
Abstract
Malignant tumors and chronic inflammatory diseases induce angiogenesis by overexpressing vascular endothelial growth factor A (VEGF-A/VPF). VEGF-A-induced pathological angiogenesis can be mimicked in immunoincompetent mice with an adenoviral vector expressing VEGF-A164 (Ad-VEGF-A164). The initial step is generation of greatly enlarged “mother” vessels (MV) from preexisting normal venules by a process involving degradation of their rigid basement membranes. Immunohistochemical and Western blot analyses revealed that versican, an extracellular matrix component in the basement membranes of venules, is degraded early in the course of MV formation, resulting in the appearance of a versican N-terminal DPEAAE fragment associated with MV endothelial cells. The protease ADAMTS-1, known to cleave versican near its N terminus to generate DPEAAE, is also upregulated by VEGF-A in parallel with MV formation and localizes to the endothelium of the developing MV. The authors also show that MMP-15 (MT-2 MMP), a protease that activates ADAMTS-1, is upregulated by VEGF-A in endothelial cells in vitro and in vivo. These data suggest VEGF-A initiates MV formation, in part, by inducing the expression of endothelial cell proteases such as ADAMTS-1 and MMP-15 that act in concert to degrade venular basement membrane versican. Thus, versican is actively processed during the early course of VEGF-A-induced pathological angiogenesis.
Vascular endothelial growth factor A/vascular permeability factor (VEGF-A/VPF) is a multifunctional growth factor and cytokine that is primarily responsible for the pathological angiogenesis of malignant tumors, healing wounds, and chronic inflammation (Dvorak 2002; Dvorak et al. 1995; Ferrara 1999). Pathological angiogenesis can be conveniently mimicked by injecting an adenoviral vector expressing VEGF-A164 (Ad-VEGF-A164) into any of several tissues of immunoincompetent (e.g., nude) mice. Such injections result in local expression of VEGF-A164 at high tumor-like levels with consequent induction of a multistep angiogenic response that closely mimics that found in tumors (Nagy et al. 2007). The initial step (1–5 days) involves the transformation of preexisting venules into greatly enlarged, thin-walled, pericyte-poor, highly permeable “mother” vessels (MV). MV formation is initiated by degradation of venular basement membrane collagen IV and laminin, with consequent pericyte detachment and several-fold expansion of vascular size (Chang SH et al. 2009). Subsequently, from 10 to 14 days, MV evolve into at least two distinctive types of “daughter” vessels—namely, glomeruloid microvascular proliferations (GMP) and vascular malformations (VM; Nagy et al. 2007). Each of these vessel types—MV, GMP, and VM—is common in autochthonous human cancers.
The breakdown of venular basement membranes, as well as the vascular hyperpermeability and resulting tissue edema associated with MV formation early in pathological angiogenesis, suggested that changes of a broader nature were taking place that might involve processing of other matrix proteoglycans such as versican. Versican is produced by vascular smooth muscle cells (Chang Y et al. 1983; Lemire et al. 1999; Schönherr et al. 1991; Yao et al. 1994) as well as by vascular endothelial cells (Cattaruzza et al. 2002; Kinsella et al. 1997) and is deposited in all three layers of the vessel wall, as well as being a component of vascular basement membranes (Bode-Lesniewska et al. 1996). Versican can be degraded by a number of proteases, including matrilysin (Halpert et al. 1996), plasmin (Kenagy et al. 2002), and members of the A Disintegrin And Metalloproteinase with ThromboSpondin motifs (ADAMTS) family (Apte 2009; Kenagy et al. 2006), especially ADAMTS-1 in human aorta (Sandy et al. 2001). ADAMTS-1 is synthesized as a proprotein and requires activation by removal of a prodomain by furin, followed by removal of the thrombospondin (TSP) repeats by matrix metalloproteinases (MMPs, mainly MMP-15/MT-2MMP; Rodriguez-Manzaneque et al. 2000; Russell et al. 2003).
To investigate the processing of versican in pathological angiogenesis, we made use of our well-established Ad-VEGF-A164 model in nude mice. We also investigated the changes of two proteases, ADAMTS-1 and MMP-15, which are known to have roles in versican processing (Kenagy et al. 2006; Rodriguez-Manzaneque et al. 2000; Russell et al. 2003; Sandy et al. 2001). We demonstrate here that vascular basement membrane versican undergoes striking degradation early in the course of Ad-VEGF-A164-induced angiogenesis during MV formation, as demonstrated both by the loss of staining for intact, microvessel-associated versican and by the appearance of a specific, ADAMTS-1-induced versican degradation product, the DPEAAE fragment, in MV endothelial cells. We also show that VEGF-A induced the expression of ADAMTS-1 and MMP-15 in cultured endothelial cells and that expression of both proteases increased in vivo in parallel with MV formation. Furthermore, ADAMTS-1 was localized to developing MV endothelial cells. Subsequently, as MV evolved into daughter GMP and VM, versican was synthesized anew. Together, these results indicate that versican degradation and subsequent resynthesis are regulated in the course of VEGF-A-induced angiogenesis and that versican processing may have a critical role in pathological angiogenesis.
Materials and Methods
Antibodies and Reagents
Ab1032 and Ab1033, rabbit anti-mouse polyclonal antibodies directed against the glycosaminoglycan (GAG)-α or GAG-β domains of versican, were purchased from Chemicon International (Temecula, CA). The antibody against the ADAMTS-1-generated fragment of versican, DPEAAE, was a rabbit polyclonal antibody to versican V0/V1 Neo (αDPE) from ABR–Affinity BioReagents (Golden, CO) (Sandy et al. 2001). Two anti-ADAMTS-1 antibodies, one directed against the carboxy terminal end and the other against the amino terminal region, were purchased from Cedarlane Laboratories Limited (Ontario, Canada). Anti-MMP-15 antibodies were from USBiological (Swampscott, MA). Chondroitin ABC lyase was purchased from MP Biomedicals, LLC (Aurora, OH).
