
Abstract
A young male Bernese mountain dog presented with neurologic abnormalities consisting of nonambulatory tetraparesis, generalized tremors, and depressed mental status. At necropsy only a mild enlargement of the lateral ventricles was seen. The histologic examination revealed the presence of eosinophilic deposits consistent with Rosenthal fibers (RFs) throughout the white matter of the central nervous system. There was also a marked proliferation of abnormally large astrocytes and limited myelin changes. RFs were most prominent in perivascular, subpial, and subependymal areas, where they were perpendicularly located, producing a pallisaded arrangement. immunohistochemically, RFs were strongly positive for glial fibrillary acidic protein (GFAP), and when they were examined ultrastructurally they appeared as electron-dense amorphous masses located within the processes of astrocytes, most particularly in the perivascular feet. The histologic and immunohistochemical findings of this canine case were consistent with the published neuropathologic descriptions of Alexander disease in humans and in a few dogs, a rare condition that in humans has been shown to be caused by dominant mutations in the GFAP gene.
Keywords
A 4-month-old male Bernese mountain dog presented to the Hospital Veterinario Rof Codina with a 3-week history of progressive tetraparesis and tremors, particularly of the hind limbs. The dog had been treated with prednisone (Dacortin, Merck Farma y Química, S. A., Barcelona, Spain) at 0.5 mg/kg per os twice a day for 3 days, but it did not respond to the therapy and the condition worsened rapidly. On admission, the neurologic examination revealed nonambulatory tetraparesis, generalized tremors and depressed mental status. Postural reactions were difficult to evaluate because of generalized weakness, but they were considered to be abnormally slow. Spinal cord reflexes were decreased in all 4 limbs, but cranial nerve function and muscle tone were preserved. The laboratory data revealed normal urine and blood values. A multifocal involvement of the central nervous system was suspected, and, therefore, sampling of cerebrospinal fluid (CSF) was performed by cerebellomedullary cisternal puncture. CSF analysis showed 5 cells/μl (reference range, <8 cells/μl), total protein concentration of 21 mg/dl (reference range, <25 mg/dl), and absence of neutralizing antibodies against canine distemper virus. Because of the deteriorated neurologic status, and at the request of the owner, the dog was euthanatized, and a complete necropsy was performed.
Gross lesions were found only in the brain, with a moderate enlargement of the lateral ventricles. The brain and the cranial portion of the cervical spinal cord were fixed by immersion in 10% buffered formalin, and then representative coronal slices were embedded in paraffin according to standard laboratory procedures. Paraffin sections (4–5 μm) were stained with hematoxylin and eosin (HE) and luxol fast blue (LFB) techniques. For immunohistochemical detection of glial fibrillary acidic protein (GFAP), paraffin sections were first digested with proteinase K (DakoCytomation, Glostrup Denmark) for 5 minutes at room temperature and then were incubated for 2 hours with a rabbit polyclonal antibody against GFAP diluted 1°3000 (DakoCytomation). Antigen–antibody complexes were detected with an anti-rabbit peroxidase-labeled polymer (EnVision+, DakoCytomation) applied for 30 minutes, and color development was achieved by exposing tissue to diaminobenzidine (DakoCytomation). Finally, the slides were counterstained with hematoxylin, dehydrated, and coverslipped.
For ultrastructural examinations, selected areas from formalin-fixed pieces of cerebellum and medulla oblongata were washed in 0.1 M cacodylate buffer, postfixed in 1% aqueous osmium tetroxide, dehydrated in ethanol solutions, and embedded in epoxy resin (EMbed 812, Electron Microscopy Sciences, Hatfield, PA), according to standard techniques. Thin sections were counterstained with uranyl acetate and lead citrate and examined under a JEOL JEM-1011 transmission electron microscope (JEOL, Tokyo, Japan).
