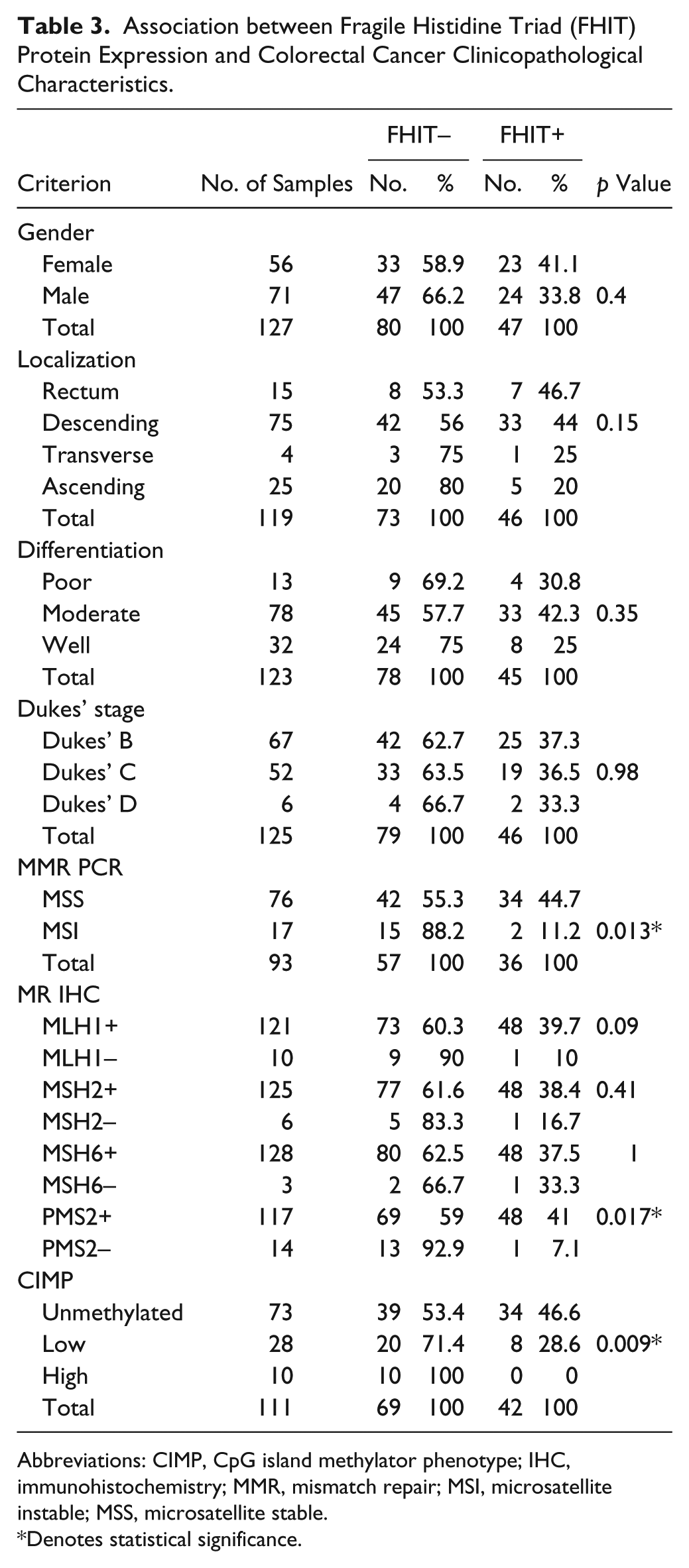
Abbreviations: CIMP, CpG island methylator phenotype; IHC, immunohistochemistry; MMR, mismatch repair; MSI, microsatellite instable; MSS, microsatellite stable.
Denotes statistical significance.
