
Abstract
A 5-month-old Hereford calf with neurologic disease was euthanatized, and a necropsy was done. No gross lesions were seen in the brain. Microscopically, neurons throughout the brain and spinal cord had distended, foamy vacuolated cytoplasm. Ultrastructure showed clear vacuoles filling the neuronal cytoplasm. A lysosomal storage disease was suspected. Sphingomyelinase deficiency was confirmed by biochemical analysis of liver and brain.
Lysosomal storage diseases comprise a group of mostly inherited diseases characterized by enzyme deficiencies affecting the degradation of proteins, carbohydrates, and lipids within lysosomes. These diseases are grouped into glycoproteinoses, sphingolipidoses, mucolipidoses, and mucopolysaccharidoses and have been reported in a variety of animal species. 3 The sphingolipidoses include sphingomyelinase deficiency, called Niemann-Pick disease, which results in the accumulation of sphingomyelin in neurons and macrophages.
A 5-month-old male Hereford calf presented with a 4-week history of neurologic signs, including ataxia, hypermetria, wide-base stance, and strabismus when the head was turned to the left. The calf was euthanatized, and a necropsy was done. The only gross lesion seen was a diffusely tan liver. Brain and spinal cord were normal.
Samples from multiple tissues were fixed in 10% neutral buffered formalin, embedded in paraffin, sectioned at 5 μm and stained with hematoxylin and eosin (HE). Samples of brain were additionally fixed in 3% glutaraldehyde, postfixed in 1% osmium tetroxide, and embedded in plastic (polybed 812). Ultrathin sections for electron microscopy were cut and stained with uranyl acetate and lead citrate.
Histopathology of the brain and spinal cord revealed swollen neurons with distended, foamy, vacuolated cytoplasm (Fig. 1). Affected neurons were located throughout the brain and spinal cord but were most numerous and notable in the cerebral and cerebellar cortices. Ultrastructurally, the cytoplasm of neurons was filled with clear vacuoles, some of which were partially membrane bound (Fig. 2). These vacuoles coalesced to fill the cytoplasm. The microscopic and ultrastructural lesions were indicative of a lysosomal storage disease.
Microscopically, hepatocytes throughout the liver were swollen and had foamy, vacuolated cytoplasm. All hepatocytes stained positive with periodic acid–Schiff (PAS) reagent but did not stain with PAS following pretreatment with amylase. This result indicates glycogen accumulation in the liver.
A lipid extraction procedure was done on liver and brain. Between 0.2 and 0.4 g of tissue was homogenized in 10 volumes of chloroform-methanol (2 : 1, by volume) using a ground glass homogenizer. The solution was filtered through a Pasteur pipette containing glass wool, and one-fifth volume of distilled water was added. After centrifugation at 1,500 × g for 10 minutes, the upper aqueous phase and lower organic phase were separated. The upper phase from the brain extract contained most of the gangliosides. Thin layer chromatography on a 0.01-ml aliquot was performed on a high performance silica gel plate using a mixture of chloroform, methanol, and 0.25 ml 2.5N NH4OH (60-35-8, by volume). After migration of the solvent to the top, the plate was air dried and sprayed with a solution of resorcinol in HCl. Following heating at 100°C while covered with another glass plate for 15 minutes, the gangliosides stained purple. Essentially a normal pattern was obtained, except for a small increase in GM2 and GM3 gangliosides.
The lower phase from the liver extract was evaporated with nitrogen and dissolved in chloroform–methanol (2 : 1) equal to the weight of the tissue sample. A 0.01-ml aliquot was spotted on a silica gel thin layer plate and placed in a tank containing a chloroform–methanol–water mixture (72-28-3.6, by volume). After the solvent ran to the top, the plate was removed, dried, and sprayed with a solution of orcinol in sulfuric acid. Following heating at 100°C for 10 minutes, lipids in the extract were detected. Although quantification of each lipid was not done, there was a great excess of sphingomyelin and cholesterol compared with normal calf liver. This would indicate a diagnosis of sphingomyelinase-deficient Niemann-Pick disease.
An enzyme analysis of a liver extract was performed to confirm the diagnosis of Niemann-Pick disease. A 0.1-g sample of liver was homogenized in 10 volumes of distilled water using a ground glass homogenizer. After determination of the protein concentration, a number of lysosomal enzymes were measured using methods described by this laboratory. 7 The tissue had normal β-galactosidase, α-mannosidase, and β-hexosaminidase activities. However, no acid sphingomyelinase activity could be measured, confirming the diagnosis of Niemann-Pick disease.
