
Abstract
We report an encephalomyelopathy in three 18-month-old Merino sheep with features of adult-onset Alexander’s disease (AD), a human primary astrocytic disorder. The signature histologic finding was the presence of numerous hypereosinophilic, intra-astrocytic inclusions (Rosenthal fibers), mainly in perivascular, subpial, and subependymal sites, especially in the caudal brain stem and spinal cord. Although AD usually results from mutations in the glial fibrillary acidic protein (GFAP) gene, no such mutation was detected in these sheep. However, the annual clinical presentation of this disorder in a few sheep in the affected flock is suggestive of a familial pattern of occurrence.
Keywords
Alexander’s disease (AD), named for the pathologist W. S. Alexander, is defined by the presence of eosinophilic intra-astrocytic inclusions termed Rosenthal fibers (RF). 2 The fibers, first described by Rosenthal, 17 have been shown to be ubiquitinated aggregates of glial fibrillary acidic protein (GFAP) and the heat shock proteins (HSPs), αB-crystallin, and HSP27.9,20 Rosenthal fibers are found in swollen astrocytic processes and perikarya throughout the central nervous system, but they are more numerous in perivascular, subpial, and subependymal sites. Rosenthal fibers are not pathognomonic for AD and are also found, albeit in greatly reduced numbers, in chronic reactive astrocytosis and low-grade astrocytomas in humans, but they are seldom encountered in animal neuropathology.8
The phenotypic expression of human AD depends on the age of onset; 3 forms (infantile, juvenile, and adult) are recognized. The infantile form, occurring up to 2 years of age, is the most severe and rapidly progressive. Marked mental retardation and spastic paresis result in death between 1 and 10 years of age. The juvenile form, presenting from 4 to 10 years of age, features spasticity, bulbar signs, and mental deterioration. Adult-onset AD is generally the mildest form, presents from adolescent to geriatric age, and is sometimes only detected at autopsy. The adult form, once considered the rarest, is more prevalent than previously thought and might even be the most common form. In adult-onset AD, lesions are predominantly in the lower brain stem, whereas in infantile or juvenile forms, supratentorial abnormalities prevail.7,8,14 In almost all human AD cases, a heterozygous missense mutation is identified in exons 1 to 8 of the GFAP gene.3,14 Thus, clinical diagnosis requires genetic confirmation in conjunction with consistent magnetic resonance imaging (MRI) abnormalities.14,21
Spontaneous encephalopathies resembling AD have been reported in 6 juvenile dogs (onset at 9 weeks to 6 months of age) of both sexes and different breeds (2 Labrador retrievers, 2 Bernese Mountain dogs, a Scottish Terrier, and a Miniature Poodle).1,4,11,16,19,22 In 2 canine cases, littermates were also affected.4,11 All affected dogs had a rapidly progressive motor degeneration. An AD-like encephalopathy has also been reported in a 4-year-old, female, White Alpine sheep that was unable to stand, and progressed to lateral recumbency with paddling of limbs and intermittent profound depression or hyperexcitability. 5 In none of these cases was the genome examined for GFAP mutations. We report here the clinicopathologic, immunohistochemical, ultrastructural, and molecular genetic investigation of an AD-like encephalomyelopathy in 3 Merino sheep.
Materials and Methods
Three 18-month-old Merino ewes were evaluated from the same farm. Case no. 1, pregnant with twin fetuses at approximately 1 month gestation, was necropsied in the field; samples of brain, spinal cord, liver, and heart were formalin-fixed and submitted for histologic examination. Case nos. 2 and 3 were necropsied by the authors; brain and spinal cord were fixed in 40% formalin for 2 weeks. Selected tissues, including eyes and optic and peripheral nerves, were fixed in 10% buffered formalin. For histologic examination, each brain was sliced coronally at 5-mm intervals, routinely processed, embedded in paraffin, sectioned at 6 µm, and stained with hematoxylin and eosin (HE). Duplicate sections of brain and cord were also stained for myelin (Luxol fast blue and Weil’s methods).