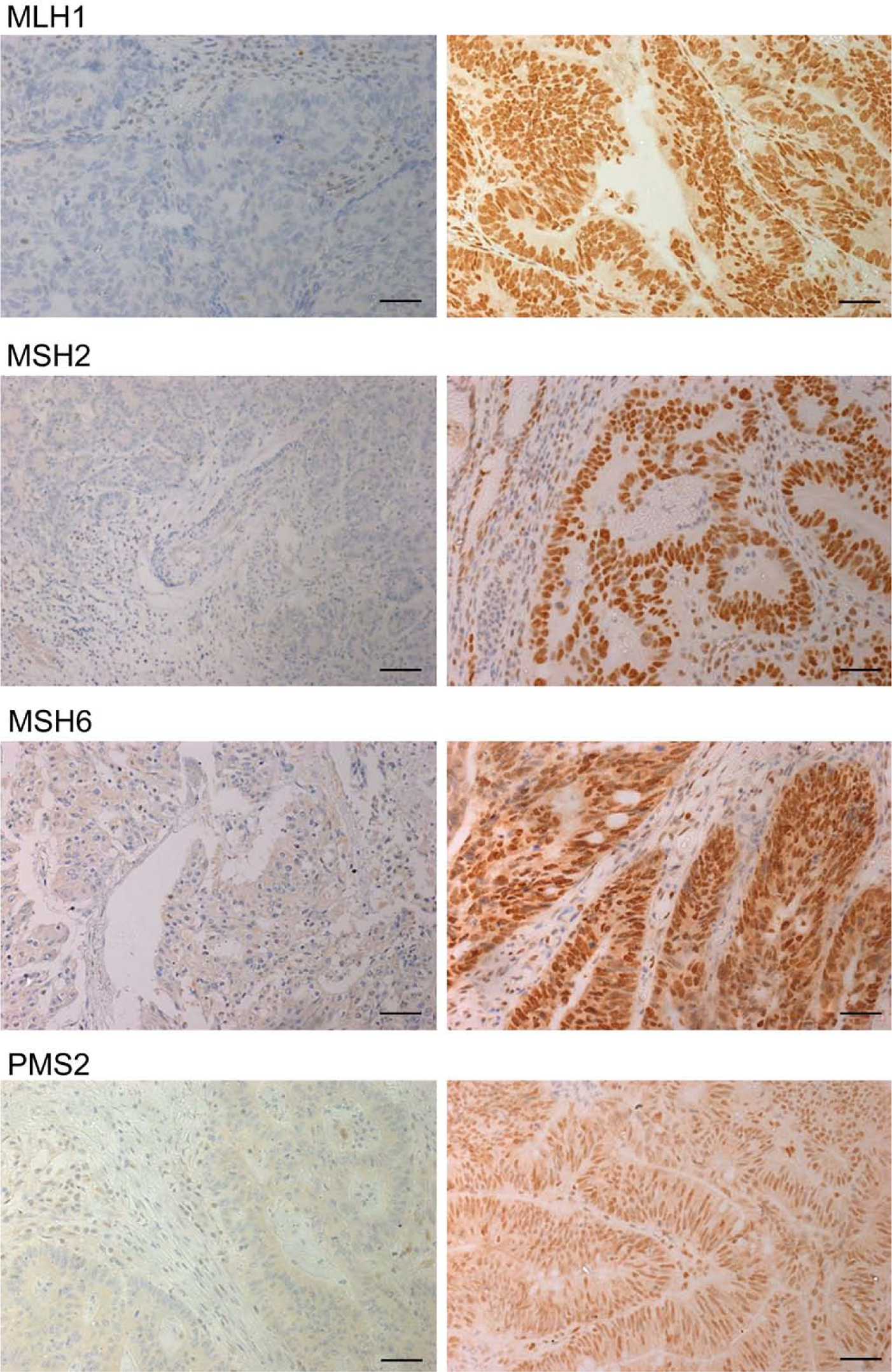
Representative images of colorectal cancer tissues immunostained for the expression of mismatch repair proteins. The panels discriminate negative (left) and positive (right) staining results for MLH1, MSH2, MSH6, and PMS2, respectively, all of which exhibit nuclear staining. Bars = 20 µm.
