
Abstract
Central core disease is a nonprogressive or slowly progressive congenital myopathy with a variable degree of hypotonia and axial and proximal muscle weakness that is histologically characterized by areas devoid of oxidative enzyme activity, resulting from an absence or low numbers of mitochondria in these regions (central core). A 10-month-old, male, pony foal was examined because of stiff gait, marked contractures of the distal portion of the limbs, flexion deformities of the hooves, and moderate hypotonia that had been present from birth. The foal had increased creatine kinase (282 U/ liter; reference interval 10-135 U/liter), lactate dehydrogenase (1,188 U/liter; reference interval 150–450 U/liter), and aspartate transaminase (377 U/liter; reference interval <290 U/liter) activities, suggesting muscle disease. Muscle biopsy was performed. In cytochrome oxidase-, succinate dehydrogenase-, and reduced nicotinamide adenine dinucleotide tetrazolium reductase-reacted sections, the dominant morphologic feature was the absence of oxidative enzyme activity in the cores. By use of immunohistochemical technique with a monoclonal antibody against desmin, the cores were clearly delineated and a desmin network was present within the cores. Ultrastructurally, the core areas were characterized by preserved sarcomeres with irregular Z-lines, with some streaming or zigzag appearance and abnormal sarcoplasmic reticulum profiles and T-tubules. Lack of mitochrondria within central cores was observed. Diagnosis of myopathy with central cores was made.
In human medicine, central core disease (CCD), is a nonprogressive or slowly progressive congenital myopathy with a variable degree of hypotonia and axial and proximal muscle weakness often pronounced in the hip girdle. 1 Skeletal involvement comprising congenital dislocation of the hips, scoliosis, and talipes equinovarus, are common. 14 Other contractures are rare, and some individuals have marked ligament laxity. Serum creatine kinase activity is normal or only moderately increased. Presentation usually occurs in infancy or early childhood, but cases with later onset have been described. 14 Most affected individuals achieve the ability to walk, although more severe variants are on record. 8 Muscle cramps when exercising are a common finding in some families. 6
In muscle biopsy specimens from patients with CCD, areas devoid of oxidative enzyme activity are seen, resulting from an absence or low numbers of mitochondria in these regions. Sarcoplasmic reticulum (SR) profiles and T-tubules are visible within the cores, but they are abnormal. 5
Central core disease is transmitted as an autosomal dominant trait, and at least one gene responsible is the ryanodine receptor gene (RYR1) at chromosome 19q13.1. Mutations in the RYR1 gene on chromosome 19q13.1 have now been detected in many cases of CCD. 7, 10 Recent data suggest that sporadic or recessive inheritance may also occur. 8, 13
In veterinary medicine, a few cases of myopathy with core-like structures have been described, 4, 9, 11 but to the authors' knowledge, no reports exist about the horse. Thus, we describe the clinical, histologic, and ultrastructural findings of myopathy in a foal that were similar to findings of CCD in humans.
A 10-month-old, male, pony foal was examined because of stiffness, marked contractures, and flexural deformity of the distal portion of the limbs and moderate hypotonia of the proximal muscles (Fig. 1). The owner reported that these findings had arisen from birth. The pony was standing on “tip toes” and was reluctant to walk. It presented difficulties in rising from sitting and ingesting food on the floor. There was flexural deformity of the metacarpophalangeal and interphalangeal joints of the limbs. Results of neurologic examinations were unremarkable. Radiography of the vertebral column and coxofemoral and stifle joints was done, but abnormalities were not found. Routine hematologic analysis revealed no abnormalities. Biochemical analysis revealed increased values of creatine kinase at 37°C (282 U/liter; reference interval 10–135 U/liter), lactate dehydrogease (1,188 U/liter; reference interval 150–450 U/liter), and aspartate transaminase (377 U/liter; reference interval <290 U/liter).