For immunohistochemistry on brain and cord, the following antisera were used: polyclonal antibodies to GFAP (Dako, Carpentaria, CA, USA, Cat #Z0334), ubiquitin (Dako, Carpentaria, CA, USA, Cat #Z0458), αB-crystallin (Novocastra, UK, Cat #NCL-ABCrys), and monoclonal antibodies to vimentin (Dako, Carpentaria, CA, USA) and HSP27 (Stressgen, USA, Cat #SPA-801), using a standard streptavidin-biotinylated immunoperoxidase technique. In brief, sections were dewaxed in xylene, rehydrated through alcohols, and subjected to antigen retrieval using citrate buffer (pH 6) in a calibrated microwave oven for 10 minutes. Slides were allowed to cool and were washed twice in phosphate-buffered saline (PBS; pH, 7.4), after which endogenous peroxidase activity was quenched. Nonspecific proteins were blocked with normal horse serum for 20 minutes. Antibody to GFAP was diluted 1:40,000; ubiquitin, 1:4,000; αB-crystallin, 1:2,000; vimentin, 1:1,000; and HSP27, at 1:3,000 at room temperature overnight. The following day, the sections were washed twice in PBS; then, either a biotinylated anti-rabbit secondary (Vector Laboratories, USA, Cat #BA-1000) or a biotinylated anti-mouse secondary (Vector Laboratories, USA, Cat #BA-2000) antibody was applied for 60 minutes at room temperature. Following 2 PBS washes, the slides were incubated 1 hour at room temperature with a streptavidin-conjugated peroxidase tertiary antibody (Pierce, USA, Cat #21127). The immunoreactions were visualized using diaminobenzidine tetrahydrochloride (DAB) as chromogen, washed, and counterstained with hematoxylin. With every antibody run, a positive control and a negative primary control were included.
For ultrastructural examination, small cubes of brain were fixed in 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.3) for 2 hours, rinsed in buffer, and postfixed in 2% osmium tetroxide in 0.1 M cacodylate buffer (pH 7.3) for 1 hour. Fixed tissues were then dehydrated in ethanol, cleared in propylene oxide, and embedded in Epon. Semithin sections (1 µm) were stained with toluidine blue for light microscopy; ultrathin sections were contrasted with uranyl acetate and lead citrate.
For genetic analyses, genomic DNA was extracted from frozen brain and liver from 2 sheep (Case Nos. 2 and 3) and 1 age- and breed-matched control sheep using the QIAamp DNA minikit (Qiagen, http://www.qiagen.com). Only a portion of the ovine GFAP mRNA sequence has been published (GenBank AJ551397.1); therefore, primers were designed to amplify the GFAP gene using conserved regions in the human (RefSeq NC_00017.9/NM_002055.3) and bovine (RefSeq NC_007317.3/NM_74065.2) genomes. Fragments corresponding to the highly conserved regions of exons 1 to 9 of the bovine GFAP gene were amplified using the primers listed in Table 1. These amplicons covered the entire protein-coding region of the human gene, including the mutation hotspots for AD. The polymerase chain reaction (PCR) sample consisted of 1 × PCR buffer II, 2.5 mM MgCl2, 1.25 U AmpliTaq Gold (all from Applied Biosystems, Foster City, CA, USA), 0.2 mM dNTPs (GE Healthcare, Little Chalfont, UK), 0.8 µM each primer (Sigma-Aldrich, Castle Hill, Australia) and 20 ng DNA, in a final volume of 25 µL. Following preincubation at 94°C for 10 minutes, touchdown PCR conditions were used. These conditions consisted of 20 cycles of 30 seconds at 94°C, followed by 30 seconds at 65°C-0.5°C/cycle and 30 seconds at 72°C, and then a further 30 cycles with the annealing temperature constant at 55° C. A final step of 7 minutes at 72°C allowed the completion of any partial extension products. The resulting products were sequenced using the primers used for amplification and standard dye terminator sequencing chemistry (Applied Biosystems) and compared to the bovine reference sequence.