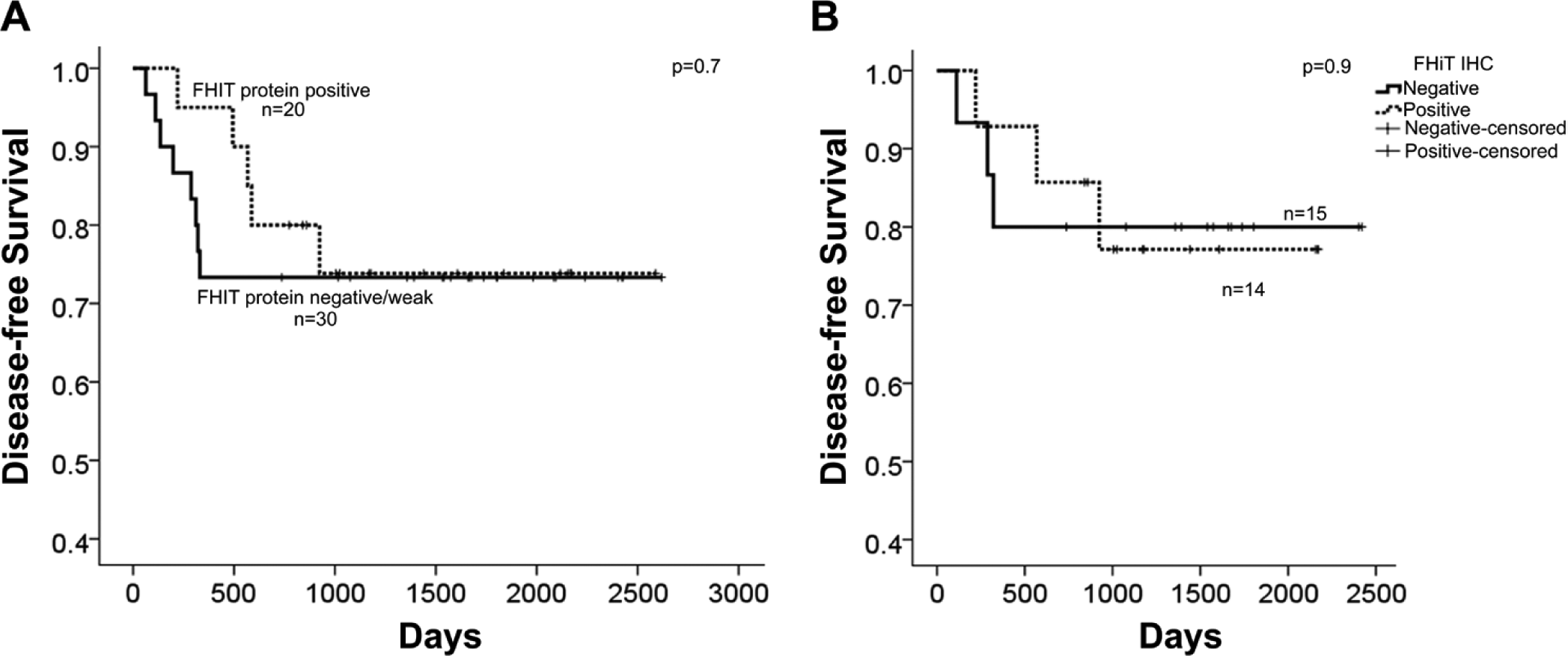
Disease-free survival of patients with colorectal cancer (CRC) in relation to fragile histidine triad (FHIT) protein expression. (A) Kaplan-Meier survival curves for 50 of the 131 cases where both FHIT protein staining and follow-up information were available. (B) Survival data limited to microsatellite stable CRC in relation to FHIT protein expression. p values indicate the Log-Rank test.
Discussion
Classification of CRCs into molecular subtypes is reported to be of prognostic value (Jass 2007; Sanchez et al. 2009; Kang 2011). In this study, we aimed to dissect the intricate genomic landscape of CRC using molecular techniques of varied sensitivities and resolutions. We set out to further catalogue recurrent markers that are distinctly associated with a CRC molecular subtype and might be of prognostic value. Here, we chose to focus our report on the FHIT protein because it has been the subject of an ongoing debate since its reported mutational discovery in cancer (Ohta et al. 1996; Virgilio et al. 1996). Mounting reports of FHIT mutations and reduced expression associated with CRC loss of differentiation and progression are contradicted by limited reports that state the opposite (Thiagalingam et al. 1996; Elnatan et al. 1999; Sarli et al. 2004; Azzoni et al. 2007; Wierzbicki et al. 2009). Our findings conform with reports of FHIT mutations and loss of expression specifically associated with MSI-CRC. Translational studies have been reported outlining a correlation between FHIT’s loss of expression and genomic instability. First, the fact that FHIT is located in a common fragile site on chromosome 3 itself contributes to genomic instability by being prone to chromosomal recombination and changes that mitigate genomic control (Debatisse, El Achkar, and Dutrillaux 2006; Ruiz-Herrera, Castresana, and Robinson 2006). Second, FHIT is a designated tumor suppressor protein that regulates apoptosis and stress responses by interacting with mitochondrial proteins (Pichiorri, Palumbo, et al. 2008) and affecting the expression of cell-cycle proteins (Pichiorri, Ishii, et al. 2008). Last, the functional association of FHIT with inflammatory responses to cancer invasion by inhibiting Prostaglandin E2 expression would limit downstream cellular proliferation events (Mimori et al. 2006). These three attributes of FHIT make it a sound target for mutations in cancer cells to escape cellular control and attain characteristic genomic instability.