Niemann-Pick disease in humans has been described in predominantly 3 forms, types A, B, and C. 2 Niemann-Pick types A and B are caused by a deficiency of acid sphingomyelinase, an enzyme catalyzing the conversion of sphingomyelin to ceramide and phosphorylcholine. 2 Niemann-Pick type A disease has an early onset in infancy of neurologic disease and an acute course culminating in death at a few years of age. Type B disease has little effect on the nervous system and pursues a chronic course with survival into adulthood. Type C disease is not a deficiency of sphingomyelinase, but a defect in 1 of 2 proteins involved in the intracellular transport of cholesterol and other lipids from lysosomes. 1 Intracellular accumulation of cholesterol as a result of these defective proteins leads to inhibition of sphingomyelinase. 5
Niemann-Pick disease type A has been reported in Siamese 3, 6 and domestic short-hair cats 3 and a Miniature Poodle dog. 3 Type C disease has been described in cats, 4 Boxer dogs, 3 and mice. 3 Niemann-Pick disease has not been reported in cattle. The findings in this calf are most consistent with Niemann-Pick disease type A.
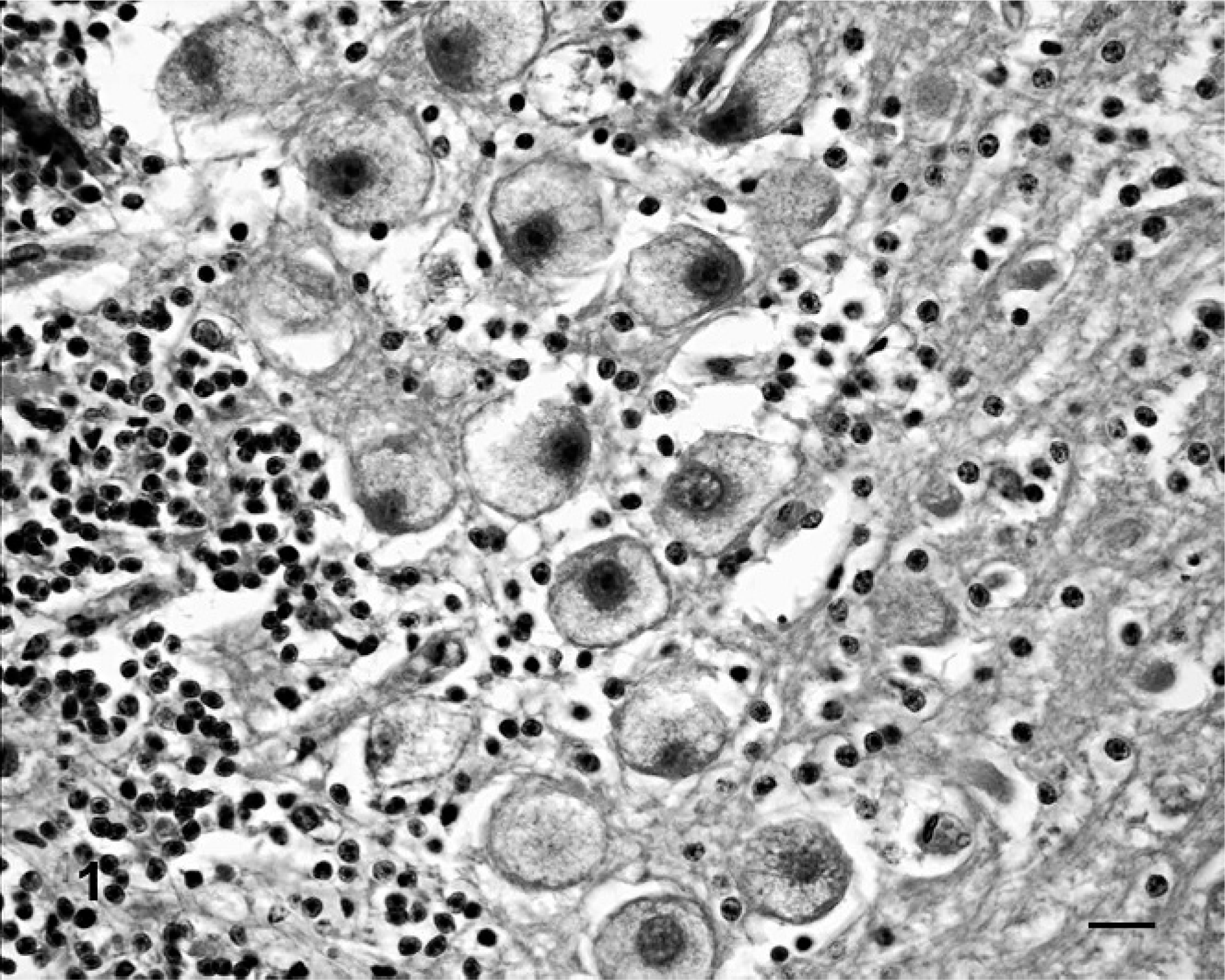
Cerebellum, calf. Neurons have distended, foamy vacuolated cytoplasm. HE. Bar = 25 μm.
