
Abstract
Obesity is a well-established risk factor for hypertension, hyperlipidemia, type II diabetes, coronary heart disease, stroke, obstructive sleep apnea, asthma, orthopedic disorders, and certain cancers. Despite this risk, the prevalence of obesity continues to increase worldwide, and there is a growing demand for safe and effective antiobesity drugs. Previous antiobesity drugs or anorexigens, particularly centrally acting agents, have poor safety records. Life-threatening safety issues led to the withdrawal of aminorex in 1968, fenfluramine and dexfenfluramine in 1997, and phenylpropanolamine in 2000. Many of the safety issues, such as valvulopathy with fenfluramine and pulmonary arterial hypertension with aminorex, were initially not predicted by routine preclinical toxicology studies. To date, there are no validated animal models or preclinical and/or toxicologic screens to accurately predict anorexigen-induced valvulopathy and pulmonary arterial hypertension in humans. This review covers the current state of antiobesity drugs and their safety concerns, and highlights new therapeutic targets and scientific advances toward the development of appropriate animal models by using novel techniques that will aid in understanding pathogenesis and pathophysiology of anorexigen-related safety issues.
Obesity is now widely accepted as a multifactorial, chronic disorder, with an alarming increase in the worldwide prevalence in both adults and children. In North America and most European countries, the obesity rates have more than doubled in the last 20 years. The International Obesity Task Force estimates that more than 300 million individuals worldwide are obese body mass index (BMI ≥ 30 kg/m2) and 800 million are overweight (BMI between 25 and 29.9 kg/m2). 46 Currently, 66% of US adults are overweight or obese, 16% of US children and adolescents are overweight, and 34% are at risk of becoming overweight. Based on the average annual increase in prevalence, Wang and Beydoun 130 projected that by 2015, 75% of US adults and nearly a quarter (24%) of US children and adolescents are expected to be overweight or obese. This increasing overweight and/or obese population is disturbing because of the well-established major health risks and chronic conditions associated with obesity, such as hypertension, hyperlipidemia, type II diabetes, coronary heart disease, stroke, obstructive sleep apnea, asthma, orthopedic disorders, and social and mental health problems. 46, 70 Several epidemiologic studies have shown that overweight and/or obesity contributes to the increase in mortality and morbidity of multiple cancer types. 8, 60 This increased risk was clearly evident for postmenopausal breast cancer and cancers of the colon and/or the rectum, endometrium, kidney, esophagus but not definitely for prostate, gallbladder, ovary, and pancreas. 4, 60 Obesity is blamed as a contributing factor in more than 300,000 deaths annually in the United States, with economic costs of the epidemic reaching more than US $100 billion per year. 16 It has become the second leading preventable cause of disease and death in the United States, second only to tobacco use. 127
Antiobesity Treatment Strategies
Current strategies for obesity management include diet, exercise, drug therapy, and bariatric surgery, either alone or in combination. Most guidelines throughout the world recommend drug therapy for obesity when the BMI is ≥30 kg/m2 or when the BMI is 27.0–29.9 kg/m2 with a major obesity-related comorbidity (e.g., hypertension, diabetes, obstructive sleep apnea). 2, 95 Bariatric surgical procedures, such as gastric banding or gastric bypass, are effective for sustained weight loss but are clearly invasive and often associated with a significant rate of complications. 34, 132 Surgical procedures are generally used for patients with morbid obesity (BMI > 40 kg/m2) and those with obesity-related complications. 9 This review focuses on antiobesity drugs that are currently available and new therapeutic targets, and the safety of antiobesity drugs, including in vivo animal models to screen their toxicity. Although several antiobesity drugs are in the US market for short-term use, only 2 drugs, sibutramine (Meridia Abbott Laboratories, North Chicago, IL, USA) and orlistat (Xenical, Roche Laboratories, Nutley, NJ, USA; and over-the-counter Alli, GlaxoSmithKline, Philadelphia, PA, USA), are currently approved by the US Food and Drug Administration (FDA) for long-term use. These drugs affect appetite, metabolic rate, and/or inhibit caloric absorption, and generally fall into 3 broad classes: 1) peripherally acting, 2) centrally acting, and 3) combination (i.e., central and peripheral acting).
Peripherally acting antiobesity drugs