The majority of assayed FHIT deletions involved exon 5, which is the first coding exon of the FHIT gene. Therefore, it is reasonable to deduce that such deletions would ultimately affect the expression of FHIT protein resulting in protein loss. However, that is not the case in our results as FHIT protein staining was positive in at least half of the cases with deletions. Possible explanation for such a discrepancy is that the FHIT gene might be influenced by nuclear control to undergo biallelic expression to combat ensuing pathogenetic events related to colon cancer progression. Thus, the second allele’s compensatory expression might be a late event and remnants of it persist in assayed tissues despite its consequent silencing. It is possible that FHIT biallelic mutations and/or silencing are a threshold CRC progression event in a subtype of CRC similar to reported MYH gene biallelic mutations in CRC (Lefevre et al. 2010). In addition, residual FHIT staining could simply be due to altered protein half-life. One intriguing result emanating from our data is that all CRC with a CIMP-high signature expressed little or no FHIT protein. No prior studies have elaborated on or studied the association between FHIT protein loss or diminution and CIMP. CIMP-high CRC with MSI is considered a predictor of good prognosis (Hawkins et al. 2002; Barault et al. 2008; Lee et al. 2008; Kim et al. 2009), whereas CIMP-high alone (Simons et al. 2013) or in association with MSS/CIN CRC genotypes bestows a poor prognostic index in patients (Kim et al. 2009; Dahlin et al. 2010). Our modest data sets showed no significant association between FHIT gene deletions or its protein loss and disease-free survival in both cohorts. The small number of cases also curtails further stratification of the data, and we therefore suggest that a larger study may need to be undertaken to confirm our findings. It may be beneficial to our understanding of CRC classification to compare the MSI/CIMP-high/FHIT-negative, MSI/CIMP-negative/FHIT-negative, MSS/CIMP-high/FHIT-negative, and MSS/CIMP-negative/FHIT-negative subgroups with each other (Fig. 5) in a large data set. Nevertheless, FHIT protein loss did not influence disease-free survival when the data were limited to the MSS CRC subtype, confirming the limited utility of FHIT protein expression as a predictor of disease-free survival. Current CIMP analyses involve a panel of genes with CpG islands in their promoter regions prone to methylation (Ogino et al. 2006; Ogino et al. 2007). According to our results and the specific FHIT promoter methylation reports of others (Dong et al. 2005; Roa et al. 2008), we suggest including FHIT protein immunohistochemistry as an additional parameter for recognizing CIMP-high CRC.