Histologic changes consisted of a marked proliferation of astrocytes with abnormally large cell bodies, as well as numerous hypereosinophilic, round, or elongated bodies distributed throughout the white matter of the brain and the cranial portion of the cervical spinal cord. These eosinophilic deposits were consistent with Rosenthal fibers (RFs), and they were especially prominent around blood vessels, where they produced a pallisaded arrangement (Fig. 1). Lesions were most pronounced from midbrain to the spinal cord, as the gray matter also was involved and many nuclei showed diffuse accumulation of RFs in the neuropil (Fig. 2). In these areas, RFs also were disposed perpendicularly below the pia matter and ependyma, and the reticular formation showed vacuolization. The enlarged astrocytes expressed increased levels of GFAP, and, in addition, RFs were found to be strongly immunoreactive for GFAP (Figs. 3, 4). Although it was not a salient feature, LFB staining revealed diminished myelin in the pyramids and in the white matter of the spinal cord. At the ultrastructural level, astrocytes showed abnormally large cell bodies and thick cytoplasmic processes, both densely packed with huge amounts of glial filaments (Fig. 5). RFs consisted of electron-dense, non–membrane-bound granular aggregates of uneven size and intimately associated with the matrix of glial filaments. They were located within the processes of astrocytes, particularly in the perivascular feet (Figs. 5, Figs. 6).
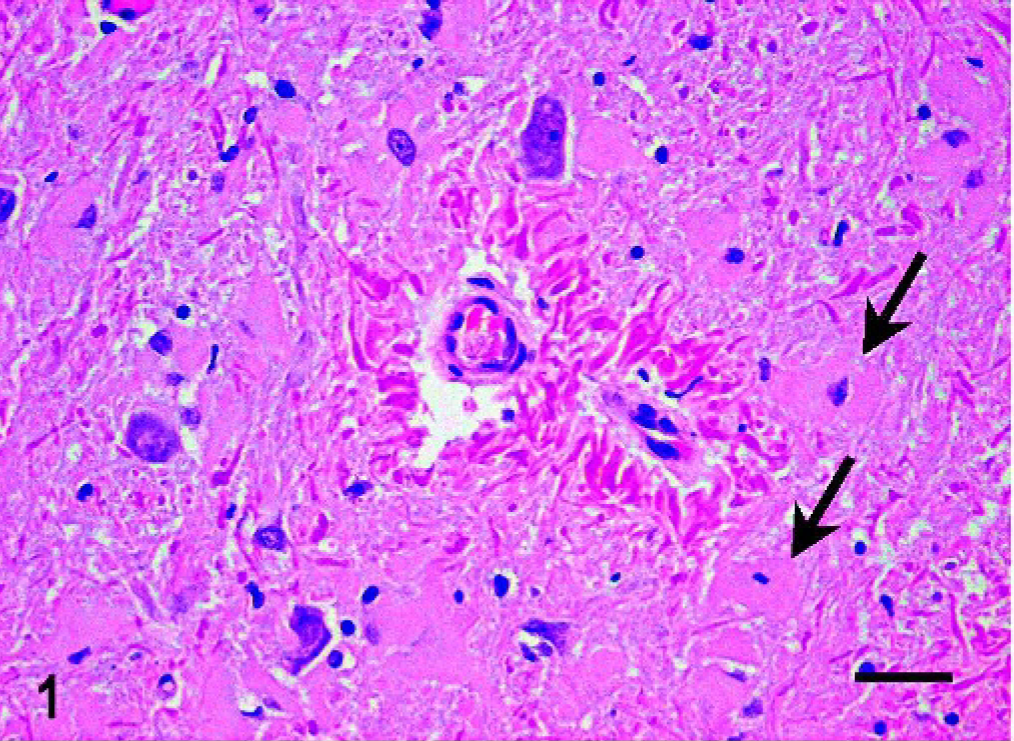
Central nervous system, medulla oblongata; dog. RFs encircling 2 small vessels in the gray matter of the cervical spinal cord. Note the presence of astrocytes with abnormally large bodies (arrows). HE. RFs = Rosenthal Fibers. Bar = 30 μm.