Peripherally acting drugs mediate their effects by reducing the calorie absorption in the gastrointestinal system or by affecting metabolic and/or control systems outside the central nervous system (CNS). Currently, the only peripherally acting antiobesity drug globally approved for long-term use is orlistat, a lipase inhibitor. Orlistat is a hydrogenated derivative of lipstatin, a naturally occurring lipase inhibitor produced by Streptomyces toxytricini. 131 The drug acts in the gut lumen by forming a covalent bond with the active serine site of gastric and pancreatic lipases. By forming the covalent bond, orlistat inhibits these lipases from hydrolyzing the ingested fat into absorbable free fatty acids and monoglycerides (Fig. 1). Orlistat has been shown to reduce the amount of ingested fat absorption by up to 30% in humans. 47, 125 The rationale behind this drug is that this decreased absorption of ingested fat leads to an overall decreased caloric absorption, in turn leading to weight loss. 3, 48, 54, 75, 78 The adverse effects of prescribed orlistat (Xenical) are typically gastrointestinal and include oily spotting, liquid stools, fecal urgency or incontinence, flatulence, and abdominal cramping. The systemic adverse effects are rare because of the lack of systemic absorption. A few cases of serious hepatic effects (cholelithiasis, cholestatic hepatitis, and subacute liver failure) were reported. 30 No effect has been observed on calcium, phosphorus, magnesium, iron, copper, or zinc balance, or on bone biomarkers. 136 Orlistat can impair the absorption of amiodarone and cyclosporin, and can potentiate the effect of warfarin. 84, 136, 137 Because orlistat may also impair the absorption of fat-soluble vitamins (vitamins A, E, D, and K), a daily multivitamin supplement is recommended. 31, 88, 91, 100, 115
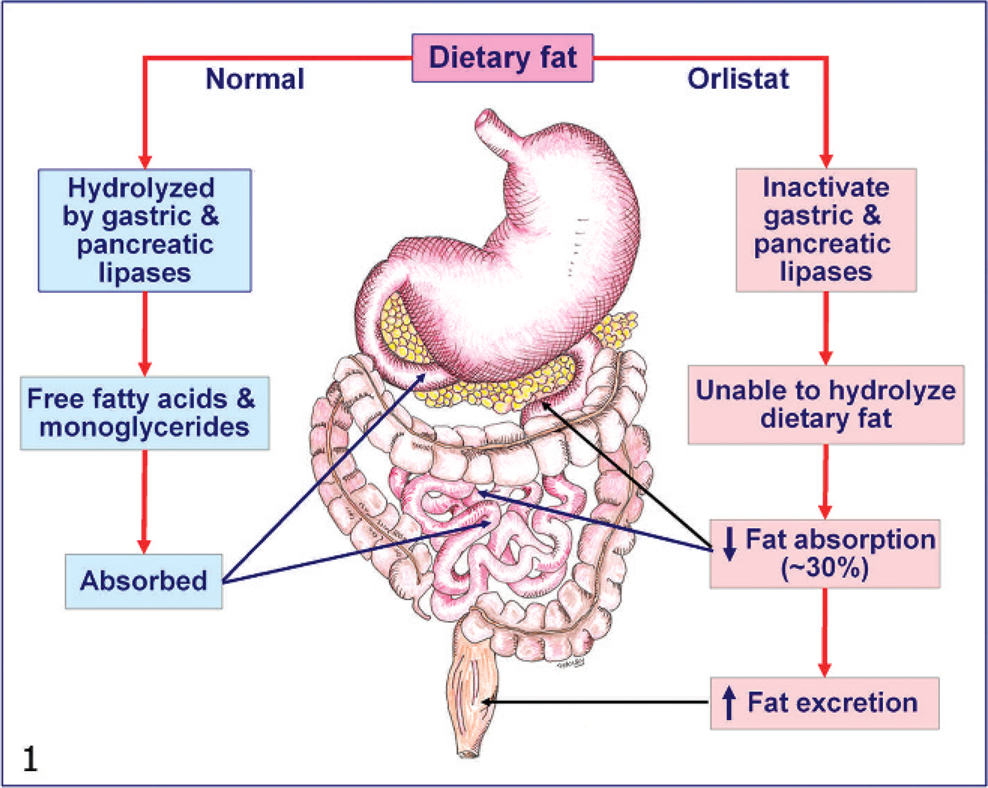
Schematic representation of orlistat's mechanism of action. Orlistat blocks the absorption of ingested dietary fat by inhibiting pancreatic and gastric lipases from hydrolyzing the ingested fat into absorbable free fatty acids and monoglycerides.
Centrally acting antiobesity drugs
These drugs act on the CNS by 3 mechanisms: catecholaminergic noradrenaline and dopamine (e.g., diethylpropion, methamphetamine, phendimetrazine, and phentermine), 5-hydroxytryptamine (5HT) mediated, or combined (e.g., sibutramine). 44 Satiety may be regulated through an effect on 5HT, noradrenaline (norepinephrine), or dopamine receptors in the hypothalamus, whereas energy expenditure may be increased directly by thermogenesis and lipolysis or through the stimulation of the sympathetic nervous system. Sibutramine is the only centrally acting drug currently approved for long-term treatment of obesity in adults. Sibutramine inhibits re-uptake of norepinephrine, 5HT, and, to a lesser extent, dopamine, which results in weight loss because of reduced hunger, increased satiety, and thermogenesis. 31, 53, 62, 74 Common adverse effects of sibutramine are dry mouth, anorexia, insomnia, constipation, headache, and hypertension and increased pulse rate. 99 In 2002, sibutramine was temporarily withdrawn from the market in Italy because of adverse cardiovascular events (mainly tachycardia, hypertension, and arrhythmias), including 2 deaths. 133 Between 1998 and 2001, sibutramine-related 397 adverse events were reported in the United States, including 143 cardiac arrhythmias and 29 deaths (19 deaths from cardiovascular causes). 59, 133 Because of negative effects on the heart, sibutramine should be used with caution in patients with uncontrolled hypertension and tachyarrythmias. 96 Although other centrally acting drugs, such as diethylpropion, methamphetamine, phendimetrazine, and phentermine, are indicated for the management of exogenous obesity as short-term adjunct, their use is restricted because of their adverse effects, including pulmonary arterial hypertension, valvulopathy, and the potential for abuse and dependency. 59
Central- and peripheral-acting drugs
Rimonabant (Acomplia, Sanofi-Aventis, Paris, France) is a selective endocannabinoid (CB) 1 receptor antagonist that acts both centrally and peripherally to inhibit food intake and to regulate metabolic functions at diverse peripheral organs, including gut, liver, adipose tissue, and skeletal muscle. 20, 35, 109 Rimonabant is approved by European Agency for the Evaluation of Medicinal Products and sold in nearly 40 countries but recently failed to gain FDA approval (for reasons described below). 126 The endocannabinoid system includes 2 major ligands (anandamide and 2-arachidonoyl-glycerol) and 2 receptors (CB1 and CB2 receptors), and constitutes a retrograde signaling system. 17, 19, 20, 56, 72, 80 The CB1 receptor is a G-protein–coupled receptor that is extensively expressed in the CNS, including areas known to control food intake. 20 Animal studies have demonstrated that endocannabinoids interact with several anorexic (appetite suppressing) and orexigenic (appetite stimulating) pathways within the CNS, including the central melanocortin (via binding with anorexigenic melanocortin-4 receptors) and mesolimbic pathways, increasing motivation to eat, and stimulating food intake. 