Primers Used to Test for Mutations in the GFAP Gene

Results
Signalment and Clinical Features
The 3 sheep were from a farm near Karoonda in the Mallee region of South Australia. In each of the past 5 years, several Merino sheep, of both sexes, in the 12–18 month age range, developed hind limb incoordination. When the flock was driven, these sheep lagged behind and eventually collapsed; after a short time, they regained their feet. Over the next 3 months, they gradually lost hind limb function and could move only using the forelimbs. They eventually became permanently recumbent and would have perished if not euthanatized. Wasting was not a feature until late in the course of the disease, when the affected sheep were unable to obtain feed.
A neurologic examination of sheep No. 3, performed by the primary author, included testing of most cranial nerves (which were adjudged normal), observation of mentation (which was likewise adjudged normal), as well as wheel barrowing, fore- and hind-limb hopping, pain perception in all limbs, and testing of patellar reflexes. These tests did not allow definitive distinction between upper motor neuron versus lower motor neuron deficits.
Pathologic Findings
No pertinent macroscopic lesions were noted at necropsy or upon trimming of fixed tissue, including brain and spinal cord. The entire spinal canal was sectioned longitudinally in sheep nos. 2 and 3; no gross abnormalities were noted.
Histologic lesions were confined to the central nervous system in all 3 sheep. In case no. 1, numerous RF were found throughout the brain and spinal cord, particularly in perivascular astrocytic end-feet (Fig. 1) and in subpial (Fig. 2) and subependymal locations where astrocytes normally form a dense meshwork of