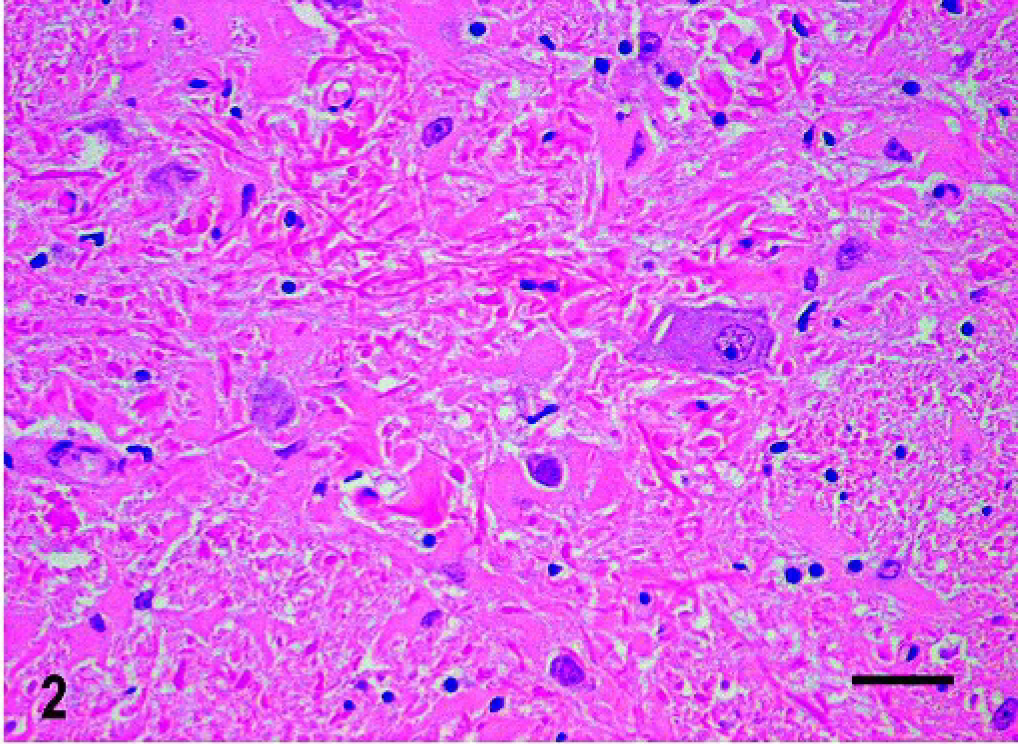
Central nervous system; dog. Diffuse accumulation of RFs in the gray matter of the medulla oblongata. HE. RFs = Rosenthal Fibers. Bar = 30 μm.
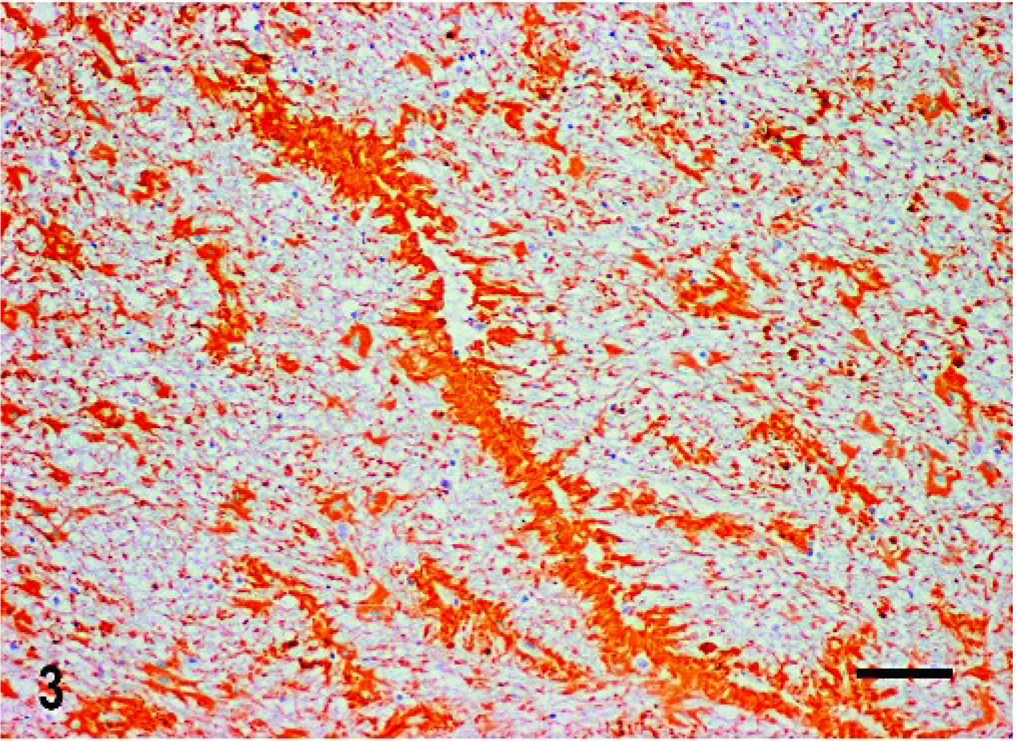
Central nervous system; dog. Immunoreactivity to GFAP in perivascularly arranged RFs and in enlarged astrocytes of the white matter of the medulla oblongata. Indirect immunohistochemical technique; hematoxylin counterstain. RFs = Rosenthal Fibers. Bar = 60 μm.