20, 55, 97, 101 Histochemistry studies 94 suggest that orexigenic cannabinoid and serotonergic systems act through gamma aminobutyric acid neurotransmission. Other rodent studies 49 showed the colocalization of CB1 receptors with dopamine (D1 and D2) and 5HT (5HT1B and 5HT3) receptors, which suggests that underlying crosstalk mechanisms exist between dopaminergic and serotonergic systems. Activation of CB1 and CB2 receptors by endocannabinoids (i.e., anandamide) was shown to increase appetite. Rimonabant works by blocking endocannabinoid binding to neuronal CB1 receptors and inhibits food intake and body-weight gain in rodents and humans. 6, 10 Peripheral mechanisms of rimonabant include enhanced thermogenesis via increased oxygen consumption in skeletal muscles, 76 diminished hepatic 98 and adipocyte lipogenesis, 15 augmentation of adiponectin concentrations, 18 promotion of vagally mediated cholecystokin-induced satiety, 20, 38 inhibition of preadipocyte proliferation, and increased adipocyte maturation, without lipid accumulation (Fig. 2). 37 The most frequent adverse effects seen in patients treated with rimonabant include nausea, diarrhea, and psychiatric disorders (depression, anxiety, suicidality, and other cognitive and neurologic symptoms). 18, 103, 115, 128 Because of neurologic and psychiatric problems, including a twofold increase in suicide, parasuicide, or clinical depression, the FDA's Endocrinologic and Metabolic Drugs Advisory Committee in June 2007 unanimously recommended against the approval of Rimonabant in the United States. 126
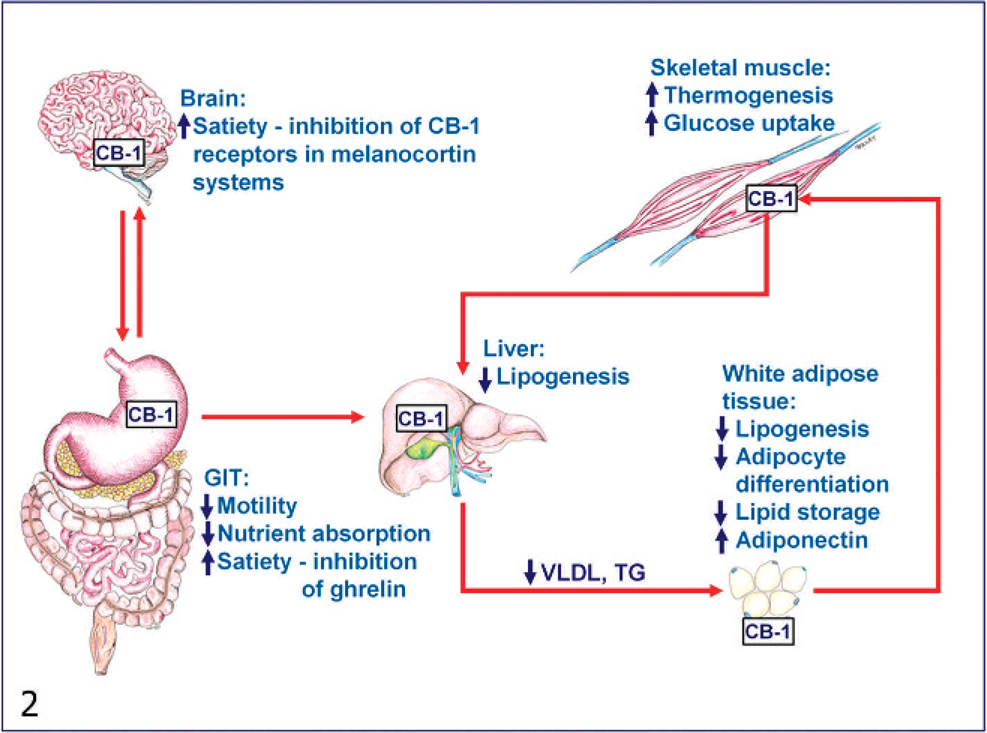
Schematic representation of the mechanism of rimonabant (central and peripheral acting drug). Rimonabant acts centrally by blocking endocannabinoid (CB) binding to neuronal CB1 receptors, which results, in increased satiety and reduced food intake. Peripheral mechanisms include enhanced thermogenesis via increased oxygen and glucose consumption in skeletal muscles; diminished hepatic lipogenesis; decreased adipocyte differentiation and lipogenesis in white adipose tissues, and augmentation of circulating adiponectin concentrations; and decreased gastrointestinal tract (GIT) motility and nutrient absorption, and increased satiety through the inhibition of gastrointestinal hormone “ghrelin.” VLDL = very low-density lipoprotein; TG = triglyceride.
New Molecular Targets of Antiobesity Drugs
Currently available antiobesity drugs are modestly effective, and, in some subjects, they are associated with unacceptable and life-threatening adverse effects. As a result, there is a growing need for pharmaceutical companies to find effective, safe, and well-tolerated antiobesity drugs. Over 100 molecular targets for potential antiobesity drugs are in various stages of preclinical and clinical development. 12, 53 These molecular targets are based on newly discovered homeostatic pathways, including gut-hypothalamic axis, anorexic, and orexigenic hormone receptors within the hypothalamus, effectors of leptin and insulin signal transduction, and central and peripheral nutrient sensing pathways. 12, 71 Some of the potential targets for new antiobesity drugs include antagonists or inhibitors (melanin-concentrating hormone receptor-1, opioid receptor-μ, ghrelin, neuropeptide Y, galanin, Agouti-related protein, orexin [A and B], acetyl CoA carboxylase-2, 11β-hydroxysteroid dehydrogenase type 1, fatty acid synthase, central carnitine palmitoyltransferase-1, corticotrophin-releasing hormone receptor, histamine receptor, dipeptidyl peptidase IV, suppressor of cytokine signaling-3 tyrosine phosphatase 1B), agonists, and/or stimulators (adiponectin, alpha-melanocyte-stimulating hormone/melanocortin-4 receptor, apolipoprotein A-IV, brain-derived neurotrophic factor, cholecystokinin-A receptor, ciliary neurotrophic factor (Axokine), cocaine and amphetamine-regulated transcript, glucagon-like peptide-1 and axendin-4, human growth hormone fragment, insulin mimetics, leptin, leptin receptor, oxyntomodulin, peptide YY, phosphatidylinositol 3-kinase, somatostatin, β3, etc.), and enzyme inhibitors and/or blockers (gastric inhibitory polypeptide) (Fig. 3). Detailed discussions of these drugs and targets are beyond the scope of this article, and the reader is referred to published reviews. 12, 14, 43, 53
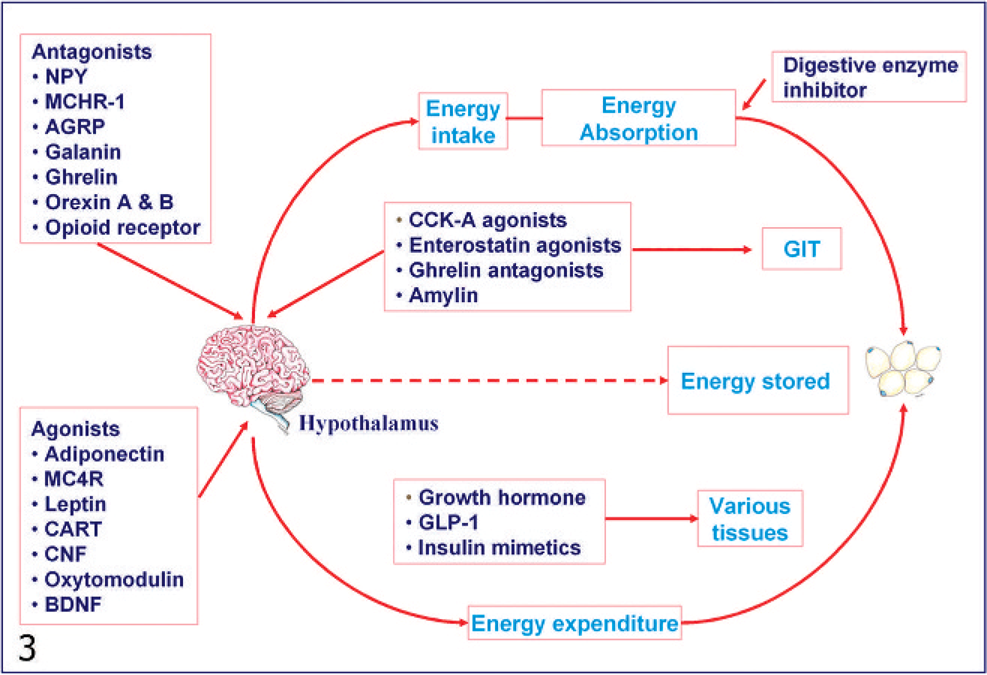
Potential antiobesity targets. Obesity arises as a result of how the body regulates energy intake/absorption, energy expenditure and energy storage. Energy intake and body weight are tightly controlled by various signaling mechanisms from the gastrointestinal tract (GIT) and adipose tissues that link with central pathways within the brain (hypothalamus). Therapies specially targeted to newly discovered appetite control centers in the hypothalamus include antagonists of appetite stimulating and agonists of appetite suppressing hormones/neuropeptides. Agents targeting peripheral biosignaling, gut-hypothalamus-axis and metabolism include cholecystokinin-A receptor (CCK-A), enterostatin, amylin, ghrelin, and human growth hormone fragment. NPY = neuropeptide Y; MCHR-1 = melanin concentrating hormone receptor-1; AGRP = Agouti-related protein; MC4R = melanocortin-4 receptor; CART = cocaine and amphetamine-regulated transcript; CNF = ciliary neurotrophic factor; BDNF = brain-derived neurotrophic factor; GLP-1 = glucagon-like peptide-1.