Animal Model and Cell Culture
The Ad-VEGF-A164 angiogenesis model was performed as previously described (Nagy et al. 2007; Nagy et al. 2008; Nagy et al. 2002; Pettersson et al. 2000). Briefly, 5 × 108 pfu of Ad-VEGF-A164 or control virus Ad-LacZ, both in 50 µl, was injected intradermally into the flank skin of female athymic nude mice (NCI). Tissues were collected at various time points after adenovirus injection and snap frozen in liquid nitrogen for RNA or protein extraction. All animal studies were approved by the Institutional Animal Care and Use Committee of the Beth Israel Deaconess Medical Center. Human dermal microvascular endothelial cells (HDMVECs) isolated from fresh human foreskin as described (Richard et al. 1998) were provided by the Center for Vascular Biology Research (CVBR) Cell Biology Core and used at passages 4 to 6.
Western Blots
Protein homogenates of flank skin were prepared by extracting finely minced tissues with 4 M GuHCL buffer (4 M guanidine HCL, 100 mM sodium sulfate, 100 mM Tris base, 2.5 mM Na2EDTA, 0.5% TX-100, pH 7.0) with protease inhibitors (5 mM benzamidine hydrochloride, 100 mM 6-aminohexanoic acid, 1 mM phenylmethyl sulfonyl fluoride) overnight at 4C. Tissue extracts were then dialyzed against 8 M urea buffer (8 M urea, 2 mM EDTA, 50 mM Tris base, 0.5% TX-100, pH 7.5) to remove the guanidine. Protein concentration was determined with the Coomassie Protein Assay kit (Pierce, Rockford, IL). Western blots and proteins in each sample were ethanol precipitated and electrophoresed directly on 8% SDS-PAGE gels with 3.5% stacking gels. Proteins were transferred to nitrocellulose and probed with Versican V0/V1 Neo, which recognizes the DPEAAE fragment of versican generated by ADAMTS cleavage (Sandy et al. 2001; 1 µg/ml; Affinity Bioreagents), respectively. Results were visualized using the enhanced chemiluminescence kit (Applied Biosystems, Foster City, CA). Actin was used as a loading control for the DPEAAE westerns.
Immunohistochemistry
Flank skin was excised, fixed in 4% paraformaldehyde, and processed for paraffin embedding. Endogenous peroxidases were blocked in 5 µm deparaffinized sections using H2O2 in methanol. For versican immunohistochemistry, 0.2 U/ml of chondroitin ABC lyase was used for the digestion to expose versican epitopes. For DPEAAE and ADAMTS-1 immunohistochemistry, the chondroitin ABC lyase digestion step was eliminated. The tissue sections were blocked in 3% Carnation non-fat milk, 2% BSA-PBS for 1.5 hr at room temperature and then incubated in a moist chamber overnight at 4C with two rabbit antiversican polyclonal antibodies that recognize the GAG-α and GAG-β domain of versican (1:100) (Chemicon) or versican V0/V1 Neo, which recognizes the DPEAAE fragment of versican (1:300; Affinity Bioreagents) or anti-ADAMTS-1 (1:100; Affinity Bioreagents) or isotype-matched control IgG. This was followed by biotinylated goat anti-rabbit IgG antibody (1:400) (Zymed Laboratories, South San Francisco, CA) for 1 hr at room temperature. After PBS washes, tissue sections were incubated with the Vector “Elite” ABC-HP kit (Burlingame, CA) in a moist chamber for 30 min followed by incubation with the Vector NovaRed substrate for 10 min at room temperature. The slides were counterstained with hematoxylin and coverslipped.
In Situ Hybridization
In situ hybridization was performed as previously described (Shih et al. 2009). Tissues were fixed in 4% paraformaldehyde, and cryostat sections were hybridized overnight with antisense or sense (control) 35 S-labeled RNA probes specific for versican as previously described (Brown et al. 1999).
Results
Versican Accumulation and Degradation in Mouse Skin after Ad-VEGF-A164 Injection (Immunohistochemistry, In Situ Hybridization, and Western Blotting)
Versican staining was present in the superficial dermis of flank skin injected with Ad-Lac-Z, as well as in association with hair follicles and small blood vessel endothelial cells and pericytes (Fig. 1A–C). Except for hair follicles and vessels, staining was largely absent from the deep dermis. An identical distribution of staining was observed in normal, uninjected flank skin (not shown). However, following injection of Ad-VEGF-A164, versican staining was strikingly lost from venules transitioning into MV (Fig. 1D,E). Subsequently, strong staining for versican reappeared as MV evolved into GMP and VM (Fig. 1F,G).