processes. Rosenthal fibers often appeared as perpendicular arrays, were more numerous in white than gray matter, and in the spinal cord and medulla oblongata than in more rostral aspects of the brainstem or cerebellum. Rosenthal fibers were irregularly shaped, elongated, or round, deeply eosinophilic intra-astrocytic aggregates that varied in diameter from 4 to 20 µm. Hypertrophied astrocyte cell bodies (Figs. 2 and 3) paralleled in density the number of RF and were thus more numerous in subpial and perivascular locations in the caudal brain stem and spinal cord. Affected astrocytes approached a diameter of 60 µm and had large nuclei, prominent nucleoli, and pale hyaline cytoplasm (Fig. 2). Scattered astrocytes were bi- or tri-nucleated; rare astrocytic nuclei contained large eosinophilic inclusions, consistent with cytoplasmic invaginations.
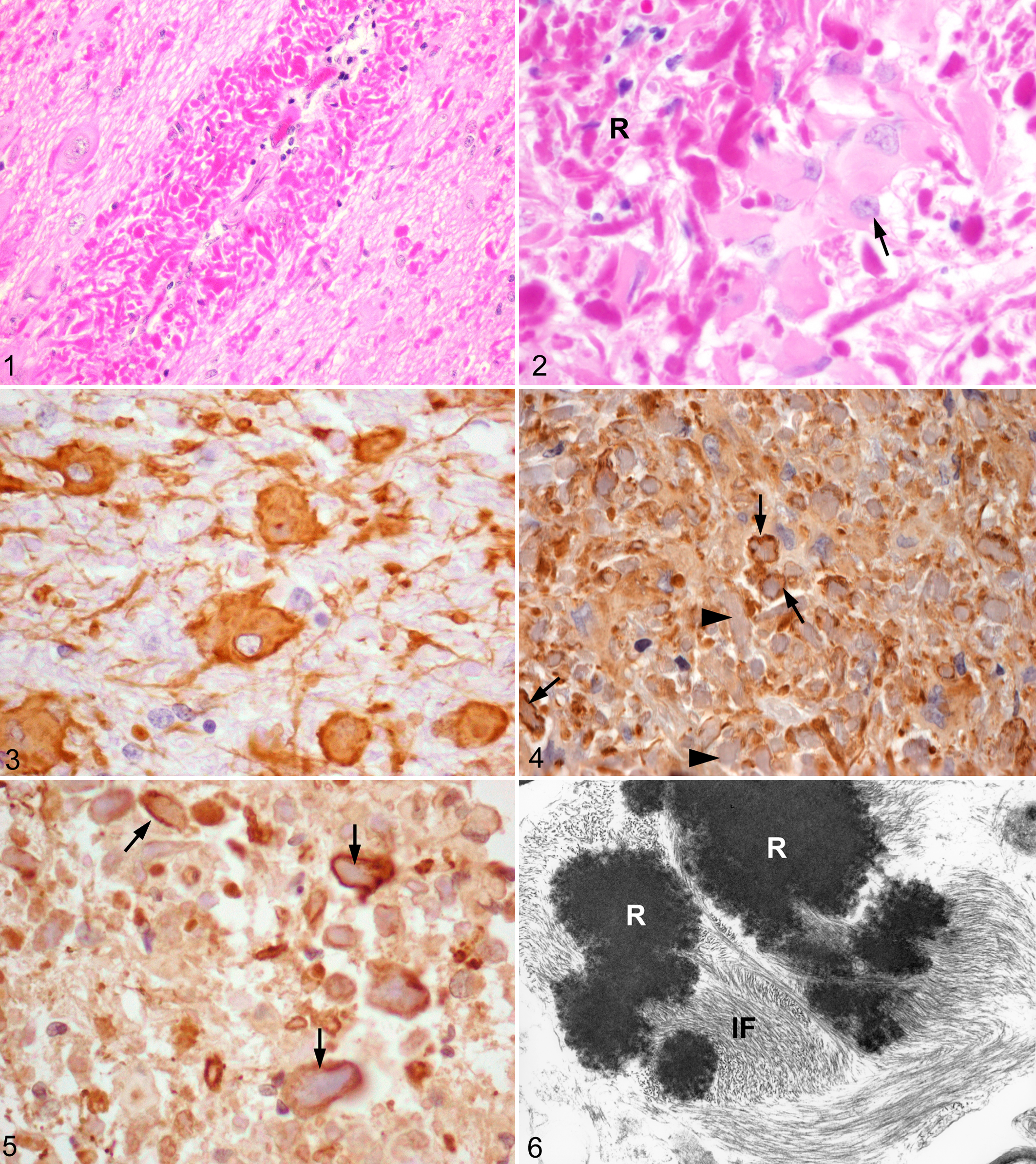
Cerebellum, sheep no. 2. Perivascular aggregates of Rosenthal fibers. Hematoxylin and eosin.
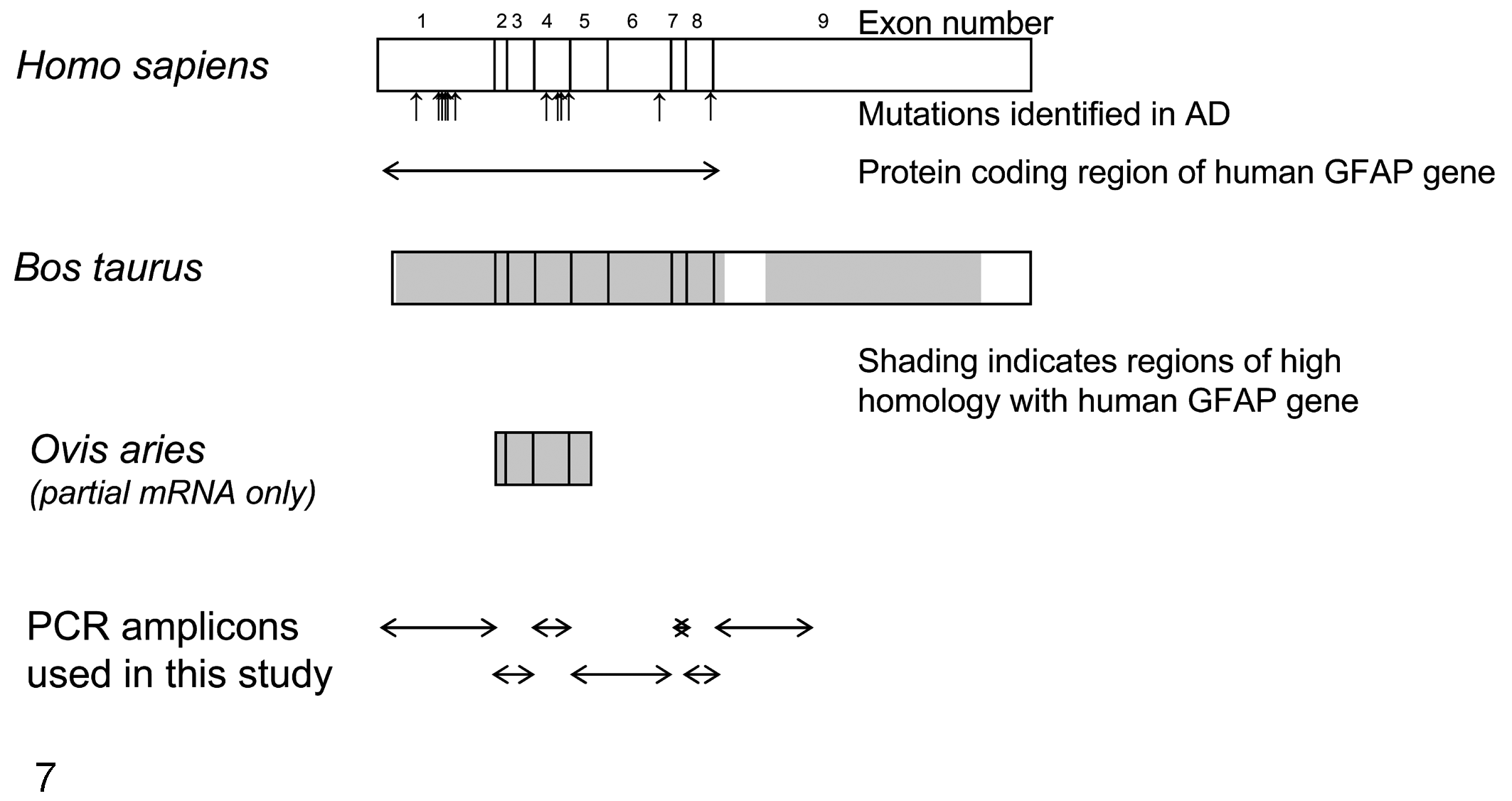
Schematic of the alignment of exons of human (NM_002055.3), bovine (NM_174065.2) and ovine (AJ551397.1) glial fibrillary acid protein (GFAP) genes. The position of mutations associated with Alexander’s disease is marked (↑) below the human gene. The human and bovine genes share 86% homology (shaded on the bovine gene). The partial sequence available for the ovine GFAP gene has 90% and 97% homology with human and bovine genes, respectively. The PCR amplicons are represented diagrammatically below the genes (see Table 1 for primer sequences).