Safety Concerns of Antiobesity Drugs
Previous antiobesity drugs have very poor safety records (Table 1). Because of the potential for widespread misuse (i.e., for aesthetic purpose), the bar for the safety of antiobesity drugs is set at a much higher level than for other chronic diseases, such as diabetes and hypertension. In fact, clinical trials for approval of antiobesity drugs are twice as long as the trials for hypertension and diabetic drugs. 39 Life-threatening complications have led to the withdrawal of aminorex in 1968, followed by fenfluramine and dexfenfluramine in 1997, and phenylpropanolamine (norephedrine) in 2000. 59 Aminorex was withdrawn from the market because of its association with the epidemic of pulmonary arterial hypertension (PAH). Reports of valvulopathy 13 in 1997 led to the withdrawal of fenfluramine and dexfenfluramine from the US market. In 2000, the FDA requested the withdrawal of all products that contained phenylpropanolamine based on reports of an increased risk of hemorrhagic stroke in women. 69 Over-the-counter dietary supplements that contain ephedra alkaloids were withdrawn because of hypertension, tachycardia, stroke, and seizures. Even for the currently approved antiobesity drugs, such as diethylpropion and phentermine, the safety data are largely lacking, and their approval for obesity management is limited to short-term use. The benefit-risk profiles of sibutramine and orlistat, currently approved for long-term use, appear positive; however, sibutramine continues to be monitored because of long-term safety concerns, including hypertension and tachycardia. Furthermore, the safety and efficacy of currently approved antiobesity drugs have not been evaluated in children and elderly patient populations. The long-term safety of current and potential new drug therapies in adults and children will require several years of postmarketing surveillance to fully elucidate their adverse effects and potential mechanism. Two complications, namely 1) pulmonary arterial hypertension and 2) valvulopathy, highlight the serious consequences of antiobesity medications, namely, aminorex, fenfluramine, and dexfenfluramine treatments, including a multibillion dollar settlement by the manufacturers of these drugs. 39, 59, 120
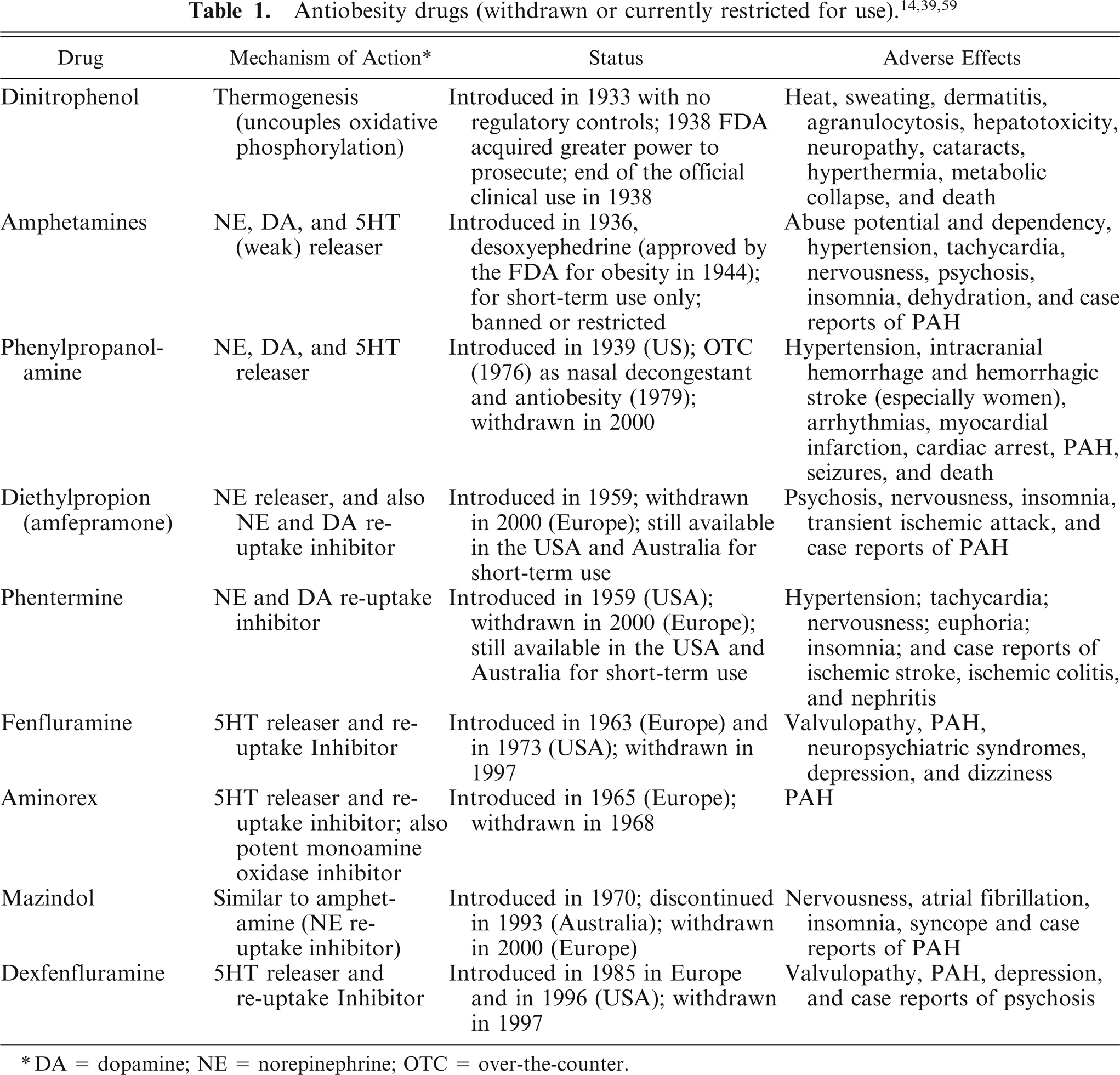
DA = dopamine; NE = norepinephrine; OTC = over-the-counter.