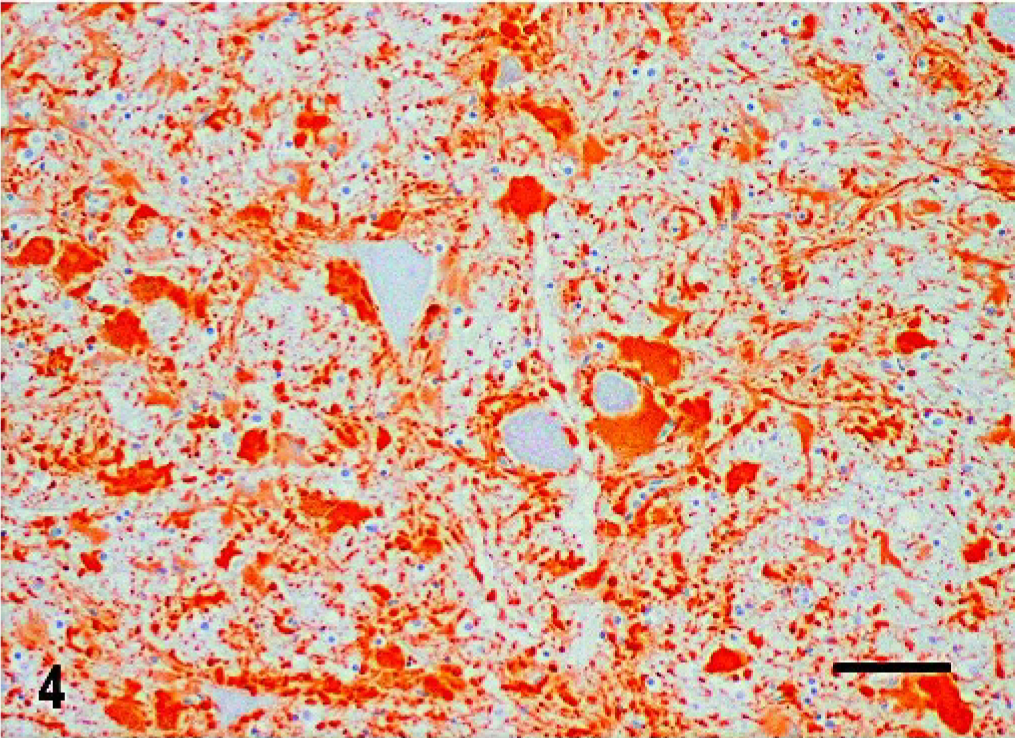
Central nervous system; dog. GFAP-positive hypertrophic astrocytes surrounding neurons in the gray matter of the cervical spinal cord. Indirect immunohistochemical technique; hematoxylin counterstain. Bar = 60 μm.
