
Abstract
Voluntary movement in animals is modulated by a number of subcortical systems. One of these resides in the basal nuclei and their associated projections and utilizes dopamine as a neurotransmitter. Apart from regulating movement, the dopaminergic axis is also involved in the control of goal-oriented behavior, cognition, and mood. Disorders of this system result in common human neurologic disorders such as Parkinson's and Huntington's diseases, as well contributing to a host of behavioral conditions, such as schizophrenia, attention deficit hyperactivity disorder, and addiction. Many individual mouse models of human dopaminergic dysfunction have been described in varying degrees of detail. However, when evaluating this region of the brain, the veterinary pathologist is confronted by a paucity of information summarizing the comparative aspects of the anatomy, physiology, and pathology of the central dopaminergic system. In this review, a systematic approach to anatomic phenotyping of the central dopaminergic system in the mouse is described and illustrated using tyrosine hydroxylase immunohistochemistry. Differences between murine neuroanatomy and comparable regions of the nonhuman primate brain are highlighted. Although the mouse is the focus of this review, conditions in domestic animals characterized by lesions within the basal nuclei and its projections are also briefly described. Murine behavioral and motor tests that accompany abnormalities of specific anatomic regions of the dopaminergic axis are summarized. Finally, we review mouse models of Parkinson's and Huntington's diseases, as well as those genetically altered mice that elucidate aspects of dopamine metabolism and receptor function.
Keywords
Appropriate and coordinated voluntary movement is essential to the well-being of all animals. Although these signals originate in the cerebral cortex, they are modulated by a number of subcortical systems. One of these is the dopaminergic (DA) axis residing in the ventral brainstem and striatum. 39 Dopamine is a highly conserved catecholaminergic neurotransmitter involved in the control of movement, goal-oriented behavior, cognition and mood. 76, 101 It is utilized predominantly in the brain, where it controls the function of the basal nuclei and their associated projections. In the peripheral nervous system, it is used as a neurotransmitter in a few peripheral neuronal groups, but primarily serves as the substrate for the synthesis of norepinephrine and epinephrine in sympathetic neurons and the adrenal medulla. 84, 85, 101
Most of our knowledge of the role of the basal nuclei in movement has been gained from study of human neuropsychiatric disorders such as Parkinson's and Huntington's diseases. 102 However, dopamine also plays a central role in behavior, particularly in the areas of attention and reward. 101 At moderately elevated extracellular levels, dopamine promotes enhanced mood and energy levels, whereas, at higher levels, it is associated with hyperactivity and compulsive and erratic behaviors. 32 Abnormal dopaminergic transmission contributes to symptoms of schizophrenia, 38, 76, 92 attention deficit hyperactivity disorder, 55, 89 Tourette's syndrome, 40 and manic-depressive syndromes. 105 Additionally, dopaminergic transmission mediates the reinforcing effect of many addictive substances. 29, 56 Dopaminergic pathways controlling voluntary movement are most easily studied, as the relevant neuroanatomy is more clearly defined and the locomotor phenotype easily discernible. Consequently, dopamine-associated movement disorders in humans have been quite comprehensively replicated using mouse models that develop lesions in the relevant anatomical areas 64, 66, 68, 87, 90 and that develop gait abnormalities, with a variable behavioral phenotype. In contrast, mice that are genetically altered for genes encoding proteins involved in dopamine synthesis, metabolism, or neurotransmission result in global dopaminergic dysfunction rather than a specific neuroanatomic lesion. These models have minimal pathology, but usually display behavioral changes 73 that replicate some features of dopamine-influenced human behavioral syndromes. Although individual mouse models are described in varying degrees of detail, currently no texts exist that describe an overall approach to anatomic phenotyping of the dopaminergic system.
Using tyrosine hydroxylase immunohistochemistry, a systematic approach to anatomic phenotyping of the central dopaminergic system in the mouse is described and illustrated. Differences between murine neuroanatomy and comparable regions of the nonhuman primate brain are highlighted. Behavioral and motor tests that accompany abnormalities of specific anatomic regions of the murine dopaminergic axis are introduced. The mouse models that illustrate dysfunction of specific anatomic regions of the basal nuclei and their attendant pathways as well as those genetically altered mice that elucidate aspects of dopamine metabolism and receptor function are reviewed.
Materials and Methods
Methods described in this section were used to illustrate the functional anatomy of the basal nuclei and its projections. These methods can also be used as a guide to obtain the needed coronal and sagittal sections to screen the major components of the central dopaminergic system.
Animals and tissues
All tissues were obtained from animals used in Institutional Animal Care and Use Committee (IACUC)-approved protocols at the Yale School of Medicine. Brains were obtained from 3-month-old male C57BL/6 mice (Mus musculus) that were used as control animals in an unrelated study. Following euthanasia by placement in a chamber prefilled with 70% CO2, mice were decapitated, the calvarium was removed, and the head placed in Fekete's acid ethanol for 48 hours. Next, brains were removed from the cranium and sectioned in coronal and sagittal planes as illustrated in Fig. 1. For nonhuman primate sections, the brain was obtained from a healthy 5-year-old male squirrel monkey (Saimiri sciureus) euthanized for unrelated reasons. The brain was provided by the investigator and had been fixed in 10% formalin for 7 days prior to sectioning. The brain was sectioned in the coronal plane at 0.3-cm intervals from the midfrontal gyrus to just caudal to the precentral gyrus. Following routine paraffin embedding, 5 µm hematoxylin and eosin-stained sections were obtained for initial evaluation.