Pulmonary arterial hypertension
PAH is a life-threatening and often fatal disease characterized by diffuse medial hypertrophy and endothelial and smooth-muscle-cell hyperplasia of small pulmonary arteries, which result in narrowing of the vessel lumen, with subsequent increases in pulmonary arterial pressure and vascular resistance. 102, 112 Elevation of pulmonary arterial pressure leads to right heart failure and death. Although PAH has been classified as idiopathic (also known as primary) (IPAH), familial (FPAH), anorexigen-induced (APAH), and secondary forms, a common pathologic finding exists in all forms. 11, 58 That is, APAH is histologically indistinguishable from IPAH or FPAH forms. 102 However, patients with APAH had poorer survival than IPAH (1-year survival 50% in APAH versus 88% in IPAH, and 3-year survival 17% in APAH versus 60% in IPAH). 108 The epidemiologic data clearly show that fenfluramines and aminorex increase the risk of developing PAH. 1, 41 An association between aminorex and PAH was first established in the late 1960s, with a 20-fold increase in the incidence of PAH among patients exposed to aminorex in central Europe. 41 A similar association between the use of fenfluramine and PAH in Europe and the United States was established in the 1990s. Abenhaim et al. 1 estimated that taking fenfluramines for more than 3 months increased the risk of developing PAH by 23-fold. Results from a more recent US study 107 demonstrated that a history of fenfluramine exposure increases the risk of PAH by sevenfold.
The mechanism by which fenfluramines and aminorex might increase the risk of developing PAH is not fully understood. However, an abundance of evidence suggests that 5HT plays an important role in the pathogenesis of PAH. 81, 92, 119 5HT has been shown to induce proliferation and hypertrophy of pulmonary artery smooth-muscle cells in vitro and may be important for in vivo pulmonary vascular remodeling. Aminorex induces the release of noradrenaline, the inhibition of 5HT metabolism by monoamine oxidase (MAO) inhibition, and the release of 5HT from platelets, which all result in an elevation in plasma 5HT levels. 32, 36, 135 Fenfluramines also cause an elevation in plasma 5HT levels by causing 5HT release from neurons and platelets, and inhibiting reuptake of 5HT. 32, 51 However, other reports have shown that fenfluramine actually lowers blood 5HT and does not increase plasma 5HT in humans. 86, 123 5HT effects are mediated extracellularly through 5HT1B and 5HT2B receptors and intracellularly via cellular capture of 5HT through the 5HT uptake transporter (5HTT). 66 A reduction in 5HT uptake has been shown to produce increased concentrations of 5HT in the pulmonary vasculature, which might contribute to the pathogenesis of lung injury. 45 Because only a small proportion of patients exposed to fenfluramines develop PAH, it is likely that some type of genetic susceptibility is also involved. Bone morphogenetic protein receptor type 2 (BMPR2) and 5-HT2B mutations were found in some patients with fenfluramine-induced PAH. 5, 57, 121 However, it is not clear if fenfluramines directly interact with BMPR2 or 5HT2BR genes in the pathogenesis of PAH.
Anorexigen-induced valvulopathy
Valvulopathy, or valvular heart disease, is a main cause of morbidity and mortality in humans worldwide. 67, 124 Anorexigens, such as fenfluramine and dexfenfluramine were linked to valvulopathy in humans. 13, 50 Connolly et al. 13 first reported that both fenfluramine and dexfenfluramine were associated with valvulopathy in 24 patients. Microscopic examination of the valve leaflets showed “stuck-on” plaques of proliferative myofibroblasts in an abundant extracellular matrix. A similar “atypical plaque” containing myxoid stroma, proliferative myofibroblasts, small vessels and lymphocytic infiltrations has been described in the subsequent larger study with valve samples from 64 patients. 129 These valvular changes resembled lesions associated with malignant carcinoid syndrome or ergotamine-induced valvulopathy. The prevalence varies considerably between studies, ranging from approximately 6% to just over 30%. One case-control study reported a prevalence of 23% in patients treated with fenfluramine. A meta-analysis of the published studies of patients treated with fenfluramines estimated that 1 in 8 patients who receive these agents for >90 days had valvulopathy. 113 It has also been shown that the duration of treatment played a role in the development of clinical valvulopathy (i.e., low prevalence rates are associated with shorter durations of therapy, and higher prevalence rates are associated with longer exposures). 64
The mechanisms responsible for the pathogenesis of the valvulopathy by anorexigens are likely a result of interactions that involve both 5HT receptors and the 5HTT. Fenfluramines have been shown to cause 5-hydroxytryptamine 2B receptor (5HT2BR) activation and/or increased circulating 5HT levels. 33, 104, 111 5HT has a direct mitogenic effect on the valvular subendocardial cells, and this mitogenic effect is mediated by 5HT receptors. 29, 105 Recent studies implicated preferential activation of 5HT2BR as a key initiating step in the pathogenesis of anorexigen-induced valvulopathy in humans. The critical role of 5HT2BR activation in the pathogenesis of valvulopathy is further highlighted by the observation that chemically similar drugs, such as lisuride and terguride, which are selective agonists for 5HT2C and 5HT2A receptors, and antagonists for 5HT2BR were not associated with valvulopathy in humans. 61, 110 Similarly, no increase in the risk of valvulopathy was observed in patients treated with the dopamine agonist pramipexole, which has a low affinity to the human 5HT2BR. 93, 114, 134 Recently, 2 large European studies independently verified the association of valvulopathy with 2 antiparkinsonian drugs, namely, pergolide and cabergoline, which are potent 5HT2BR agonists. 114, 134 As a consequence, it has been strongly recommended to screen future drugs with serotonergic activity and their metabolites at the 5HT2BR comprehensively before launching clinical trials. 110, 111, 116, 117