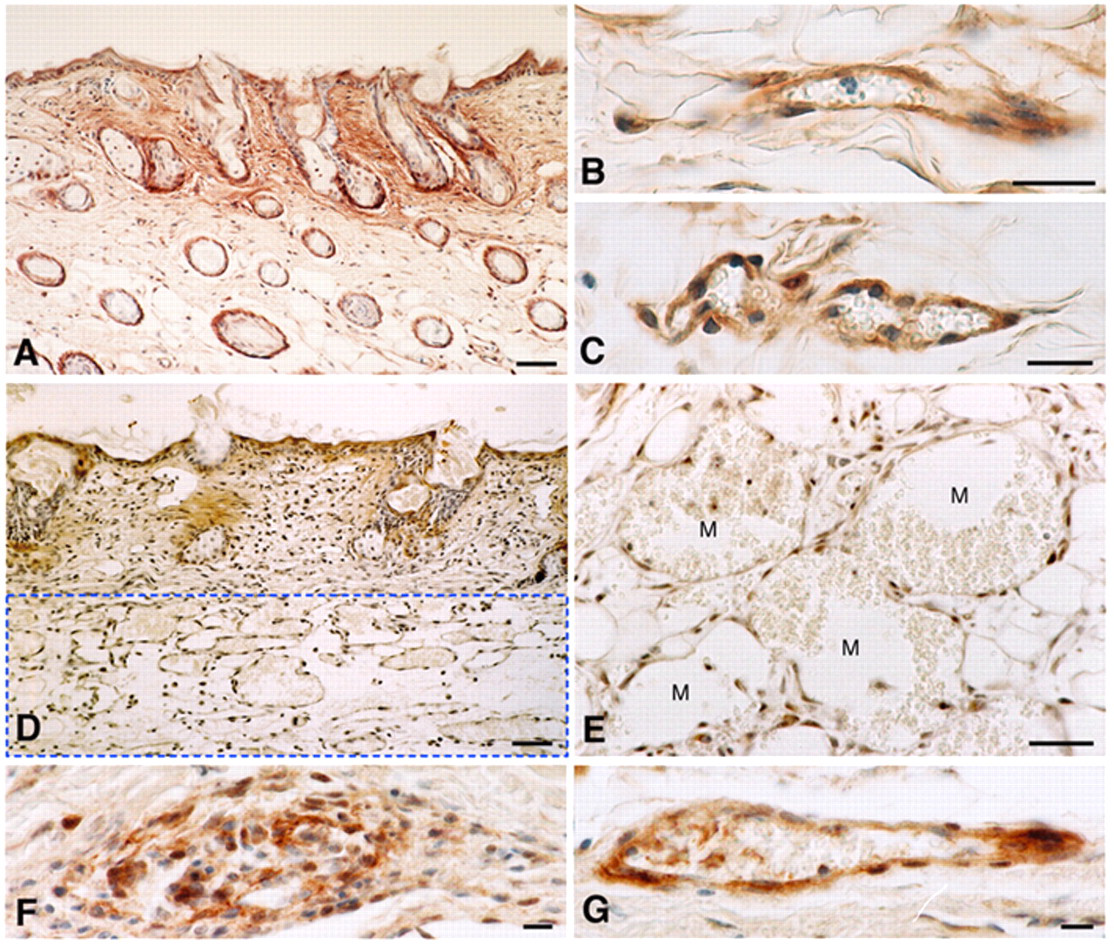
Immunohistochemistry of mouse flank skin with the anti-versican antibody that recognizes the major versican isoforms, V0 and V1. (A–C) Skin 3 days following injection of Ad-LacZ resembles that of control skin with versican staining of hair follicles, papillary dermal extravascular matrix, and deep and superficial microvasculature. (D, E). Flank skin 3 days after injection of Ad-VEGF-A164. Note reduced staining of papillary dermis and total loss of staining of MV (M) that develop primarily in the reticular dermis (rectangular zone blocked out by dashed line in D). (F, G) At 28 days after Ad-VEGF-A164 injection, daughter vessels, a glomeruloid microvascular proliferation (F) and a vascular malformation (G), stain strongly. Magnification bars: A, D = 50 µm; B, C, E–G = 20 µm.
We employed in situ hybridization to identify the cells that were making versican. Low-level labeling of hair follicles, dermal stromal cells, and preexisting microvessels was observed in control skin sites (Fig. 2A). In sites injected with Ad-VEGF-A164, increased labeling was observed in papillary dermal stromal cells 1 day after injection (Fig. 2B), but MV endothelial cells did not label (Fig. 2C). However, at later times, GMP and VM that developed from MV, as well as stromal cells, displayed strong labeling for versican mRNA (Fig. 2D,E).
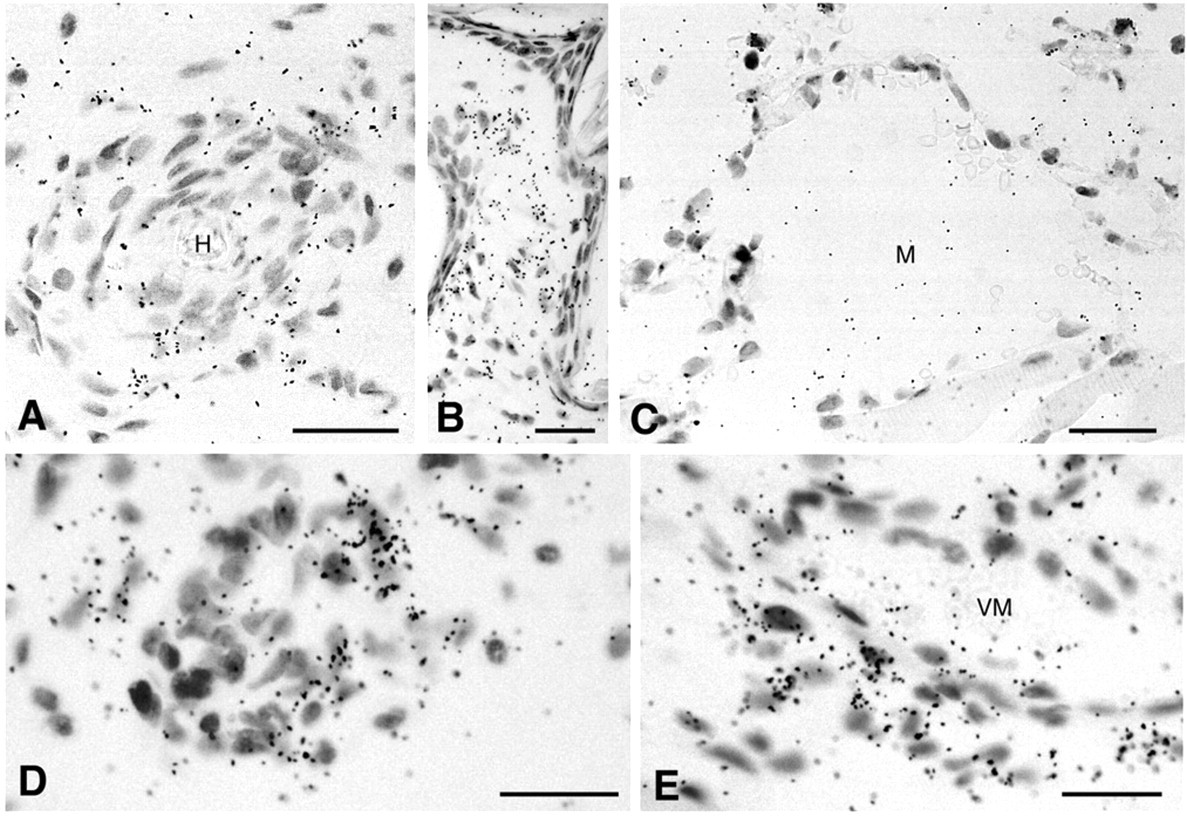
In situ hybridization of flank skin sites with an antisense probe against versican at various intervals after injection of either Ad-Lac Z (A) or Ad-VEGF-A164 (B–E). (A) Low-level labeling of hair follicle cells (H) and adjacent papillary dermis. (B) Increased labeling of papillary dermal cells 1 day after Ad-VEGF-A164. (C) Entirely negative mother vessels (M) at 5 days. (D, E) Labeling of cells in daughter vessels that derive from mother vessels, a glomeruloid microvascular proliferation in D, and a vascular malformation (VM) with associated stromal cells in E. Magnification bars: 25 µm.