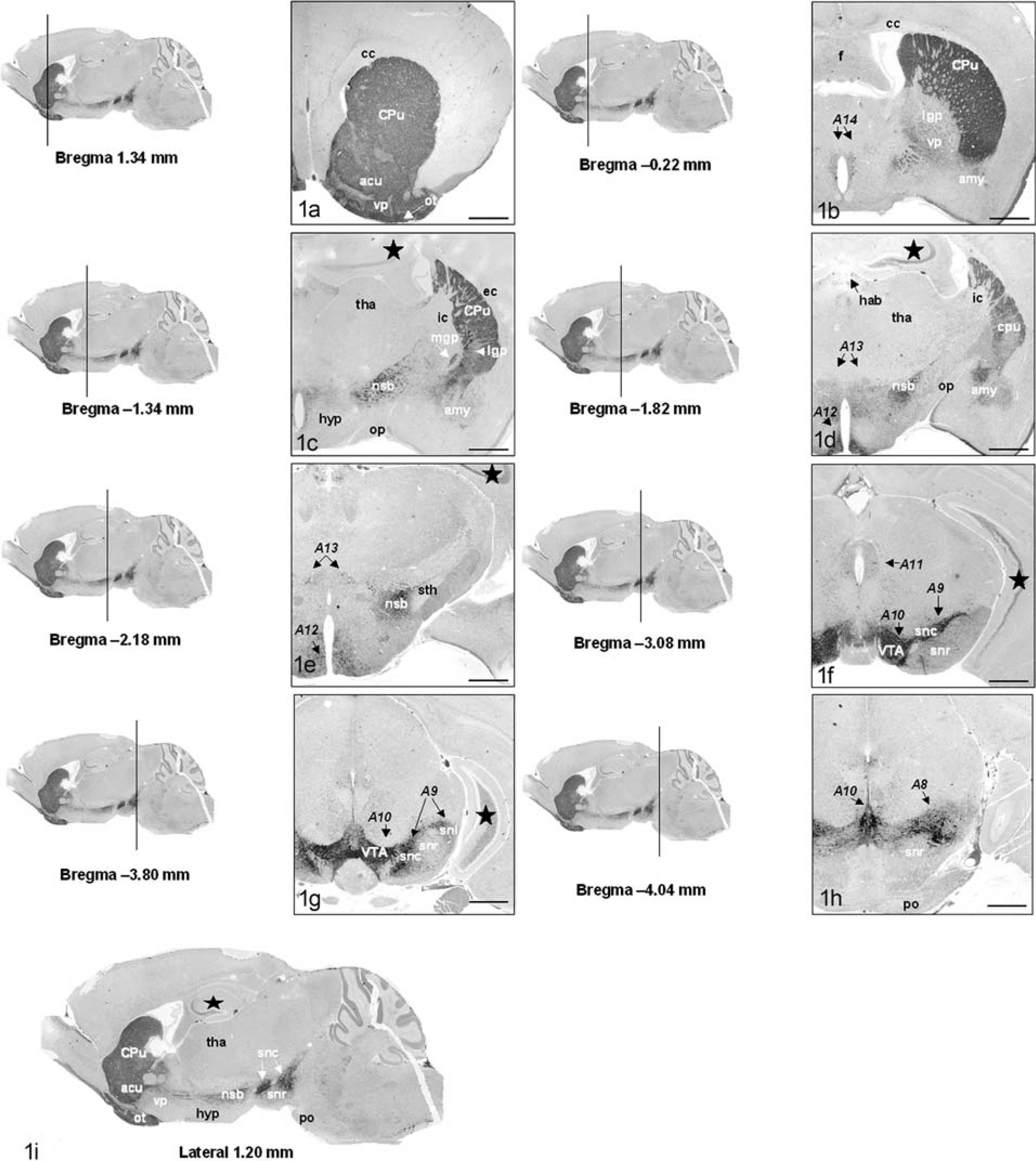
Tyrosine hydroxylase immunohistochemistry of the mouse brain.
Sections needed to examine the central dopaminergic system
A list of coronal and sagittal sections needed to initially assess the dopaminergic system is given in Table 1. The sections identify the key functional components of this system and primarily focus on the dorsal and ventral mesostriatal systems. More detailed evaluation of specific regions may require serial sectioning of that segment of the system. The olfactory bulbs are often damaged when the brain is removed. If this region must be examined, fixing the entire head in Bouin's solution may be most appropriate. Bouin's solution will fix and decalcify simultaneously (approximately 7–10 days for an adult mouse) and can be used for sectioning where the skull is retained. Dopaminergic neurons in the retina and olfactory bulb can be examined in the same section. This can be obtained by cutting a coronal section through an intact decalcified head through the middle of both eyes. The olfactory bulbs can be visualized between the eyes.
Histologic sections needed to assess the dopaminergic system in mice.