Translatability Challenges of Drug Effects from Animals to Humans
The translatability of the pharmacologic effects of antiobesity drugs from animals to man for both efficacy and toxicity has historically been poor. For example, agents that affect energy expenditure translate very poorly from rodents or dogs to humans because of differences in regulation of metabolism. However, weight loss induced mostly by reduced food intake tends to translate better, with a notable exception for leptin. 52, 122 Although several targets have been investigated in animal models as potential targets for the antiobesity treatment, a very small number of agents actually have moved into the proof-of-concept stage of human clinical testing. 118 Of these agents tested, only a fraction of them showed meaningful weight-loss efficacy in humans, despite demonstrated efficacy in animal models. On the toxicity side, antiobesity agents that work extremely well may have complicated issues in the safety evaluation (i.e., weight loss restricts dosing and reduces the therapeutic window for safety). Many of the safety issues, such as valvulopathy with fenfluramine and PAH with aminorex were initially not predicted by routine preclinical toxicology studies. To date, there are no validated animal models or preclinical and/or toxicologic screens to accurately predict anorexigen-induced valvulopathy and PAH in humans. Furthermore, there is no good way to predict rare and apparently human-specific adverse effects. Rare effects are likely to be identified only through the conduct of extremely large postmarket clinical trials or extensive monitoring efforts. Given the serious consequences of a market-released drug that is later withdrawn because of serious adverse effects, including overall patient safety, loss of a company's reputation and consumer confidence, and associated litigation, there is an ever growing need for sensitive and specific preclinical screening tools to predict safety liabilities in humans.
Experimental Models of PAH and Valvulopathy
Experiments using appropriate animal models are invaluable in understanding the pathogenesis and pathophysiology of drug-induced toxicities and also in exploring new potential safety biomarkers and in identifying therapeutic targets. A range of candidate models has been developed based on one or more genetic defects and signal transduction abnormalities. The following discussion covers animal models and the application of genomic, transcriptional, and histomorphometric techniques to study anorexigen-induced PAH and valvulopathy in humans.
Experimental models of PAH
Major advances have recently been made toward understanding the pathogenesis and pathophysiology of PAH by using experimental animal studies (Fig. 4). These include 1) 5HT signaling pathway and 2) specific gene mutations. The 5HT signaling pathway has been mechanistically implicated for PAH in humans; 5HT contributes to PAH by acting on extracellular and intracellular sites of pulmonary vascular cells. Also, 5HT effects are mediated extracellulary through 5HT1B, 5HT2B, and 5HT2A receptors and intracellularly via cellular capture of 5HT through the 5HTT. 29, 66, 105 5HT is both a vasoconstrictor and a mitogen that play a role in vascular smooth-muscle cell (VSMC) hyperplasia and hypertrophy. In rats, 5HT-induced pulmonary vasoconstriction is primarily mediated by the 5HT2A receptor. However, in chronic hypoxic rats, increased vasoconstriction in response to 5HT is mediated by both 5HT2A and 5HT1B receptors. 83 Studies in 5HT1BR knockout mice revealed a special role played by 5HT1BR in the pathogenesis of PAH. Vascular remodeling and increased pulmonary arterial contraction in response to 5HT and chronic hypoxia were decreased in 5HT1B −/− mice. 68 Animal models of PAH also support a pathogenetic role of 5HT2BR. In a chronic hypoxia model in mice, treatment with 5HT and dexfenfluramine facilitated both pulmonary VSMC hyperplasia and vascular remodeling mediated through the activation of 5HT2BR. Further evidence suggests that 5HT2BR is required for VSMC hyperplasia and structural remodeling of pulmonary arteries, and the blockade of 5HT2B receptor has been shown to prevent PAH in mice. 73 Similarly, in rats, 5HT plays a role in the development of pulmonary vascular remodeling. 22, 24, 65 5HT increases PAH in rats subjected to long-term hypoxia, and this effect can be blocked by a 5HT antagonist. 23
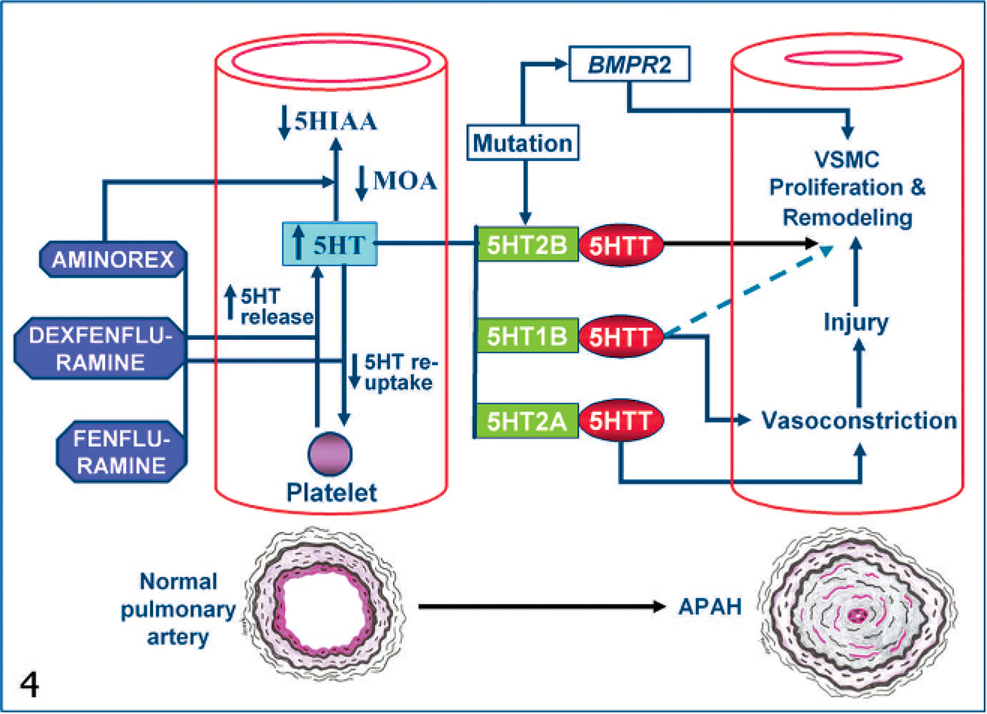
Schematic representation of genetic and 5HT signaling pathways involved in the pathogenesis of APAH. Fenfluramine and dexfenfluramine cause an elevation in plasma 5HT levels by 5HT release from platelets and neurons, and inhibiting reuptake of 5HT by platelets. Aminorex also induces release of 5HT from platelets and inhibition of 5HT metabolism (i.e., metabolic conversion of 5HT to 5-hydroxyindole acetic acid (5HTIAA) by MAO inhibition, which all result in an elevation in plasma 5HT levels. 5HT effects are mediated extracellularly through 5HT1B, 5HT2B and 5HT2A receptors and intracellularly via 5HTT, and include vasoconstriction, VSMC proliferation and remodeling, with resultant narrowing of pulmonary vessel lumen. BMPR2 (bone morphogenetic protein receptor 2) and 5HT2BR mutations act as a trigger/risk factor in genetically predisposed individuals.