Within the cerebral cortex, RF were in subpial locations and were attended by low numbers of hypertrophied, and occasionally multinucleated, astrocytes with stout fibrillar extensions into the pial membrane. Subcortical and central white matter did not contain RF, although there were a few hypertrophied astrocytes. However, RF density was high in the periventricular and subependymal regions around the third ventricle, and to a lesser extent in the subependymal region of the temporal horns. The white matter of the hippocampus and corpus callosum contained a few RF and mild astrocytic reaction. Perivascular RF were observed in the central gray matter.
The molecular layer of the cerebellum had small numbers of subpial RF, which, as in the cerebral gray matter, were attended by a few hypertrophied astrocytes. The granular layer, and particularly the folial and deeper white matter, contained numerous RF and associated hypertrophied astrocytes.
In the pons and medulla oblongata, prominent subpial, subependymal, and perivascular RF aggregates were accompanied by numerous hypertrophied astrocytes, with fewer in the white matter. Ependymal cells were hypertrophied.
Lesion intensity and distribution was consistent at all levels of the spinal cord, with florid RF formation, generally greatest in perivascular and subependymal locations. Gray matter contained nearly as many RF as did white matter. There were also moderate to large numbers of hypertrophied astrocytes, few swollen axon cylinders, and scattered axonal degeneration.
In the hypertrophied astrocytes, the pattern of immunoreactivity for GFAP was variable but generally located around RF (Fig. 4), whereas most RF themselves were immunonegative. Immunopositivity for αB-crystallin was generally peripherally in and around RF (Fig. 5), though some RF were nonreactive. Rosenthal fibers were labeled with antibodies against HSP27 and ubiquitin, but not vimentin.