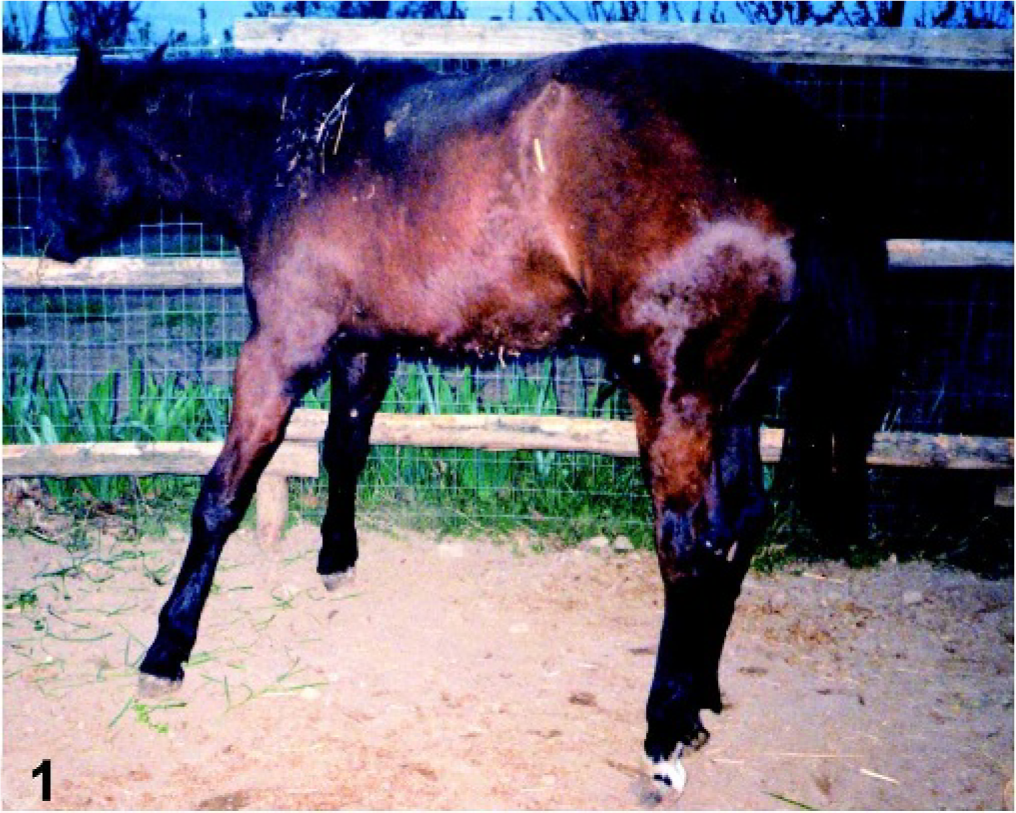
Pony foal. Marked contractures and flexural deformity of the distal portion of the limbs, with characteristic “tip toe standing.”
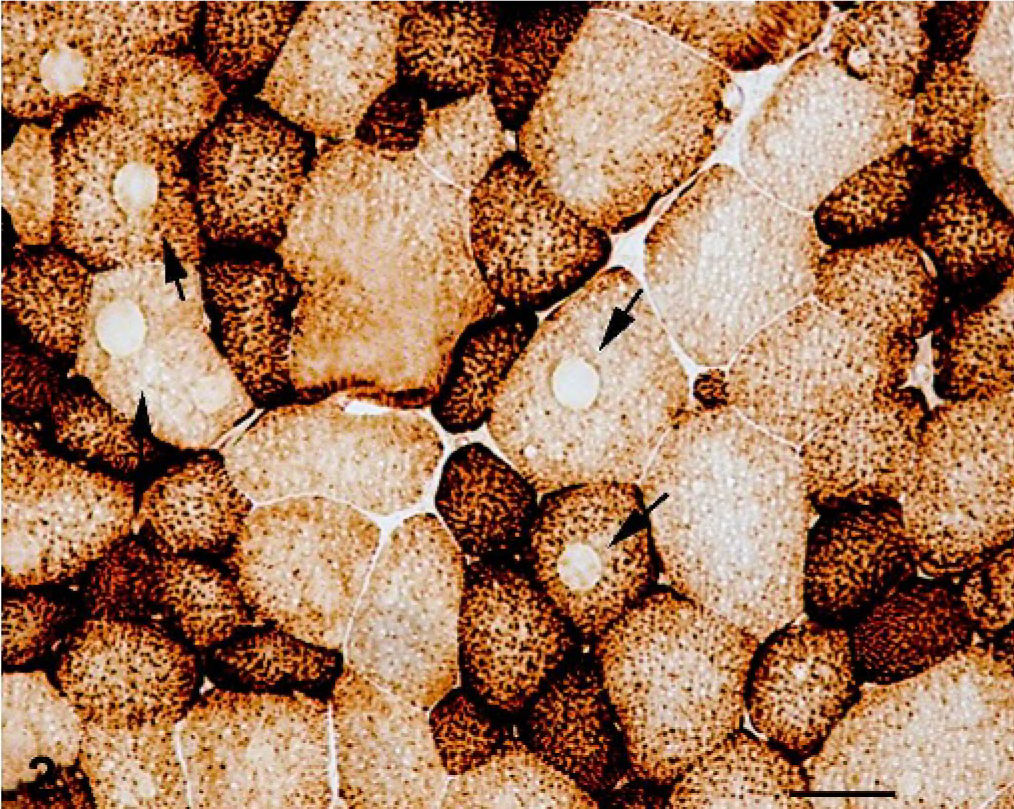
Skeletal muscle; foal. Absence or decrease of oxidative enzyme activity in several muscle fibers (arrows). COX stain. Bar = 55 µm.