The intracellular role of 5HTT is supported by the observation that 5HTT gene overexpression resulted in spontaneous development of PAH in mice in the absence of hypoxia. 40, 82 Administration of the 5HTT inhibitors (e.g., citalopram and fluoxetine) partially reduced increased pulmonary vascular resistance in chronically hypoxic mice. This finding was supported by another experiment, in which hypoxic pulmonary hypertension was attenuated in 5HTT-knockout mice. 63, 85 These findings, together with the demonstration of functional 5HT1BR, 5HT2BR, and 5HTT in human pulmonary arteries, 81, 82 suggest that comparable mechanisms exist in humans and animals, and that expression of 5HT1BR, 5HT2BR, or 5HTT may be used as a diagnostic criterion for susceptibility to PAH. Evidence of 5HT1BR, 5HT2BR, and 5HTT involvement in the pathogenesis led to the suggestion of the use of selective 5HT blockers in PAH therapies 68 in humans.
It is known that 5HT can also influence other regulatory pathways involved in PAH progression through the inhibition of bone morphogenetic protein (BMP) signaling. In vitro and in vivo experiments suggested that 5HT inhibits BMP signaling via modulation of downstream Smad proteins and the expression of bone morphogenetic protein–responsive genes. Long et al. 77 demonstrated that deficiency of BMP receptor-2 (BMPR2) increases susceptibility of PAH induced by 5HT in mice. For example, exposure to increased 5HT led to worsened pulmonary hypertension in mice deficient in BMPR2 (BMPR2 +/− mice). 77 5HT may also regulate the angiopoietin-1/Tie2 signaling pathway, another potential pathogenic contributor of PAH. These findings implicate the interaction between serotoninergic and BMP pathways, perturbation of which may be critical to the pathogenesis of PAH. However, the definitive in vivo effects of BMPR2 mutations have been inconsistent in rats. Adenoviral overexpression of wild type BMPR2 appears to ameliorate pulmonary hypertension in a hypoxic rat model, 106 but overexpression of BMPR2 fails to attenuate chemically (monocrotaline) induced PAH in a rat model. 89 Fenfluramine plus phentermine may increase the risk for PAH by interacting with otherwise silent BMPR2 mutations in humans. 79 More than 20% of patients with fenfluramine-induced PAH have germline BMPR2 mutations. 57, 121 However, it is not known if appetite suppressants, such as fenfluramine and dexfenfluramine, directly interact with BMPR2 or similar receptor proteins. Therefore, fenfluramines may act as a trigger and/or risk factor in genetically predisposed individuals. Despite these advances, a clear mechanistic explanation of the impact of BMPR2 mutations on pathogenesis is still lacking.
Experimental models of anorexigen-induced valvulopathy
The first evidence of anorexigen-induced valvulopathy in animals was reported in 1999, 2 years after the withdrawal of fenfluramine and dexfenfluramine from the US market in 1997. Bratter et al. 7 first demonstrated the thickening of mitral valves in prenatal rats coadministered with dexfenfluramine and phentermine for 14 days, but no histopathology was performed. It is not known whether mitral-valve thickening in rats 7 has similar microscopic features to that of a human valvulopathy induced by dexfenfluramine and phentermine. With knowing that dexfenfluramine and fenfluramine are potent 5HT releasers, as well as re-uptake inhibitors and that phentermine inhibits MAO (an enzyme responsible for degradation of 5HT), the major emphasis was placed on 5HT, 5HT2BR, and 5HTT, and their role in the pathogenesis of anorexigen-induced valvulopathy. More specifically, 5HT2BR activation and/or increased circulating 5HT have been associated with the development of clinically significant valvulopathy in humans. Gustafsson et al. 42 for the first time showed that 3-month 5HT injections in Sprague-Dawley (SD) rats resulted in valvular changes that are morphologically and echocardiographically similar to those seen in human carcinoid heart disease. Another recent study also showed a good correlation between valvular regurgitation and valvulopathy in rats treated with 5HT and pergolide (a potent 5HT2BR agonist) for 5 months. 21