ADAMTS-1 cleaves versican at the Glu441-Ala442 bond to generate a 70-kDa fragment termed DPEAAE. DPEAAE can be recognized by an antibody that is specific for a neoepitope generated by this cleavage (Kenagy et al. 2006; Sandy 2006; Sandy et al. 2001). Loss of versican staining in developing MV as seen in Figure 1 was accompanied by the appearance of significant immunostaining for the versican breakdown fragment, DPEAAE (Figure 3). DPEAAE appeared at 1 day after Ad-VEGF-A164 injection in some, but not all, endothelial cells of venules that were enlarging to form MV (Fig. 3A). Staining became much stronger on days 2 and 3 and by then involved nearly all MV endothelial cells (Fig. 3B,C). Thereafter, DPEAAE staining was lost as MV developed further on days 4 and 5 (Fig. 3D,E). However, DPEAAE staining reappeared in focal regions of developing GMPs (Fig. 3F).
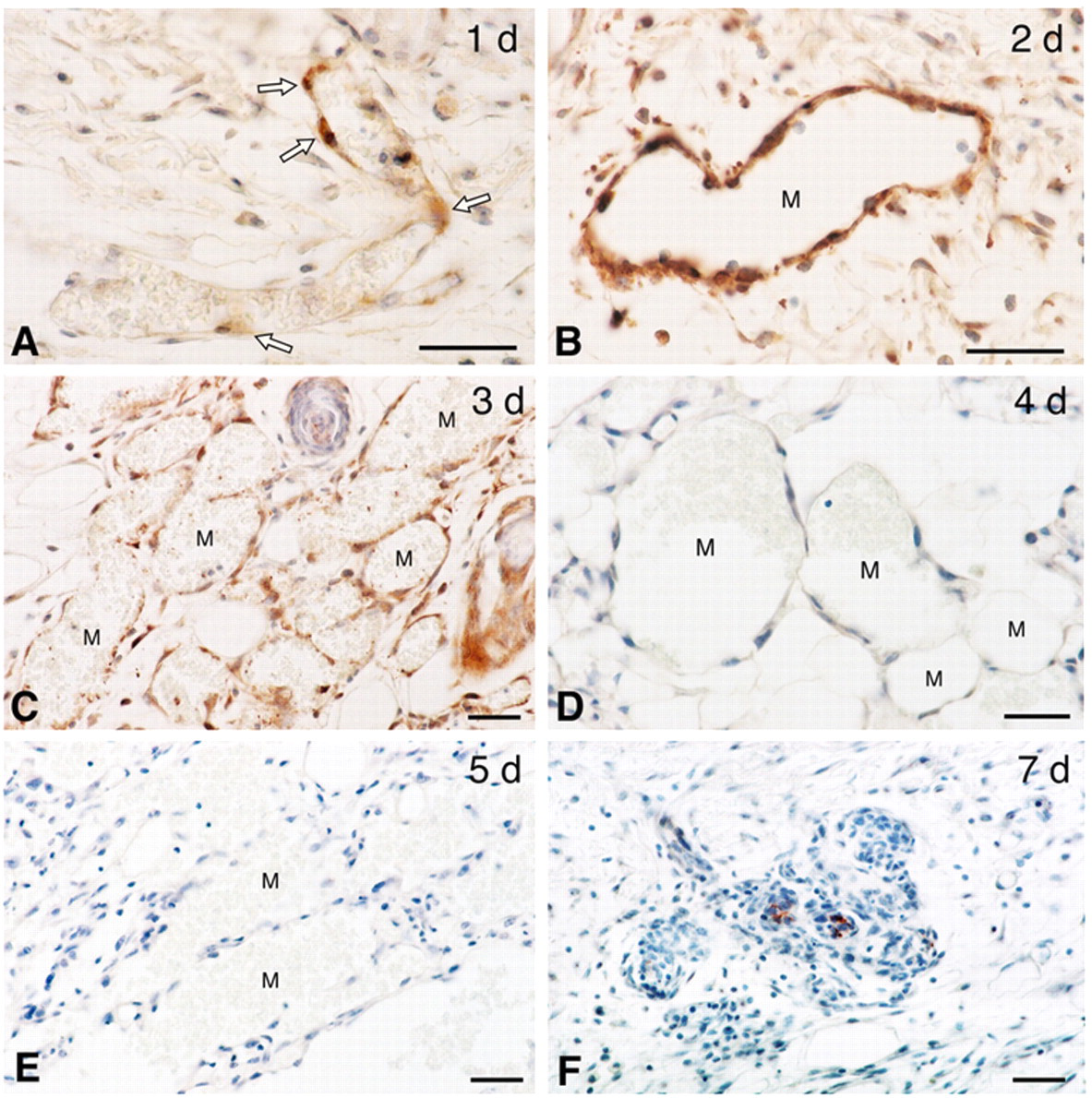
Immunohistochemistry of mouse flank skin with antibody directed against the versican degradation fragment DPEAAE, at indicated times after Ad-VEGF-A164 injection. (A) Venule at an early stage of transition into a mother vessel (MV). Some (arrows), but not all endothelial cells stain. (B) At 2 days, all endothelial cells of developing MV stain intensely. (C) More numerous MV at day 3 stain somewhat less intensely than at 2 days. (D, E) DPEAAE staining is entirely negative in fully developed MV at days 4 and 5. (F) A developing glomeruloid microvascular proliferation shows focal DPEAAE staining. M, mother vessel. Magnification bars: 20 µm.