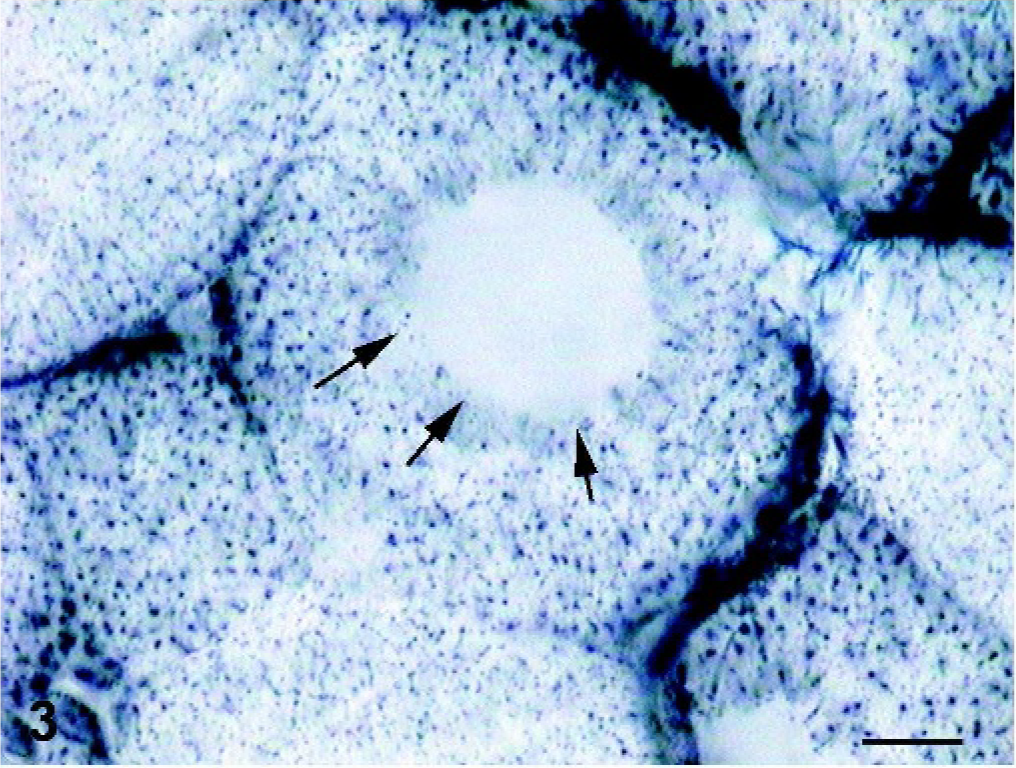
Skeletal muscle; foal. The core areas are circular in the shape and contrast sharply with the normal enzyme activity in the rest of the muscle fiber (arrows). SDH stain. Bar = 20 µm.
A diagnosis of skeletal myopathy of unknown cause was made, and biopsy specimens were taken from the gluteus medius muscle for histologic examination. The specimens were frozen in isopentane precooled in liquid nitrogen. Sections were stained by use of the following histologic and histochemical techniques: hematoxylin and eosin (HE), modified Gomori trichrome, periodic acid–Schiff (PAS), cytochrome oxidase (COX), succinate dehydrogenase (SDH), and reduced nicotinamide adenine dinucleotide tetrazolium reductase (NADH-TR). For immunohistochemical analysis, a commercial kit (Dako LSAB K680; Dako, Burlingame, CA), with peroxidase-conjugated streptavidin and a mixture of biotinylated goat antirabbit and antimouse immunoglobulins as the link antibody, was used. Monoclonal antibody against desmin was obtained from Novocastra Laboratories Ltd (Newcastle upon Tyne, UK). The immunolabeling procedure included negative-control sections incubated in phosphate-buffered saline (PBS) without primary antibody. We used a sample of the gluteus medius muscle from another foal as a control for histoenzymatic and immunohistochemical stains.