Immunohistochemistry
Tyrosine hydroxylase-stained tissues are best suited to general orientation and identification of dopaminergic anatomy, as well as cell counting and morphometry. As tyrosine hydroxylase is the first enzyme in catecholamine synthesis, immunohistochemical detection identifies all catecholaminergic neurons, i.e., those producing dopamine, norepinephrine, and epinephrine. Catecholaminergic neurons are found in the brainstem (from medulla oblongata to the diencephalon) and in the olfactory bulb and retina. The nomenclature used to describe the distribution of catecholaminergic neurons originated with Dahlstrom and Fuxe, 16 who assigned numbers to catecholaminergic cell groups in rat brain beginning with A1 in the lower medulla oblongata and extending rostrally. The first seven groups are adrenergic and noradrenergic; further identification of these can be done using dopamine beta-hydroxylase as an adrenergic marker. 101 The remaining groups constitute dopaminergic neurons. The initial classification system concluded with A12 in the basal hypothalamus. 16 This nomenclature has subsequently been expanded in a rostral direction with groups A13–A15 in the hypothalamus 10, 31 group, A16 in the glomerular layer of the olfactory bulb, and last, dopaminergic amacrine cells within the retina. 9, 42 Lesions identified at this stage may be examined further using immunohistochemistry and histochemical stains for specific neurotransmitters in affected regions. These tests often require frozen sections. Neurochemical mapping of the dopaminergic system is comprehensively reviewed in Gerfen. 33
In this study, unstained 10-µm sections of appropriate regions were used for immunohistochemistry. Antigen retrieval was performed by boiling sections in a microwave oven for 10 minutes in 10 mM sodium citrate. Sections were allowed to cool for 20 minutes, prior to a 5-minute endogenous peroxidase block in 5% H2O2. Immunohistochemistry was performed using a rabbit anti-human polyclonal antibody against tyrosine hydroxylase at a concentration of 1:400. A biotinylated goat anti-rabbit secondary antibody combined with horseradish peroxidase reagent was then applied (DAKO-USA, Carpinteria, CA), followed by development with diaminobenzidine. Negative controls were incubated with normal rabbit serum instead of the primary antibody. Using this protocol, we have been able to obtain comparable results in tissues fixed with Bouin's fixative, Fekete's acid ethanol, or 10% formalin.
Image processing
Slides were examined with a Zeiss Axioplan light microscope and digital images were collected at each location illustrated in Figs. 1 and 2 using an Axiocam digital imaging camera and Zeiss Axiovision software (Carl Zeiss MicroImaging, Inc., Thornwood, NY). For nonhuman primate sections, multiple images were collected and montages assembled in Adobe Photoshop 7.0 (Adobe, San Jose, CA).
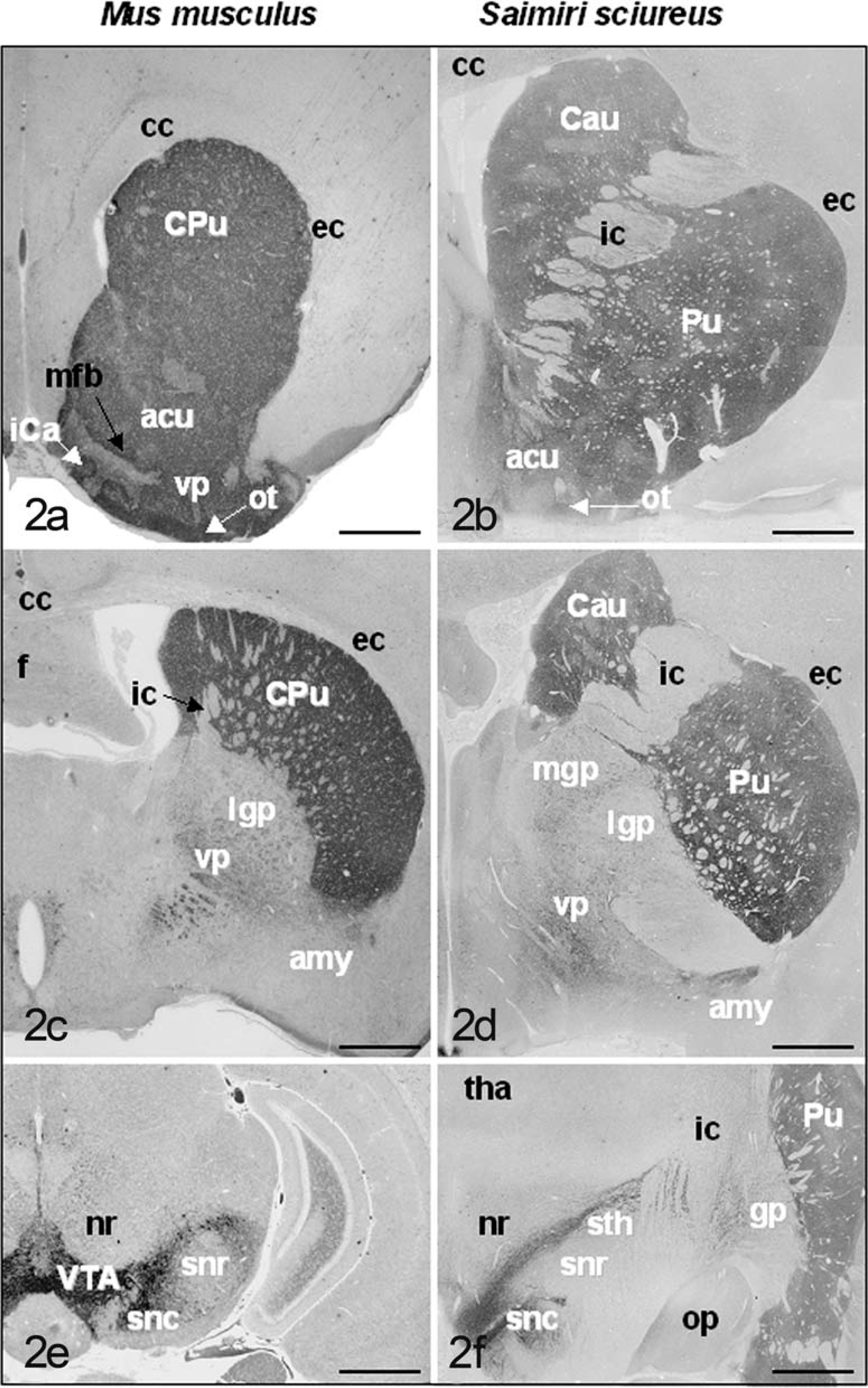
Comparative nigro-striatal neuroanatomy of mouse (Mus musculus) and squirrel monkey (Saimiri sciureus). Differences in neuroanatomical distribution of various elements of the nigrostriatal system is illustrated in Fig. 2, however some reference to structures in Fig. 1 is made in this legend. The caudate nucleus (Cau) and putamen (Pu) are clearly separated by the internal capsule (ic) in primates; in the mouse they are combined. In this species, the internal capsule is never as prominent and appears at a relatively more caudal location, at which point the lateral globus pallidus (lgp) can be visualized (
Neuroanatomic reference materials
Nomenclature of murine structures demonstrated in coronal and sagittal sections is based on the system described by Paxinos and Franklin. 82 Superimposition of dopaminergic components on these sections relies heavily on descriptions of tyrosine hydroxylase immunohistochemistry in the rat. 9, 45 Nomenclature of non-human primate sections is based upon the atlas of the squirrel monkey brain by Gergen and MacLean. 34 Nomenclature used to describe location of structures will be that used in quadripeds rather than in humans, i.e., rostral-caudal (instead of anterior-posterior), and dorsal-ventral (instead of superior-inferior).