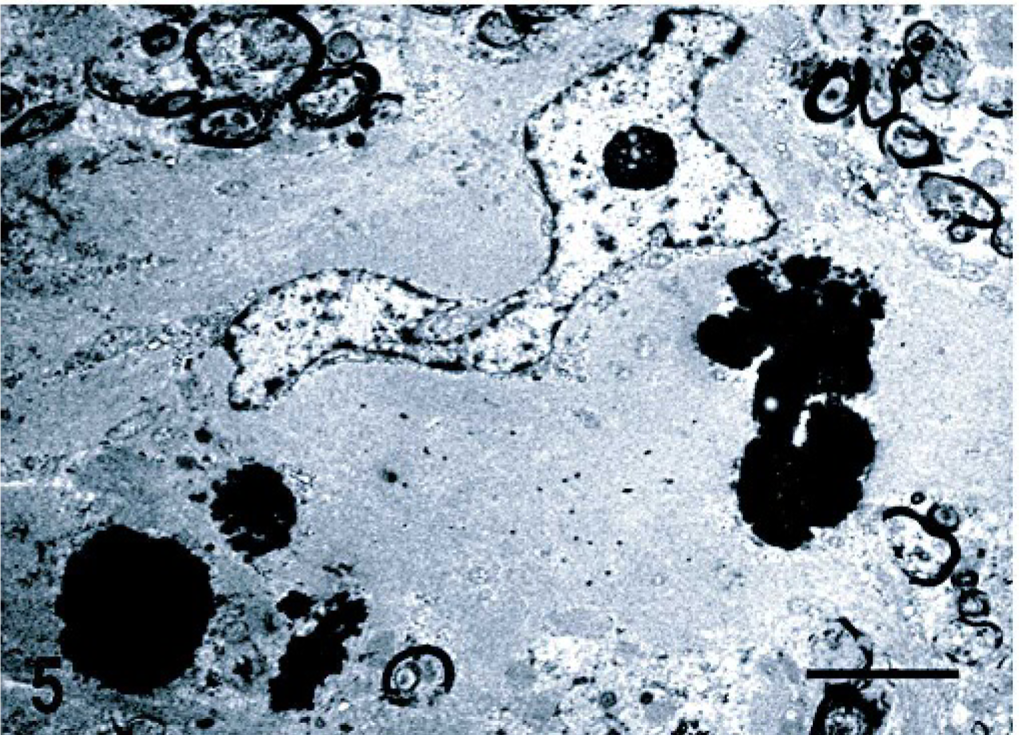
Central nervous system, cerebellar white matter; dog. Enlarged astrocyte with a cytoplasm densely packed with glial filaments. The electron-dense deposits are located within the processes of adjacent astrocytes and consist of granular aggregates intimately associated with the matrix of glial filaments. Transmission electron microscopy; uranyl acetate and lead citrate stain. Bar = 2 μm.
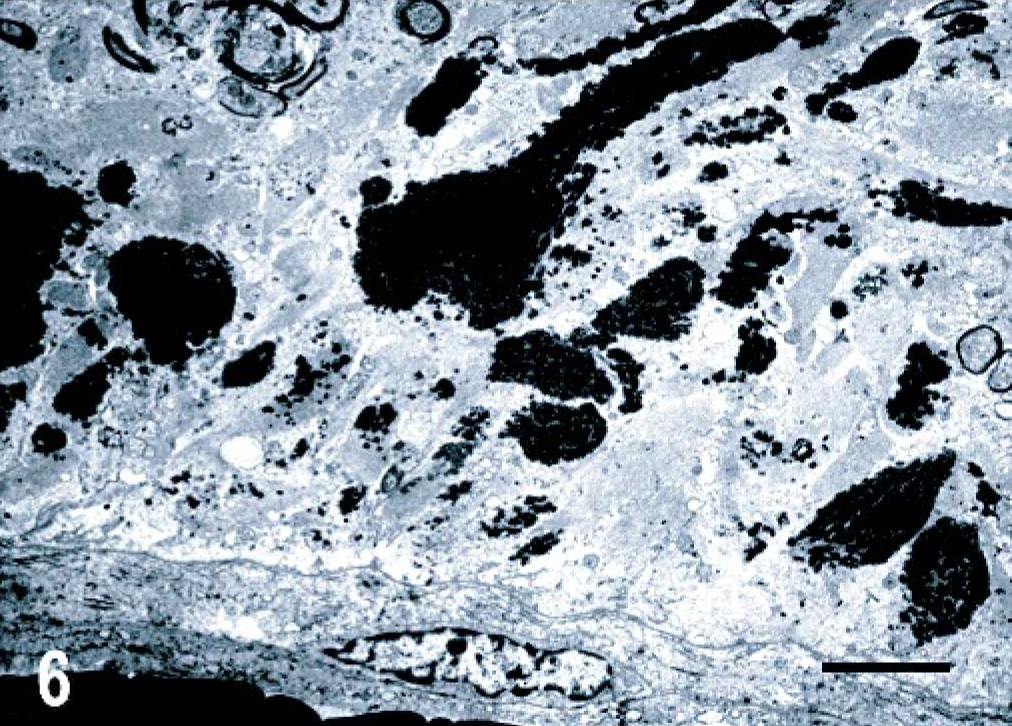
Central nervous system, cerebellar white matter; dog. Astrocyte perivascular feet containing large electron-dense aggregates. Transmission electron microscopy; uranyl acetate and lead citrate stain. Bar = 3 μm.
The histologic and immunohistochemical findings of this canine case were consistent with those of the reported descriptions of Alexander disease (AD) in humans, a rare and devastating neurologic condition characterized pathologically by the widespread accumulation within dystrophic astrocytes of protein inclusions called RFs. 3, 4, 6 It has been shown that the major chemical components of RFs are GFAP, small heat shock proteins (αB-crystallin and hsp27), and ubiquitin. 3 Although unified by the predominant presence of RFs, based on the age at onset and the type of clinical signs the disease was divided into infantile, juvenile, and adult forms. The infantile type was the first to be described, and, because it typically presents with accompanying severe myelin changes in the frontal lobes, AD was classified as a leukodystrophy. However, in both the juvenile and adult forms pathology is primary confined to the brain stem, and demyelinating lesions may or may not be present, making it difficult to label these 2 AD forms as leukodystrophies. 4, 6 Recent investigations have demonstrated that dominant missense mutations of the GFAP gene account for nearly all infantile, juvenile, and adult phenotypes of the disorder, and, therefore, that AD is a primary astrocytic disease that induces secondary dysfunction of oligodendrocytes. 