Western blotting of both the control (LacZ) and VEGF-A164-treated flank skin extracts revealed that the DPEAAE fragment appeared most prominent at day 3 compared to the LacZ controls (Fig. 4A,B), confirming the immunohistochemical observations (see Fig. 3).

Western blots of flank tissue extracts for the DPEAAE fragment of versican from (A) control (Ad-Lac Z) or (B) Ad-VEGF-A164 injected samples at days 1, 3, and 5.
To investigate the involvement of ADAMTS-1 in the generation of the DPEAAE fragments of versican, flank skin sections were stained with antibodies against ADAMTS-1. Immunohistochemistry demonstrated an increase in ADAMTS-1 accumulation associated with the endothelial cells of developing MV as early as 1 day after injection of Ad-VEGF-A164, and by 2 and 3 days, nearly all MV endothelial cells stained strongly for this enzyme (Fig. 5B–D).
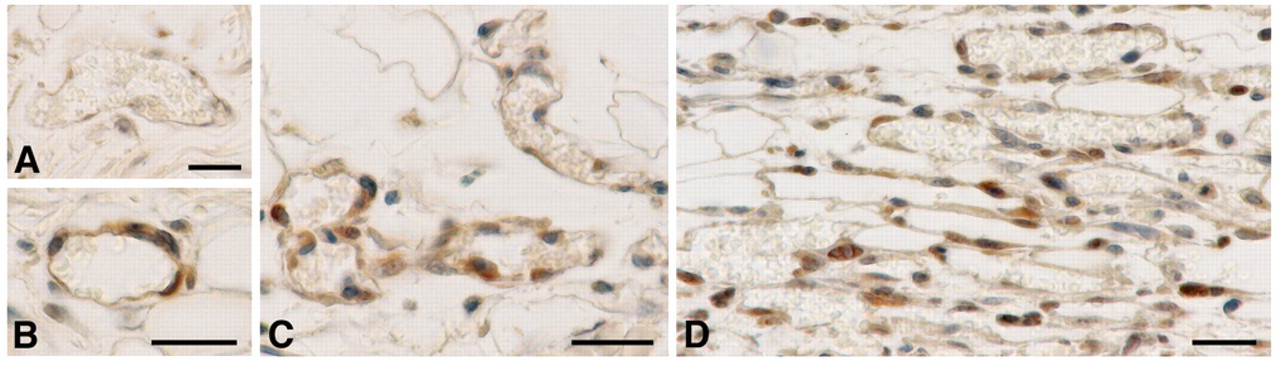
Immunohistochemical staining for ADAMTS-1 in flank skin at successive times after injection of Ad-VEGF-A164. On day 1, many microvascular endothelial cells were negative (A), although a few were positive (B). Endothelial cell staining for ADAMTS-1 reached maximal intensity on day 2 (C) and remained strong on day 3 (D). Magnification bars = 20 µm.
VEGF-A164/5 Induces ADAMTS-1 and MMP-15 Expression In Vitro and In Vivo
Earlier studies demonstrated that VEGF induced ADAMTS-1 expression in cultured human umbilical vein and retinal endothelial cells (Xu et al. 2006). We confirmed these findings in HDMVEC with VEGF-A165 (the human homologue of murine VEGF-A164). As shown in Figure 6A, ADAMTS-1 mRNA expression was strongly and progressively upregulated over the period of 30 min to 16 hr. Of interest, however, the effect of VEGF-A165 on ADAMTS-1 mRNA expression was inhibited by cycloheximide (Fig. 6B), indicating that the ADAMTS-1 upregulation induced by VEGF-A165 was indirect, likely reflecting the induction by VEGF-A165 of transcription factors that, in turn, induced ADAMTS-1 gene expression. ADAMTS-1 protein levels were also increased 30 min after VEGF-A165 treatment and reached peak levels at 1 hr (Fig. 6C).