Central Dopaminergic System: Overview of Physical Structures
The basal nuclei constitute key brain structures, the primary function of which is to receive, integrate, and project motor and sensory information. They receive input from two primary sources: the motor cortex (glutaminergic input) and the mesencephalic dopaminergic system (dopaminergic input). Neurons from the basal nuclei project to the thalamus, which in turn project to the motor cortex to modulate voluntary movement. 3, 39 In this section, physical structures comprising the dopaminergic system will be discussed under two main headings: those structures comprising the basal nuclei and related nuclei and the brainstem cell groups, which constitute the origin of the major dopaminergic projections to the basal nuclei. Understanding the anatomy of these circuits is important to accurately attach morphologic to behavioral phenotype in individual mouse models.
Dopaminergic neurons in the brainstem constitute the origin for the extensive mesotelencephalic projection system. These circuits define systems that primarily control motor activity (dorsal mesostriatal system) or primarily influence behavior (ventral mesostriatal system). 9, 45 Mouse models of human locomotory disorders typically develop histologic lesions of neuronal degeneration and loss in the substantia nigra and caudate-putamen. 64, 66, 68, 87, 90 In contrast, mice that model the dopaminergic constituents of motivated behaviors rarely demonstrate visible pathology. In these models, investigators are more reliant on neurochemistry, immunohistochemistry, and behavioral phenotyping for their characterization. 23
1) The basal nuclei and major associated nuclei
The term basal nuclei refers to a conglomerate of forebrain structures arising from the basal telencephalon. 33, 39 These include the caudate nucleus, putamen, nucleus accumbens, globus pallidus, ventral pallidum, and olfactory tubercle. Major associated nuclei that are functionally integral to striatal circuitry include the subthalamic nucleus, ventral tegmentum, and substantia nigra. The substantia nigra is the origin of the striatal system—the subthalamic nucleus is a relay nucleus, and the ventral tegmentum projects to many regions, including the striatum. 3, 15, 39
The striatum
The caudate and putamen nuclei are, together, referred to as the striatum and constitute the input side of the basal nuclei. 3, 4, 15 Their major inputs are from the cerebral cortex, and they project mainly to the globus pallidus. In the mouse, the striatum (caudate nucleus and putamen) is a large structure occupying the region between the cortex and the lateral ventricles (CPu: Fig. 1A–C). 82 Comparable sections of this region in the mouse and primate present a slightly different anatomic arrangement (Fig. 2A–D). Whereas the caudate nucleus and putamen are clearly separated by the internal capsule in primates, 34 they are combined in the mouse. 82 In the mouse, the internal capsule is never as prominent and appears at a relatively more caudal location, at which point the globus pallidus can be visualized (Fig. 2C,D). Although the striatum lacks clear cytoarchitectural subdivisions (Fig. 3A), its constituent neuronal subpopulations are organized into distinct functional compartments. 33 Medium spiny neurons constitute the vast majority (90–95%) of neuronal types within the striatum. 109 They receive dense dopaminergic innervation from the ventral tegmentum and substantia nigra (Fig. 3A) and utilize gamma amino butyric acid (GABA) as their neurotransmitter. 51 About 2% of the striatal neurons are cholinergic—these are interneurons, which influence dopamine release. 112 Acetylcholinesterase histochemistry applied to the mouse brain clearly delineates the striatum. 82

Comparative histology of the striatum and substantia nigra in the mouse. Striatal neurons (
Dorsal and ventral pallidum
The globus pallidus (or dorsal pallidum) is an output region of the basal nuclei: it receives input from the caudate and putamen, participates in other circuits involving components of the basal nuclei, and projects to the thalamus, which projects in turn to the motor and premotor cortex. 3, 15, 40 Through this route, the basal nuclei exert some control over movement by acting through descending pathways originating in the cerebral cortex. Like the striatum, neurons of the globus pallidus are also GABAminergic. 33 The globus pallidus in primates is divided into lateral and medial portions (Fig. 2D). 3, 15, 33 In coronal sections of the mouse brain, the lateral globus pallidus (lgp: Fig. 1C) lies ventromedial to the striatum, occupying the region between the striatum and the internal capsule. 81 In rodents, the medial globus pallidus is sometimes referred to as the endopeduncular nucleus and can be visualized within the internal capsule at Bregma −1.06 to −1.58 (mgp: Fig. 1C). 81 The ventral pallidum (vp: Figs. 1A,B and 2D) receives input predominantly from the accumbens nucleus and projects to a diverse array of midbrain and pontine structures. 9, 40
The accumbens nucleus
This nucleus lies at the junction of the caudate and putamen at their anteroventral limit. 34, 39 In the mouse, it lies directly ventral to the striatum (acu: Fig. 1A). 82 The nucleus accumbens and the ventromedial parts of the caudate and putamen are sometimes referred to as the ventral striatum, the part of the basal nuclei that receives input from the limbic system. 3, 9, 45 This region is considered to have a cognitive function rather than a strictly motor function. 3
The amygdala nuclear complex
An important component of the limbic system, amygdalar neurons (amy: Figs. 1B-D and 2C,D) project to the basal nuclei in addition to the hypothalamus and other components of the limbic system. 39 This dual role of this multinuclear complex is reflected by its location between the basal nuclei and hippocampus. In the mouse, it is located directly ventral to the striatum. 82