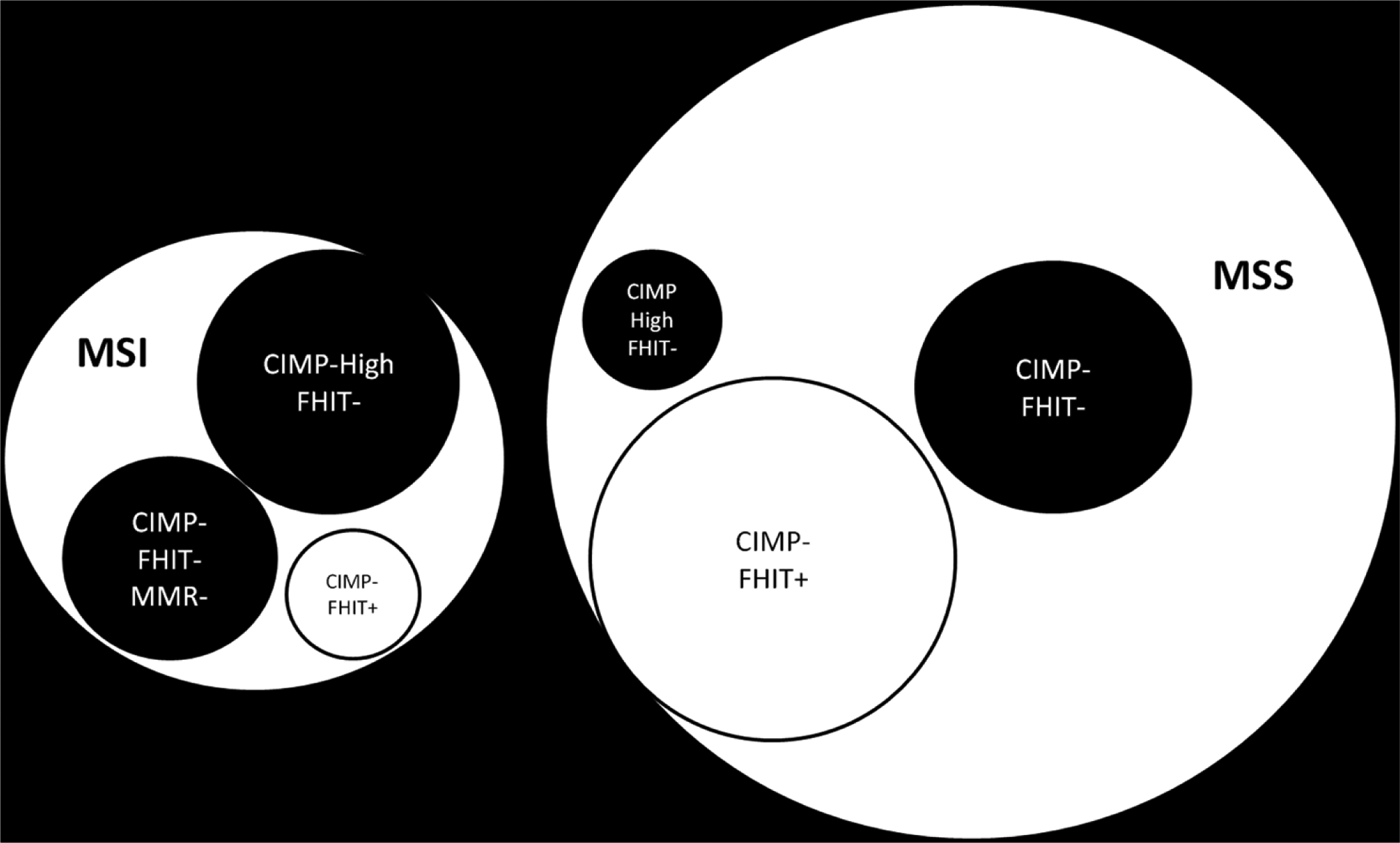
Diagrammatic representation of various subclasses of colorectal cancer (CRC) in relation to microsatellite stability, CpG island methylator phenotype, and fragile histidine triad status. The circle sizes reflect the percentages of the subclasses seen in our cohort and black parametric circles represent different subclasses of CRC that may be of particular interest to follow-up on with a larger study.
An interesting finding was the significant correlation between PMS2 and FHIT expression. Previous reports have shown a correlative expression of MSH2 and MLH1 with FHIT (Mori et al. 2001; Andachi et al. 2002; Azzoni et al. 2007), a finding that we could not reproduce in our sampled cohort. PMS2 heterodimerizes with hMLH1 and functions at replicative forks to screen for mismatched pairing and to facilitate repair when needed by making single stranded breaks. Recruitment of Polymerase III to the site of repair is also believed to be guided by the MMR system through direct interaction with the PMS2-hMLH1 heterodimer. Failure to repair damages would instigate apoptosis, a process that is regulated by FHIT, as loss of FHIT relieves apoptotic stress. It is possible that FHIT plays a critical role in gastrointestinal (GI) cellular lineages’ DNA replication control, since p53 does not seem to be effective in warding off the effects of replicative stress in the pathogenesis of GI cancer (Kitamura et al. 2001; Gorgoulis et al. 2005; Yasugi et al. 2008). The specific association of high methylation with FHIT low expression would allow for the accumulation of aggressive genomic silencing that would provide the basis for tumor progression and invasion. Our data provide supportive evidence for using FHIT in cancer therapy to target specific molecular subtypes of CRC (Ishii et al. 2001).
Footnotes
Acknowledgements
We dedicate this work in memory of Dr. Mahmoud Yousef Abdul Rahim Al-Ali, a colleague and friend who supported researchers worldwide limitlessly and selflessly.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Kuwait Foundation for the Advancement of Sciences grant 2011-1302-06.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.