For transmission electron microscopy, additional muscle specimens were clamped and fixed in 2.5% glutaraldehyde, further fixed in osmium tetroxide, and embedded in low-viscosity Spurr resin. Ultrathin sections were cut, counterstained with 0.5% uranyl acetate and lead citrate, and examined using a Zeiss Electron Microscope 902.
Sections stained with HE had few abnormalities. Nuclei were mainly subsarcolemmal, but some fibers had internal nuclei. There was no proliferation of endomysial or perimysial connective tissue or fat and no signs of inflammation. In COX-, SDH-, and NADH–TR-reacted sections, the dominant morphologic feature was the absence of oxidative enzyme activity in the center of many fibers. These areas were circular in shape (cores) and contrasted sharply with the normal enzyme activity in the rest of the muscle fibers (Figs. 2, 3). We used the intensity of histochemical reaction in the NADH-TR and SDH staining for fiber typing differentiation (type 1 = strong; type 2A = intermediate, type 2B = weak staining). 1 Cores were evident in type-1 and type-2 fibers, and no type predominated. With PAS staining, the cores appeared as light zones, and other abnormalities were not evident. Other abnormalities were not detectable using the modified Gomori trichrome stain.
Using the antibody against desmin, cores were more easily visible, and the desmin network appeared markedly modified, with sharp delineation of the cores and abnormal distribution of desmin in the cores (Fig. 4).
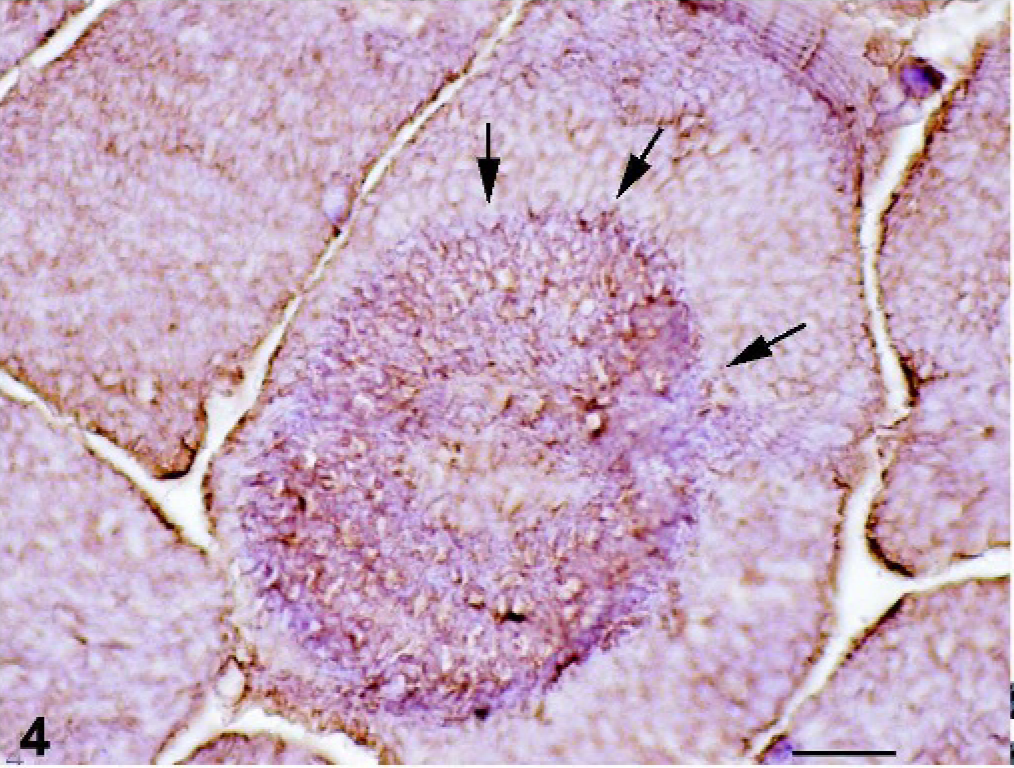
Skeletal muscle; foal. With the antibody against desmin, the core appears sharply delineated and abnormal distribution of desmin also is evident (arrows). Avidin-biotin-peroxidase complex method. Mayer's hematoxylin counterstain. Bar = 20 µm.