The substantia nigra and subthalamic nuclei
The substantia nigra constitutes a prominent aggregate of dopaminergic neurons occupying the ventral aspect of the mesencephalon. 9, 39, 45 It consists of two parts: the pars compacta (snc: Fig. 1F,G), which contains the majority of dopaminergic cells projecting to the dorsal striatum (Fig. 3B), and the pars reticulata (snr: Fig. 1F–H), which receives input from the subthalamic nucleus (sth: Figs. 1E and 2F) and projects primarily to the thalamus. 3, 15 In primates, the substantia nigra and subthalamic nucleus can be easily visualized in the same section (Figure 2F). 34 In the mouse, the subthalamic nucleus occupies a slightly more rostral position than the substantia nigra (Fig. 1E). 82 A prominent tract known as the nigrostriatal bundle (nsb: Fig. 1C–E) carries afferent dopaminergic fibers from the substantia nigra to the striatum. 9, 45 The substantia nigra is an important part of the mesencephalic dopaminergic system and constitutes the origin for the extensive afferent dopaminergic projection system, the mesotelencephalic projection system. 9, 45 This system is discussed in more detail below.
2) Afferent dopaminergic cell systems in the brainstem
The striatal complex receives afferents from two main regions—the mesencephalic dopaminergic system and various regions of the cortex. 9, 45 The mesencephalic DA system (groups A8, A9, and A10: Fig. 1F–H) contains the largest concentration of dopaminergic neurons and constitutes the origin for the extensive mesotelencephalic projection system. Components of this system are the most intensively studied, as they control voluntary motor activity (dorsal mesostriatal system) and motivated behaviors (ventral mesostriatal system). The diencephalic DA system (groups A11–A14) is smaller and gives rise to intradiencephalic projections as well as the descending diencephalospinal pathway. 9, 45 It will not be discussed in any detail in this review.
i) The mesencephalic dopaminergic cell system
The mesencephalic dopaminergic neurons form an extensive neuronal system in the ventral midbrain. 9 A distinction is made between nigral (A9) and nonnigral (A8 and A10) neurons. The A8–A10 cell groups give rise to the predominant dopaminergic system in the brain, the mesotelencephalic pathway. The nigral neurons (A9: Fig. 1F,G) are confined to the pars compacta of the substantia nigra and project to the striatum. 16 The nonnigral dopaminergic neurons are located rostral, medial, and caudal to the substantia nigra. The dopaminergic neurons of the A8 group are located in the ventrolateral tegmentum caudal to the substantia nigra and also project to the striatum (A8: Fig. 1H). The medially located neurons (A10: Fig. 1F–H) occupy the ventral tegmental area (VTA: Fig. 1F,G). 9, 16, 45 In addition, several minor dopaminergic pathways have been described, originating in the A11–A14 cell groups (Fig. 1B–H) and constituting the diencephalic dopaminergic system. 9, 31
The mesotelencephalic projection system comprises two major subsystems: 9, 45 the mesostriatal DA system, which includes projections to the entire striatum, and the mesolimbocortical DA system, which projects to limbic and cortical areas. The mesostriatal system is in turn divided into dorsal and ventral portions, the dorsal and ventral mesostriatal systems. 9, 36, 45, 91, 100 Neurons constituting the dorsal mesostriatal system (also known as the nigrostriatal system) originate in the A9 region of the substantia nigra (with lesser contributions by the A8 and A10 regions) and project to the dorsal striatum (caudate nucleus and putamen in primates, cats and dogs, or dorsal caudate-putamen in rodents), as well as the subthalamic nucleus. The dorsal striatum projects to the globus pallidus. 9 The globus pallidus is divided into distinct anatomical and functional portions—the external (lateral) and internal (medial) globus pallidus. 39 This pathway modulates voluntary motor activity, and degeneration of distinct components occurs in Parkinson's and Huntington's diseases in humans. 66, 104 The ventral mesostriatal system (also known as the mesolimbic system) 9, 36, 45 originates predominantly in the A10 group in the VTA and projects to the ventral striatum (ventral caudate-putamen, accumbens nucleus, and part of the olfactory tubercle) and the ventral pallidum (or substantia innominata). This system is thought to influence motivated behaviors, particularly those related to reward. 54
The mesolimbocortical DA system originates predominantly in the A10 cell group, with lesser contributions by the A8 and A9 groups. 9, 45 These neurons project to a diverse array of cortical areas associated with limbic function as well as the olfactory bulb, amygdala, lateral septal nucleus, lateral habenular nucleus, and locus coeruleus. It is involved in aspects of learning and memory. 60
The terminology describing these three functional networks (dorsal and ventral mesostriatal and mesolimbocortical systems) can be confusing as different (old and new) terms are used interchangeably by different authors. The more recent terminology is that listed above; 9 however, current publications will refer to the dorsal mesostriatal system as the nigrostriatal system, the ventral mesostriatal system as the mesolimbic system, and the mesolimbocortical system as the mesocortical system. 67 Nevertheless, the rationale behind the classification of these networks is determined by where their neuronal origins lie, where they project to, and what functions they control. These are described below and summarized in Table 2.