We previously reported the immunostaining and quantitative transcript levels of 5HT2BR and 5HT1BR in normal valve leaflets (mitral, aortic, tricuspid, and pulmonary valves) of SD rats and Cynomolgus monkeys, 2 commonly used preclinical models. 27 The 5HT receptor expression in 4 heart valves is comparable among Cynomolgus monkeys, rats, and humans, and, therefore, the potential exists to gain mechanistic insight to anorexigen-induced valvulopathy from animal studies. An association between spontaneous mitral valvulopathy (SMV) and an increased number of 5HT2BR-positive cells in valve leaflets were described in SD rats, which suggest that 5HT2BR may play a role in its pathogenesis. 28 Similarly, 5HT2BR may play a role in the exacerbation of SMV in Fischer 344 rats by dl-amphetamine treatment. 28 Amphetamine analogs, such as 3,4-methylenedioxymethamphetamine (“Ecstasy”) and 3,4-methylenedioxyamphetamine, were shown to induce prolonged mitogenic response in human valvular cells through in vitro activation of 5HT2BR, similar to those induced by fenfluramine in vivo. 116 It was shown that interference with 5HT transmembrane processing via knocking out 5HTT resulted in valvulopathy in mice, which established a link between 5HTT and the development of cardiac fibrosis and valvulopathy in vivo. The absence of transmembrane processing (via knocking out 5HTT), may result in increased and persistent 5HT receptor interactions, and increased valvular mitogenic activity and extracellular matrix production in the 5HTT-knockout mice. 90
Nodular or segmental thickening and subendocardial fibromyxoid proliferation, noted in SMV in rats, were similar to those seen in human valvulopathy induced by anorexigens (e.g., fenfluramine and dexfenfluramine), ergot alkaloids (ergotamine and methysergide), and carcinoid syndrome. 25 Anorexigen-induced valvulopathy has been shown to exhibit distinctive microscopic features in humans. McDonald et al. 87 demonstrated that anorexigen-exposed valves contain more glycosaminoglycans (GAG) than normal or floppy (also known as myxomatous valvular degeneration) valves. In humans, normal valves are composed of roughly equal amounts of GAGs and collagens, and rarely harbor leukocytes and blood vessels, even as they age. In contrast, valvular lesions associated with carcinoid disease in humans have GAG-rich fibromyxoid tissue and contain a large number of leukocytes and vessels per square millimeter of tissue area. 87 Although blood vessels are more prominent compared with floppy valves, anorexigen-exposed valves were still less vascular but more GAG rich than carcinoid valves. Such features are indicative of a distinctive pathologic process in anorexigen-exposed valves. 87 SMV has a strikingly similar morphology, as well as composition to anorexigen-induced valvulopathy in humans. Valve leaflets with SMV exhibited a greater valve thickness, a higher amount of GAGs, and a lower amount of collagen (5a, b). 28
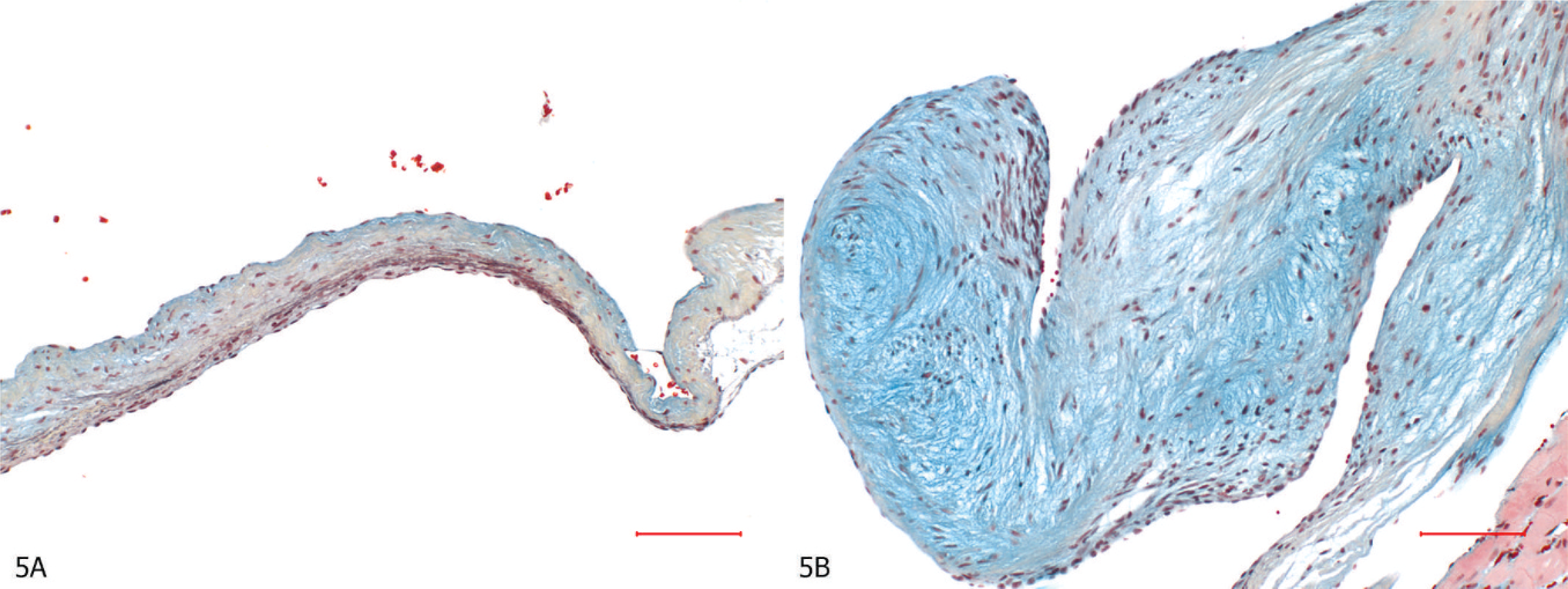
Mitral valve leaflets, Movat's pentachrome stain.
Recently, we investigated whether 7-day 5HT subcutaneous injections would lead to structural and compositional abnormalities in conjunction with transcriptomic modulation of 5HT2BR and 5HTT genes in the aortic and mitral valves of SD rats. Subcutaneous injections of 5HT for 7 days resulted in thickening and compositional alteration of aortic and mitral valves in SD rats, specifically, valve leaflets from 5HT-treated rats had greater valve thickness, a higher amount of GAGs, and a lower amount of collagen. These compositional alterations were associated with upregulation and downregulation of 5HT2BR and 5HTT genes, respectively. 26 This study suggests that the activation of 5HT2BR and inhibition of 5HTT played a significant role in the pathogenesis of 5HT-induced valvulopathy in SD rats. In contrast to 3-month or 5-month studies, 21, 42 we were able to recognize a distinctive 5HT-related valvular matrix alteration in 7 days in the aortic and mitral valve leaflets, thus providing a possible short-term investigative or screening approach to study in vivo drug-induced valvulopathy by serotonergic compounds and as a way to determine or support in vitro selectivity data. Thus, these findings further highlight the necessity and utilization of animal models to screen potential valvular effects of serotonergic compounds.
In conclusion, the worldwide obesity prevalence continues to increase, and there is a growing demand for effective and safe antiobesity drugs. Previous antiobesity drugs, particularly centrally acting agents have poor safety records. Life-threatening safety issues led to the withdrawal of number of antiobesity drugs (e.g., fenfluramine and dexfenfluramine in 1997 and phenylpropanolamine in 2000) from the US market, including a multibillion dollar settlement by the manufacturers of these drugs. These effects were not initially predictable from routine preclinical toxicity studies. However, major advances have recently been made toward the development of appropriate animal models by using novel techniques that will aid in understanding pathogenesis and pathophysiology of anorexigen-induced PAH and valvulopathy.
Footnotes
Acknowledgements
The author thanks Denise Frailey for illustrations, and Drs. Richard T. Miller, Rick A. Adler, Elizabeth Romach, and Roger H. Brown for their critical review of the manuscript.