On electron microscopy (EM), the core areas were characterized by preserved sarcomeres, with some having slightly shorter length than that of the peripheral myofibrils; the Z-lines were irregular, with some streaming or a zigzag appearance (Fig. 5), but sometimes were completely disrupted “structured cores.” Sarcoplasmic reticulum profiles and T-tubules were visible within the cores, but they were abnormal, with dilated and vacuolated cisternae (Fig. 6); furthermore, the mitochondria were decreased in number or absent in the cores, but an accumulation of them was evident in the subsarcolemma.

Skeletal muscle; foal. Electron microscopy reveals irregular Z-lines, with some streaming or zigzag appearance (arrows). Bar = 2.5 µm.
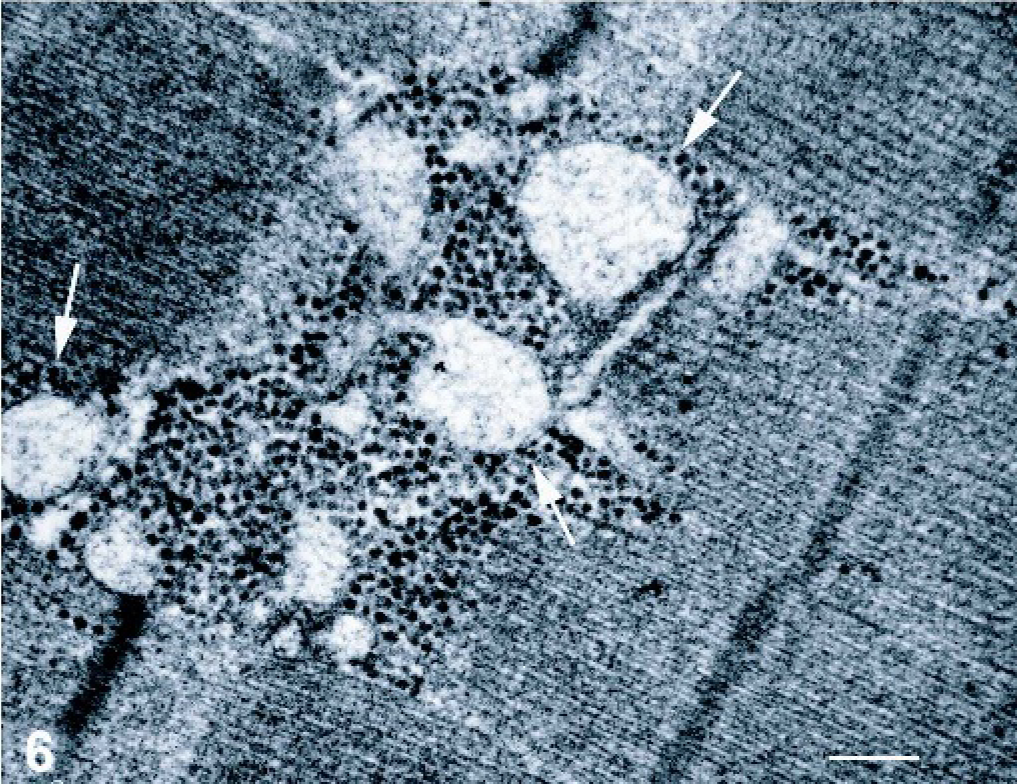
Skeletal muscle; foal. Electron microscopy reveals abnormal sarcoplasmic reticulum profiles and T-tubules within the cores, with dilated and vacuolated cisternae (arrows). Bar = 2 µm.
In human medicine, CCD is a clearly defined clinical condition with striking pathologic changes that facilitate the diagnosis; it is characterized by predominantly central, core-like areas observed with use of oxidative stains. 1, 2 Typically, the cores involve type-1 muscle fibers that predominate, but variable histopathologic pattern and involvement of both type of fibers are described. 1, 14 The degree of histopathologic changes can be variable and sampling and age of the patient must be taken into account. 6 On EM, the cores are sharply delineated, but are not membrane bound. The cores can be “structured” or “unstructured.” 1, 2 In the first case, the sarcomeres are preserved, but their length is shorter than that in the peripheral myofibrils and the Z-lines are irregular, with some streaming or a zigzag appearance. With ATPase reaction, they appear strongly reactive. In the second case, the sarcomere organization is more disrupted, with large zones of Z-band streaming; with ATPase reaction, they can appear like an unstained central zone. Structured and unstructured cores may coexist in the same biopsy specimen. 1, 2 The mitochondria are decreased in number or absent from the cores, and SR reticulum and T-tubule abnormalities are evident within cores. 1, 2 This entity should be differentiated from other abnormalities in myofibers leading to localized loss of oxidative activity.