Origins and projections of mesotelencephalic projection system.

Functional Circuitry of the Basal Nuclei
The striatal complex constitutes the interface between cortical and dopaminergic afferents, and efferent projections to associated nuclei, thalamus, and cortex. Information flow through the basal ganglia is separated into five distinct parallel circuits. 3, 39, 77 These are the motor loop, oculomotor loop (control of orientation and gaze), dorsolateral prefontal loop and lateral orbitofrontal loops (both related to cognitive processes), and limbic loop (emotional and visceral functions). The motor loop is involved in somatosensory and somatomotor control and is exclusively the subject of the text in this section. All five circuits are controlled by simultaneous and coordinated modulation of two parallel pathways, the direct and indirect pathways. 3, 15, 39, 77 These function primarily through disinhibition, i.e., appropriate release of inhibitory tone. 3, 39, 77 The basic circuitry of the direct and indirect pathways constituting the motor loop is illustrated in Fig. 4 and described below.
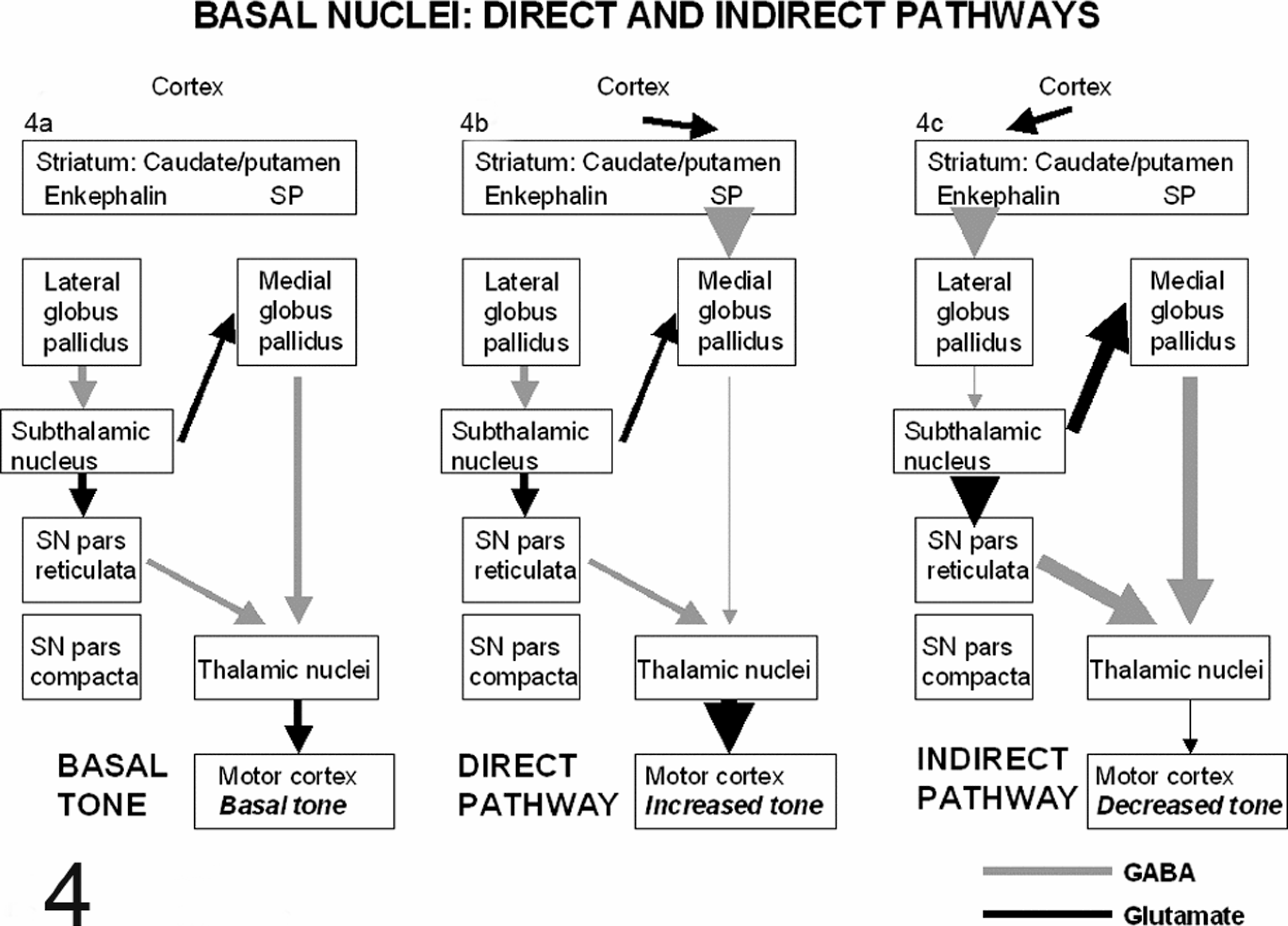
Functional circuitry of the basal ganglia. Inhibitory, GABAminergic signals are indicated by gray lines and excitatory glutaminergic signals by black lines. The thickness of the line denotes the strength of the signal.