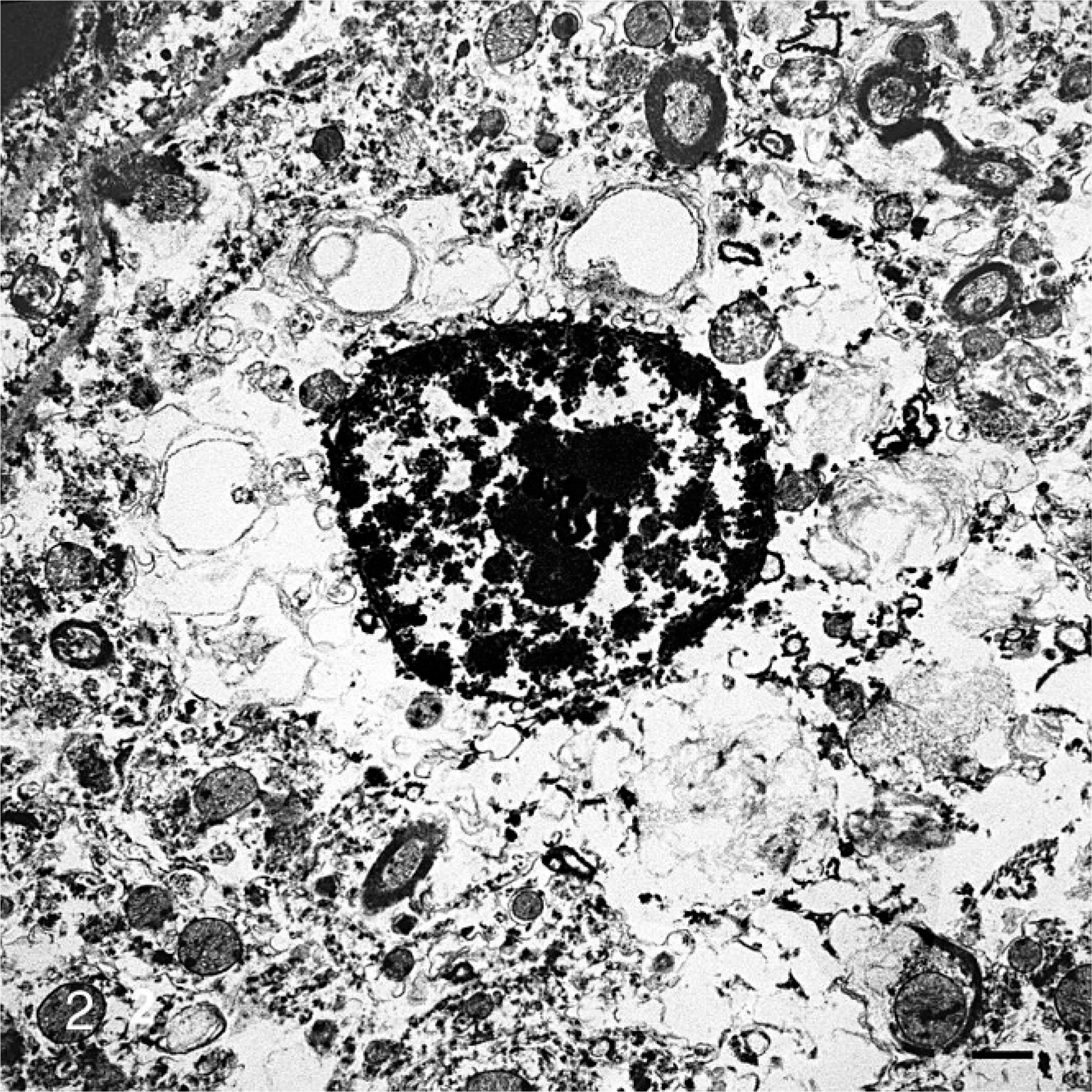
Brain, calf. Neuronal cytoplasm contains clear vacuoles, some of which are membrane bound. Uranyl acetate and lead citrate. Bar = 2 μm.
Footnotes
Acknowledgement
This research was supported by NIH grant DK38795.