Moreover, in multi-minicore disease (MMD), characterized by multifocal core-like areas by use of oxidative stains, the minicores are found in type-1 and type-2 fibers, and typically affect only a few sarcomers along the longitudinal axis of the muscle fiber. 6 On EM, the lesion appears as foci of myofilamentary disintegration, with Z-line streaming and running over a few sarcomers and with sharp limits from the adjoining normal sarcomeres. 1, 2 They are characterized by a decrease or absence of mitochondria. Recent studies indicate that, in some individuals, minicores may evolve into central cores over time, suggesting a histopathologic continuum between MMD and CCD. 3
Other morphologic changes in the architecture of fibers similar to central cores are in target fibers, most commonly in biopsy specimens from patients with chronic, peripheral neuropathies or more acute, recovering neuropathies. 1
On sections reacted for oxidative enzyme detection, target fibers consist of 3 zones: a central zone lacking oxidative activity, an intermediate zone of increased activity, and a peripheral zone of normal activity. 1 On EM, the innermost zone is characterized by filaments markedly disrupted with indistinct or absent myofibrils, the normal banding pattern is lost, and there is marked smearing of the Z-band. In the intermediate zone, the filament arrangement is only mildly disordered, and the outermost area consists of normal-appearing muscle. 1, 2
Little is known about the development of the cores. Their similarity to target fibers found with denervation and reinnervation suggests that a neurogenic factor might operate in CCD. However, many investigators have emphasized the structural differences between the core and target formations. 14 Furthermore, targets are generally single and central in the fiber, but cores can be multiple and eccentric. At this time, there is only weak evidence for a neurogenic origin of CCD. 14
The cores are a phenomenon secondary to a defect in a protein that resides in the membrane of a SR reticulum at the triadic junction. However, their origin in time, evolution, and morphogenesis remain obscure. 14 Core-like structures have also been described as a striking feature of a congenital, progressive myopathy of merino sheep, 9 of a myopathy affecting adult Meuse-Rhine-yssel cattle, 4 and of a myopathy with core-like structures in a dog. 11
Diagnosis of CCD is based mainly on the clinical phenotype and the pathologic assessment of the muscle biopsy specimen. 14 Linkage may be noninformative and gene analysis is laborious, especially of large genes such as RYR1, which has 106 exons. 14 Identification of the primary gene defect may be difficult if the defining feature is absent, or in cases in which dual pathology exists. 14
Immunohistochemical analysis can help to document the presence of cores. Previous reports illustrate the value of study of desmin, 14, 15 but accumulation of desmin is not specific to CCD and can also develop in target fibers and in association with other disorders such as desminopathies. 14
Because desmin can be absent from cores, as well as accumulating within them or at their periphery, several possible explanations for alterations in desmin distribution can be proposed. Disruption of desmin might relate to the ultrastructural appearance of misaligned myofibrils, as desmin links the myofibrils at the Z-line, or might relate to alterations in the distribution of mitochondria. 12
Electron microscopy indicated that an early abnormality is the breakdown of the Z-lines, but accumulation of Z-line material can also occur. Loss, but not accumulation of Z-line material could result from the activation of enzymes by calcium. 12
Little is known about the pathology of the SR and T-tubules in CCD. Hayashi and co-workers described the abnormalities of the SR and T-tubules, represented by large lateral cisternae forming junctional complexes with T-tubules. 5
Currently a satisfactory explanation is not available for the disordered mechanism in CCD which leads to the weakness, and since it is known that the SR and T-tubules are central to the control of the contractile system within the muscle cell, it is tempting to speculate that the distorted anatomy of the SR and T-tubules somehow leads to dysfunction in excitation-contraction coupling, and hence, the weakness that the patients experience. 2
In conclusion, we have reported a case of congenital myopathy in a foal with appreciable contractures, and flexural deformity of the distal portion of the limbs, moderate hypotonia, increased serum CK activity, with muscle biopsy findings of central core changes. We propose that this condition represents a severe clinical and pathologic condition of myopathy with central cores described here for the first time in a horse.
Footnotes
Acknowledgements
We thank Janis Mc Ferrin for proofreading of the manuscript and Raffaele Ilsami for technical assistance.