Basal motor tone
Basal cortical motor tone is controlled by glutaminergic, excitatory projections from thalamic nuclei (Fig. 4A). The extent of thalamic activity is increased or decreased by proximal components of the basal nuclei, predominantly the medial globus pallidus (mgp) and substantia nigra pars reticulata (
The direct pathway
The net outcome of predominance of the direct pathway is to increase motor tone via disinhibition of thalamic nuclei. 3, 15, 77 The direct pathway (Fig. 4B) begins as an excitatory, glutaminergic projection from the cerebral cortex to the striatum. Striatal neurons utilize GABA and inhibit their targets—they are further subdivided into two primary populations determined by whether they utilize substance P (for the direct pathway) or enkephalin (for the indirect pathway). Striatal neurons in the direct pathway use substance P and exert an inhibitory effect on the mgp. This results in reduced inhibition of the thalamus by the mgp (thalamic disinhibition), with consequent cortical excitation. Thus, the function of the direct pathway is to release the thalamus from its pallidal inhibition and increase cortical motor activity. 15, 77
The indirect pathway
The indirect pathway (Fig. 4C) also begins with excitatory, glutaminergic projections from the cerebral cortex to the striatum. 3, 15, 77 To engage the indirect pathway, striatal neurons that utilize enkephalin and GABA are activated. These project to and inhibit the lgp. As the lgp inhibits activity in the subthalamic nucleus, inhibition of the lgp results in increased subthalamic nuclear activity (subthalamic disinhibition). 39, 77 Consequently, increased glutaminergic stimulation of subthalamic targets in the mgp and snr occurs—these nuclei in turn increase their inhibitory tone, thus reducing cortical stimulation via the thalamus. Therefore, the net effect of the indirect pathway is to decrease activity of the thalamus and consequently decrease activity of the cerebral cortex. Dopamine has opposite effects on the direct and indirect pathways. 77 It is excitatory to cells feeding into the direct pathway and inhibitory to those feeding into the indirect pathway. Consequently, dopamine promotes increased cortical tone and its depletion, as in Parkinson's disease, results in hypokinesis. 77, 78
Imbalance of direct and indirect pathways
The motor deficits seen in human and animal basal nuclei syndromes result from neurochemical imbalance within the striatal complex. 3, 15, 77 Imbalance within direct and indirect pathways results in either increased (hyperkinetic) or reduced (hypokinetic) motor activity. 3, 77 Although motor disturbances are most easily appreciated in these syndromes, they are accompanied by associative memory and limbic dysfunction as well. 77
Hyperkinetic disturbances take the form of dyskinesias. 77 They result from dysfunction of the indirect pathway with resulting dominance of the direct pathway due to reduced inhibition of thalamic neurons and increased cortical activity. Striatal neurons (medium spiny neurons) that project to the lgp are selectively lost in Huntington's disease. 104 This results in reduced activity of the indirect pathway, with relative dominance of the direct pathway and hyperkinetic motor disturbances. These can also be seen in Parkinsonian patients treated with L-DOPA. 14
Hypokinetic disorders are characterized by akinesia (impaired initiation of movement) and bradykinesia (reduced velocity and amplitude of movement). Both are characteristic of Parkinson's disease. 77, 78 Hypokinetic disorders originate in lesions resulting in loss of connection between the striatum and mgp, resulting in relative dominance of the indirect pathway. Parkinson's disease results from loss of dopamine-producing neurons in the substantia nigra pars compacta. 66 As dopamine is excitatory to cells feeding into the direct pathway, its loss results in relative dominance of the indirect pathway. Thus, tonically active mgp neurons continuously inhibit their thalamic targets, resulting in reduced motor cortical tone. 77
Hypokinetic syndromes resulting from dopaminergic dysfunction have been reported in the mouse and the horse. Hypophagia is a characteristic of the dopamine-deficient mouse. 113 In the horse, nigropallidal encephalomalacia occurs following prolonged ingestion of yellow star thistle (Centaurea solstitialis) or Russian knapweed (Centaurea repens). Lesions are characterized by malacia of the globus pallidus and substantia nigra 30, 59, 111 and result in a hypokinetic syndrome. Affected horses are inactive and somnolent. A typical sign is an inability to take hold of food, while the capacity to swallow is retained. 111
Mouse Models of Dopaminergic Physiology and Dysfunction
Genetic alterations resulting in neuropathology of the dopaminergic system induce clinical abnormalities in mice that can be appreciated by simple observation. Easily noted changes include alterations in body weight, posture, gait, and spontaneous activities. Motor abnormalities can be quantitated using tests of motor function, such as the rotarod, balance-beam test, vertical pole test, and painted paw test. 13 In contrast, those genetically altered mice in which components of dopaminergic neurotransmission (dopamine synthesis, transport, reuptake, and receptor binding) have been targeted exhibit more pronounced behavioral changes and less profound gait abnormalities. 95 These behaviors can be quantitated by assessing baseline open-field locomotion, rearing, and stereotypy. 13 Some models 6, 22, 106, 110, 113 also demonstrate the role of dopamine in the vegetative functions, such as feeding, drinking, and blood pressure. In genetic models characterized by neuropathology, bilaterally symmetrical lesions result in bilateral gait deficits. In contrast, unilateral loss of the substantia nigra and nigrostriatal bundle using toxic agents such as 6-hydroxydopamine results in circling toward the lesioned side. 75, 100 Treatment with dopaminergic agonists, such as dopamine, apomorphine, and amphetamine, induce contralateral circling due to receptor denervation supersensitivity after a unilateral lesion. 26, 99