Ultrastructurally, within astrocytes, RF were electron dense (Fig. 6), rounded or elongated, coarsely granular structures of variable size with irregular contours. They were surrounded by densely aggregated sheaths of intermediate filaments, some of which appeared to be continuous with the central osmiophilic mass of the fiber (Fig. 6).
All neuronal populations in the brain and spinal cord, including those in cortical, central, brain stem, and spinal gray matter, as well as the pyramidal and granule cells of the hippocampus and cerebellar Purkinje and granule cells were morphologically normal. Oligodendroglial nuclei also appeared normal.
In sheep nos. 2 and 3, RF were greater in number and density but had similar distribution to that of case no. 1. There was mild, axonal degeneration in the spinal cord, with ellipsoids (digestion chambers) that contained axon fragments and, focally, macrophages. Myelin ellipsoids were in all funiculi but were focally more prominent in, or largely restricted to, the ventral funiculi.
Molecular Genetics
Analysis of the sequencing data revealed no heterozygous GFAP mutations in the brain of case no. 2 or case no. 3. Numerous homozygous nucleotide variations were identified in both the affected and a control sheep in comparison to the published bovine sequence, but these were considered to represent interspecies variations. Genetic analysis of case no. 1 was unsuccessful, probably owing to the extended time the brain was immersed in formalin.
Discussion
The age of these 3 sheep, neuroanatomical distribution of RF, prominent involvement of the medulla oblongata and spinal cord, and paucity of myelin loss was similar to adult-onset AD in humans.6,7,14,18 The previously reported AD-like condition in a sheep also had prominent RF formation in subpial, subependymal, and perivascular sites, particularly in the cerebellar white matter. 5 Variable myelin rarefaction was found, especially in the cerebellar folial white matter, but lipid-laden macrophages were not observed, and axons appeared normal. The spinal cord was not examined. 5 In contrast, the AD-like encephalopathies reported in dogs resembled the juvenile form of human AD.4,11,16,19,22
In human AD, pathologic and radiologic changes are most pronounced in the infantile form. Modern imaging techniques permit demonstration of leukoencephalopathy and macroencephalopathy, which may be used as diagnostic criteria. In long-term brain imaging studies, white matter abnormalities progressively spread from periventricular to subcortical regions and from frontal to posterior lobes. Histologically, myelin loss has a similar distribution and progression, hence the categorization of AD as a leukodystrophy. In adult-onset cases, radiologic and histologic changes are most obvious in the brain stem, cerebellum, and upper spinal cord, and myelin loss may be absent, suggesting that AD in older patients should not be classified as a leukodystrophy.14,21
The human GFAP gene is on chromosome 17q21 and contains 9 exons spanning almost 10 kb (Fig. 7). In over 90% of human cases, AD is an autosomal dominant disorder resulting from mutations in the GFAP gene, which encodes for the intermediate filament protein GFAP, which is specific to astrocytes. However, there is substantial phenotypic variability and age of onset for the same mutation (http://www.waisman.visc.edu/alexander/). New mutations are regularly found, and, in most cases, the parental genes do not contain the mutation. Accordingly, though AD is a dominant disorder, there are so many mutations that the pattern of inheritance resembles a sporadic condition. A familial pattern is also occasionally demonstrable in the adult form when patients survive long enough to reproduce. Moreover, although most GFAP mutations arise in the parental (usually paternal) germ line, there is some evidence for a somatic origin in a few cases.3,10,11,15
Mutations in the GFAP gene frequently occur de novo, especially in infantile cases, whereas in adult-onset AD, both sporadic cases with de novo mutations and familial cases have been reported. The mutations described to date generally involve highly conserved domains. However, since an occasional mutation has been associated with all 3 forms, the genotype-phenotype relationship is complex. Nearly all mutations in AD have been heterozygous missense mutations, with hotspots at R79 in exon 1 and R239 in exon 4. There are currently 20 different GFAP mutations associated with adult-onset AD. It is unclear why the lesion distribution and age of onset differs among AD patients, but varying mutation sites might differently affect GFAP production. Moreover, other genetic or as yet unknown environmental factors may influence phenotype and might have been operative in the ovine cases presented here. Mutations of other genes, perhaps in those encoding for a protein involved with GFAP, may also account for the small number of AD cases without GFAP mutations. Males and females with identical mutations often have different clinical presentations, suggesting that sex may modify disease progression. Furthermore, clinical signs can vary between affected members in the same family, implying that modifier genes and other factors might play a significant role in the ultimate expression of the clinical phenotype.14,15,18