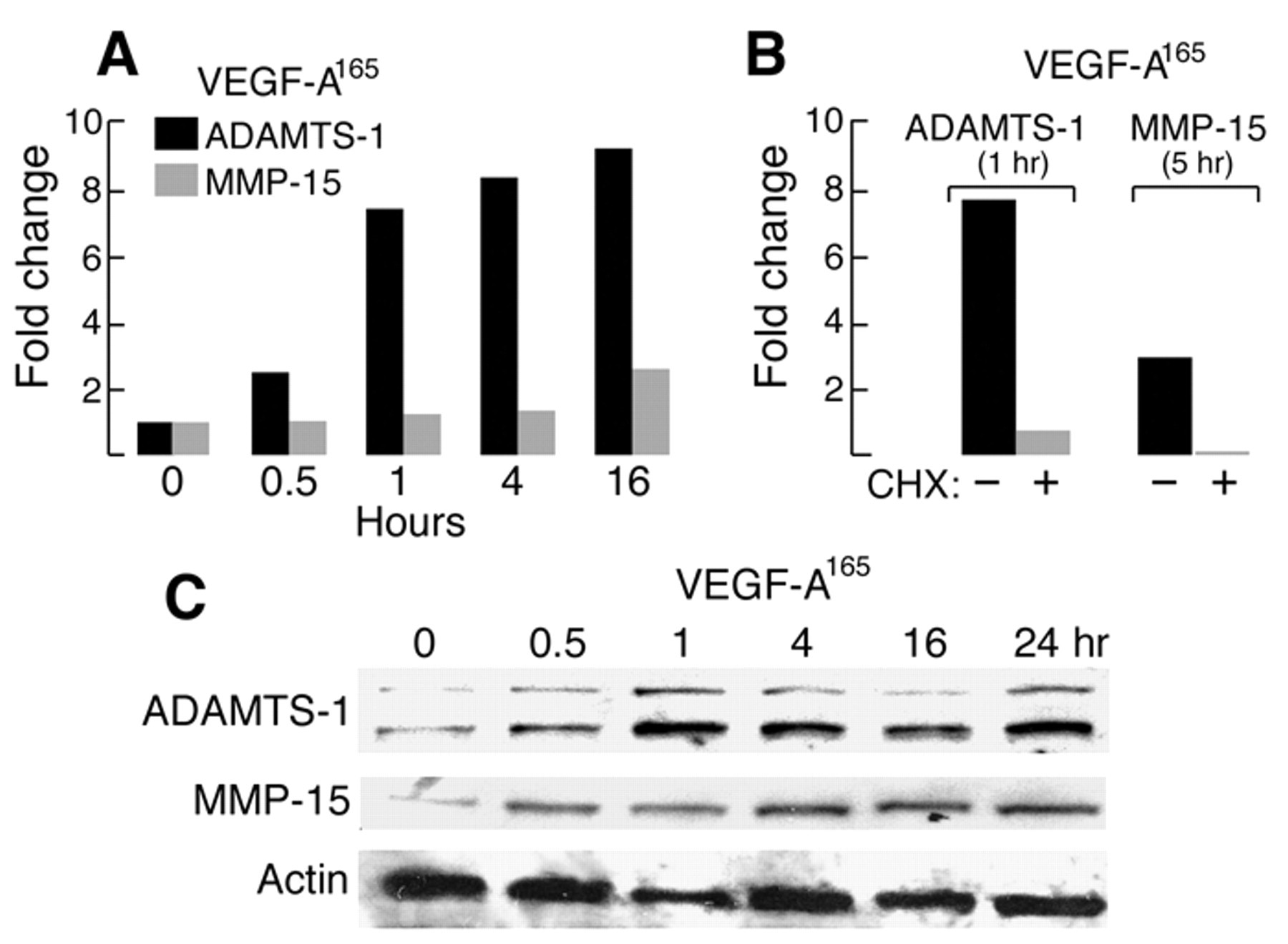
Effect of VEGF-A165 (10 ng/ml) on ADAMTS-1 and MMP-15 expression in cultured HDMVEC. (A) ADAMTS-1 and MMP-15 mRNA expression at indicated times after addition of VEGF-A165. (B) Effect of cycloheximide on VEGF-A165-induced ADAMTS-1 and MMP-15 mRNA expression. (C) Western blot of ADAMTS-1 and MMP 15 protein expression in response to VEGF-A165.
ADAMTS-1 requires posttranslational processing for its activation, and furin and MMP-15 are thought to have important roles in this process (Rodriguez-Manzaneque et al. 2000). MMP-15 expression was induced in HDMVEC stimulated with VEGF-A165, increasing by nearly 3-fold by 16 hr; as with ADAMTS-1, this effect was blocked by cycloheximide (Fig. 6A,B). MMP-15 protein levels also increased detectably within 30 min of VEGF-A165 stimulation and increased further at later time points (Fig. 6C). Other studies show that MMP14 (MT1-MMP) is involved also in angiogenesis (Chun et al. 2004; Genis et al. 2006), and our PCR data did show that MMP14 (MT1-MMP) mRNA is approximately doubled in the course of MV formation from already high baseline levels (data not shown), indicating that more than one MT-MMP may be involved in versican degradation in this system.
VEGF-A164/5 Induces ADAMTS-1 and MMP-15 Expression In Vivo
We next used Western blotting to determine whether the findings observed with VEGF-A-stimulated endotheilial cells in vitro also occurred in vivo in Ad-VEGF-A164-induced angiogenesis in flank skin. As already noted, ADAMTS-1 is produced initially as a proenzyme that undergoes two-step processing to form the mature enzyme. First, a prodomain is removed by furin, and then the N-terminal TSP repeats are removed by proteases, particularly MMP-15 (Rodriguez-Manzaneque et al. 2000). Activation of ADAMTS-1 typically results in three major bands on Western blots for ADAMTS-1: the proform at ~110 kDa and the further processed forms at 87 kDa and 65 kDa. As shown in Figure 7A, major bands were found at 110, 87, and 60 kDa in extracts at the early time points following Ad-VEGF-A164. These data correlate nicely with immunohistochemistry data (Fig. 5) and with the appearance of the versican DPEAAE fragment as determined by Western blot (Fig. 4) and immunohistochemistry (Fig. 3). All three dominant ADAMTS-1 bands increased within a day following Ad-VEGF-A164 injection (Fig. 7A).