3, 4, 6 AD cases can be either sporadic or familial: in the former case they are likely caused by de novo dominant GFAP mutations arising from germinal or postzygotic events; in the latter, by transmission of a mutant GFAP allele from a heterozygous parent to his or her children. 3, 4, 6
In animals, RFs have very rarely been observed, being only reported in cases clinically and morphologically identical to AD in humans. They have been described in 5 dogs of different breeds (2 Labrador retrievers, 1 Scottish terrier, 1 Miniature poodle, 1 Bernese mountain dog) 1, 5, 7, 8, 10 and in 1 sheep. 2
As has been suggested by other authors, 9 if we take into account the age at onset and the type and topography of lesions in the central nervous system, all canine cases, including this dog, could be considered juvenile forms of AD, whereas the adult sheep that showed RFs and limited myelin changes could be viewed as an adult-onset form of AD. Infantile AD cases, if present in veterinary medicine, are likely to go unnoticed, as the affected individuals would die soon after birth. On the other hand, the condition in dogs seems to be sporadic, even though the possibility of congenital transmission cannot be ruled out, since there have been 2 descriptions of canine AD in which the littermates also showed neurologic abnormalities: in one case the disease was diagnosed histologically in 2 littermate Labrador retriever dogs, 5 but in the second instance 3 littermates of a Scottish terrier with AD were euthanatized with no pathologic confirmation of the disease. 1 Unfortunately, no information was available on the familial history of the dog described here. Finally, it is important to stress the fact that the condition in dogs is morphologically identical to human AD, but it is still necessary to demonstrate a similar pathogenesis, as in canine cases the GFAP gene has not been studied.