In AD, dominant gain-of-function mutations result in an overexpression of abnormal GFAP with filament disorganization (impaired polymerization and assembly), decreased solubility, and possibly ineffective degradation and clearance of the aberrant protein. There is probably a threshold for GFAP expression, beyond which the accumulation of the abnormal protein proves toxic to astrocytes. Precipitation of mutant GFAP protein as RF may be initiated by stress, which also results in the production of ubiquitin and heat shock proteins, including αB-crystallin. This step could be followed by microglial activation with concomitant loss of neurons and/or synaptic density. Moreover, there is a link between astrocytes and myelin formation and maintenance, with astrocytes also regulating oligodendrocyte survival. The toxic accumulation of GFAP in AD patients may thus perturb these astrocytic functions, affecting myelin formation and leading to its loss. Astrocyte derangement might also produce excess extracellular glutamate, which is excitotoxic to neurons and oligodendrocytes.13,14
Protein aggregates, morphologically and immunohistochemically indistinguishable from RF, can be induced by overexpression of human GFAP in transgenic mice, 12 but the mice lack overt white matter abnormalities, indicating only partial modeling of the AD pathophysiology. 12 The pathogenesis, therefore, appears complex and probably involves interaction among astrocytes, other glia, and neurons.13,14
Neurologic examination was performed in only 1 of the sheep reported here, and results did not facilitate correlation between clinical signs and lesions; however, the hind limb incoordination with progression to paralysis resembled the clinical signs in some canine cases 22 and in the single reported ovine case of Rosenthal fiber encephalopathy. 5 Tetraparesis/tetraplegia probably would have eventually developed in affected sheep if the clinical course had been longer, which is suggested in the farmer’s history, and by the difficulty sheep no. 2 had in supporting weight on its front legs (in contrast to sheep no. 3).
In human adult-onset AD, a diagnosis based upon clinical criteria is problematic, for the course of the disease is unpredictable. 14 However, AD is a primary astrocytic disorder and, because astrocytes provide metabolic support to both neurons and oligodendrocytes and facilitate neurotransmitter recycling, functional perturbations of astrocytes would be expected to have deleterious effects on neurons and/or oligodendrocytes. These functional changes may not manifest as structural alterations.
Because the entire ovine GFAP gene has not been sequenced, we used highly conserved regions to amplify and analyze the sequence corresponding to the entire protein-coding region of the human GFAP gene (Fig. 7). As in occasional AD patients, mutations were not found in coding regions of GFAP in either of the 2 tested sheep. We must therefore consider the possibilities that mutations are localized to noncoding regions, affect other gene targets that indirectly influence GFAP turnover, or enhance the level of GFAP expression. Moreover, we must consider the possibility that environmental factors contribute significantly to disease manifestations.
Though the authors suspect that the disease in these sheep has a genetic basis, inadequate breeding records and the often yearly introduction of new rams precluded a detailed evaluation of the pedigree. However, the farmer indicates that this syndrome is a recent occurrence, and that AD is not found on neighboring sheep farms. The availability of additional cases from this farm will contribute to molecular and pathologic evaluations, so the possible genetic basis of the disease may be further investigated. If proved, this animal model might also lead to a better understanding of the genetic basis of AD and the mechanisms by which GFAP mutations produce neurologic dysfunction.
Footnotes
The authors declared that they had no conflicts of interest with respect to their authorship or the publication of this article.
The authors declared that they received no financial support for their research and/or authorship of this article.