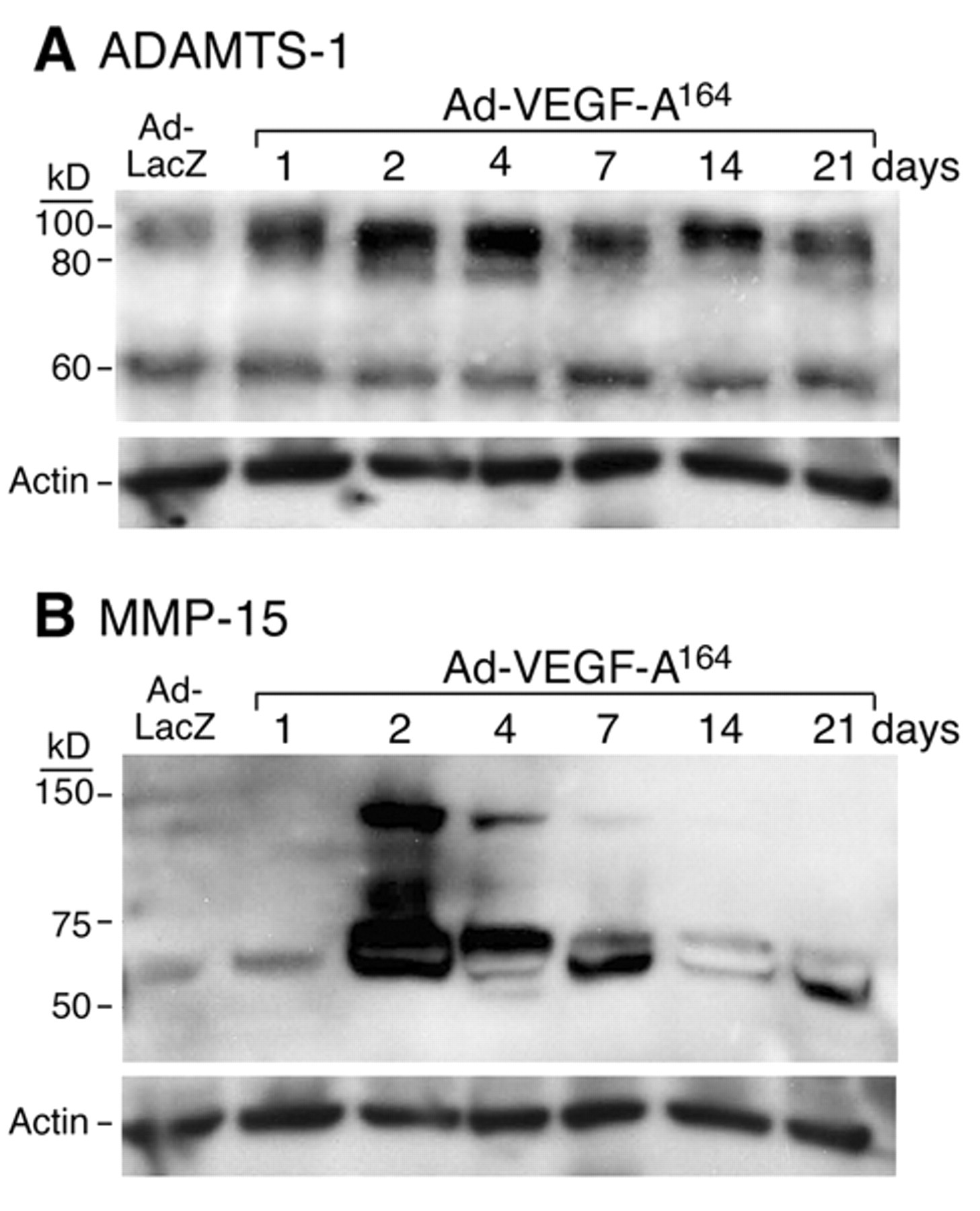
Western blots of (A) ADAMTS-1 and (B) MMP-15 expression in flank skin at successive intervals following injection of Ad-Lac-Z or Ad-VEGF-A164, as indicated. Description of the banding pattern is found in the text.
Ad-VEGF-A164 also induced a striking increase in MMP-15 protein (~68 kDa) by day 2, compared with day 1 and Lac-Z control (Fig. 7B). Expression of a larger, undefined protein (approximately 140 kDa) on day 2 was observed, which gradually decreased and became undetectable after day 7.
Discussion
Ad-VEGF-A164 induces an angiogenic response that mimics that of the pathological angiogenesis induced by tumors and chronic inflammation (Nagy et al. 2007). MV are large, thin-walled, pericyte-poor, hyperpermeable sinusoids and are the first new vessel type to develop and do so from preexisting normal venules (Nagy et al. 2007). Venular basement membrane breakdown is a first essential step in MV formation. Vascular basement membranes are rigid structures that exhibit little elasticity and can enlarge only by ~30% when exposed to increased intravascular pressures (Swayne et al. 1989). Therefore, for venules to increase 4- to 5-fold in cross-sectional area in the course of MV formation, basement membranes must be degraded. We recently demonstrated that two major components of vascular basement membranes, laminin and collagen IV, are indeed degraded in the course of MV formation (Chang SH et al. 2009). In this report, we investigated another vascular basement membrane component, versican, and found that it was also processed during the course of pathological angiogenesis.
MV developed primarily in the lower (reticular) dermis and were characterized by a striking loss of versican as demonstrated by immunohistochemistry (Fig. 1D,E); furthermore, in situ hybridization demonstrated a lack of versican mRNA expression (Fig. 2C). As additional evidence of versican degradation in the course of MV formation, we found a striking increase in the versican DPEAAE fragment by western blots (Fig. 4) and localized DPEEAE to developing MV endothelial cells by immunohistochemistry (Fig. 3).
ADAMTS-1 is one of the enzymes known to degrade versican, for example, in human aorta (Sandy et al. 2001). Therefore, we investigated whether VEGF-A165, the human homologue of mouse VEGF-A164, might induce ADAMTS-1 expression in endothelial cells. We found that VEGF-A165 induced a rapid and dynamic increase in ADAMTS-1 expression in cultured HDMVEC; peak levels were attained by 1 hr (Fig. 6), in agreement with earlier studies showing that VEGF upregulates ADAMTS-1 in other types of endothelial cells (Xu et al. 2006). Previous studies have shown that ADAMTS-1 expression is induced in endothelial cells under a variety of different conditions, for example, by shear stress (Bongrazio et al. 2000) and hypoxia (Hatipoglu et al. 2009).
ADAMTS-1 is synthesized as a proenzyme that requires activation by removal of its prodomain and the N-terminal TSP repeats. Furin is thought to be responsible for the former activity, but furin mRNA expression was not altered by Ad-VEGF-A164 during the course of these experiments (data not shown). MMPs, especially MMP-15 (MT2-MMP), are believed to be responsible for the removal of ADAMTS-1’s TSP repeats (Rodriguez-Manzaneque et al. 2000). We found here that VEGF-A165 also induced MMP-15, an enzyme that activates ADAMTS-1, but with slightly slower kinetics (Fig. 6). Furthermore, both ADAMTS-1 and MMP-15 protein expression were upregulated in the course of MV formation in flank skin following injection of Ad-VEGF-A164 (Fig. 6), and ADAMTS-1 was localized to the endothelial cells of developing MV (Fig. 5). Transient expression of a larger MMP-15 protein band (~140 kDa) was noted from day 2 to day 7 with the highest level on day 2. The nature of this protein is unclear, but the time course of its expression corresponds to that of MV formation. At later times, versican synthesis was renewed and was localized to the GMP and VM that evolved from MV (Figs. 1 and 2). Taken together, these data make a strong case for the MV endothelial cells as the source of ADAMTS-1 in the processing of versican.
These new data can be compared with our earlier study of vascular basement membrane collagen IV and laminin degradation, which was also induced during the course of Ad-VEGF-A164-induced MV generation (Chang SH et al. 2009). Although degradation of these proteins and versican occurred in parallel, different proteolytic enzymes and different cells as a source of these enzymes were apparently involved. Collagen IV and laminin degradation was associated with increased expression of cathepsins B, S, and L, along with a parallel loss of the cysteine protease inhibitors stefin A and cystatins B and C, known inhibitors of cathepsin function (Chang SH et al. 2009). Furthermore, whereas the loss of cysteine protease inhibitors was apparent in both endothelial cells and pericytes, cathepsin activity was localized exclusively to pericytes (Chang SH et al. 2009). The present data indicate that both ADAMTS-1 and MMP-15 are expressed by endothelial cells, and it would not be surprising to find that they also contribute to the degradation of vascular basement membrane components other than versican. For example, MMP-15 has been reported to degrade laminin (Somerville et al. 2003) and ADAMTS-1 basement membrane nidogens (Canals et al. 2006).
It should also be noted that a number of other proteases are capable of degrading versican, including MMPs (Halpert et al. 1996; Kenagy et al. 2006; Passi et al. 1999; Sandy 2006) and serine and cysteine proteases (Kenagy et al. 2002). Therefore, it is not clear whether ADAMTS-1 is essential for the versican degradation associated with Ad-VEGF-A164-induced angiogenesis. Use of ADAMTS-1 null mice will help to address this question. Eliminating specific proteases has an impact on VEGF-induced angiogenesis. For example, Ad-VEGF-A164-induced angiogenesis was reduced by about 50% in cathepsin B null mice (Chang SH et al. 2009). However, it is not clear whether this protease can degrade versican. Taken together, it is clear that a variety of different proteases are expressed by different vascular cells in the course of angiogenesis, and these may well have overlapping functions in degrading different vascular basement membrane components.
ADAMTS-1 and versican have been implicated in other examples of blood vessel remodeling. For example, ADAMTS-1 expression is elevated in wound healing, where it regulates migration of fibroblasts and endothelial cells (Krampert et al. 2005) and during endothelial tubulogenesis in fibrin gels (Lafleur et al. 2002). In ADAMTS-1 null mice, there is a defect in vascular formation in the adrenal glands, which is manifested as a disrupted reticular network of capillaries in the medulla with cavities containing red blood cells (Yokoyama et al. 2002). Modified expression of ADAMTS-1 resulted in altered blood vessel morphology in prostate tumor xenografts (Gustavsson et al. 2010). Low expression levels of ADAMTS-1 were associated with small-diameter blood vessels, whereas high levels of ADAMTS-1 were associated with larger vessels. Versican has also been implicated in the embryonic development of the aorta and pulmonary arteries (Mjaatvedt et al. 1998; Yamamura et al. 1997). More recently, Kern and colleagues (Kern et al. 2007) showed that increased versican degradation was also part of outflow tract development and localized to the developing aorta, along with ADAMTS-1. Such observations suggest that versican processing by ADAMTS-1 is involved in the development of blood vessels in the embryo, just as here it is involved in Ad-VEGF-A164-induced pathological angiogenesis.
That ADAMTS-1 is here implicated as having a proangiogenic role may seem surprising in that it has generally been thought of as an antiangiogenic protein (Iruela-Arispe et al. 2003). ADAMTS-1 inhibits endothelial cell proliferation in vitro and blocks the neovascular response induced by growth factors in vivo (Luque et al. 2003; Vazquez et al. 1999). It apparently acts by binding and so reversibly inactivating VEGF through its TSP repeats, but its catalytic domain also apparently has a role in its antiangiogenic function (Iruela-Arispe et al. 2003). Recently, another protein thought to have an exclusively antiangiogenic function, thrombospondin-1, has been found, paradoxically, to have a potentially proangiogenic role. Thrombospondin-1 null mice exhibit decreased vascular endothelial growth factor (VEGF) receptor phosphorylation in response to VEGF stimulation, both in vitro and in vivo (Zhang et al. 2009). Such results illustrating both agonist and antagonist roles for single molecules in cellular and metabolic events involved in angiogenesis highlight the complexity in the control of the angiogenic response, a complexity that no doubt involves selectivity of molecular interactions and a dose, spatial, and temporal sequence for pro- and/or antiangiogenic signaling. Sorting out the common players and the requirements for these opposing activities is the challenge for the future.
Footnotes
The author(s) declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research and/or authorship of this article: This work was supported in part by National Institutes of Health (NIH) grants P01 HL 18645 and 5 R01 HL 064387 to TNW and by grants to HFD as follows: NIH grant P01 CA92644 and by a contract from the National Foundation for Cancer Research.