I. Mouse models with neuropathology of the dopaminergic axis
The majority of these models were developed to investigate the two most common human disorders of the dopaminergic axis, namely Parkinson's and Huntington's diseases. In addition, spontaneous mutants (the weaver mouse) 94 and mice genetically altered for genes originally not suspected to result in striatal pathology have been described. 27, 70 The specific neuropathology in these models is relatively subtle and ranges from loss of specific cell groups 27, 70, 87 neurodegeneration 66 and accumulation of intracytoplasmic or intranuclear inclusions. 18, 44, 66, 90 These are best visualized using antibodies specific for the relevant protein, e.g., alpha-synuclein, 66 Huntington, 44, 90 or ubiquitin. 18 Electron microscopy may be required to illustrate dendritic lesions. 57
1) Parkinson's disease
Parkinson's disease is caused by loss of dopamine-containing neurons in the substantia nigra pars compacta that project to the striatum. Several lines of evidence suggest its mixed environmental and genetic origin. 66 It usually affects patients in midlife (over 50) and is characterized by tremor at rest, increased muscular tone, akinesia, bradykinesia, disturbances of eye movements (reduced blinking), and a flexed posture. 77, 78 At autopsy, the substantia nigra pars compacta is devoid of dopaminergic neurons, accompanied by intracytoplasmic inclusions known as Lewy bodies and gliosis. 79 Lewy bodies are composed of aggregated proteins, notably alpha-synuclein and ubiquitin. Alpha synuclein misfolding appears to be a central feature of the variously triggered forms of Parkinson's disease. 66 In a small fraction of cases, dominantly inherited mutations in the human alpha-synuclein or parkin genes result in early-onset Parkinson's disease. 52 Interaction between environmental neurotoxicants (herbicides, other pesticides, and industrial chemicals) and genetic susceptibility is likely to result in presynaptic damage to nigral cells, resulting in cell injury and abnormal protein folding. 12, 69, 103 Progressive injury induced by disposal of possibly toxic abnormally folded proteins is believed to evolve over years. In addition to familial forms caused by mutations in alpha-synuclein itself, mutations in two additional proteins involved in proteasomal degradation of misfolded proteins support the notion that accumulation of aberrantly folded proteins plays a role in the disease. 62
Mouse models
Both toxin-induced and genetic mouse models of Parkinsonism exist. 79 The latter group focus primarily on the role of alpha synuclein in the pathogenesis of the disease. Mouse models of Parkinson's disease typically display hypokinesia and bradykinesia, similar to their human counterparts.
a) Toxin-induced models
The toxin 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine (MPTP) is the most common chemical agent used to mimic Parkinson's disease in the mouse 87 as well as in nonhuman primates, 79 sheep, 8 and pigs. 72 MPTP toxicity was first recognized in a group of patients who developed Parkinsonian symptoms at an early age (twenties) after taking heroin contaminated with this compound. 58 MPTP is metabolized via monoamine oxidase B to the active agent MPP+, which inhibits mitochondrial complex I. This is taken up into dopaminergic neurons via the dopamine transporter, leading to malfunction or loss of dopaminergic neurons, without, however, the Lewy body formation characteristic of Parkinson's disease. 35 MPTP given to mice selectively kills dopaminergic neurons, particularly in the substantia nigra and ventral tegmentum. 36 Strain-specific differences in susceptibility to MPTP exist, with C57/BL6 mice being more susceptible to toxin-induced neuronal death than other strains. 43, 91 These findings may be related to higher intrinsic dopamine levels in the striatum of C57/BL6 mice. 36, 43, 91 MPTP administration in nonhuman primates results in tremors, rigidity, and akinesia characteristic of Parkinson's disease. Cell loss is limited to the substantia nigra pars compacta and, like MPTP-intoxicated mice, no Lewy bodies occur. 79 In mice, MPTP-induced hypokinesia is reflected by reduced locomotor activity in the open field test 91 and reduced capacity to remain on a rotarod. 87 In both mice and nonhuman primates, motor deficits are relieved by administration of L-DOPA. 79, 87
b) Transgenic models
Transgenic models have been used to overexpress wild-type or mutant human alpha-synuclein. 66 Lesions depend largely on the promoter used—promoters such as Thy1, 49 platelet-derived growth factor-beta, 71 and the prion promoter, 61 which drive widespread expression results in alpha-synuclein accumulation and neuronal damage (accompanied by motor deficits) in multiple brain regions. A more appropriate model limits wild-type or mutant human alpha-synuclein overexpression to the dopaminergic system by using the tyrosine hydroxylase promoter. 83 Mice expressing the mutated form develop age-related bradykinesia and hypokinesia, consistent with excessive or inappropriate function of the mutated protein and resulting in cellular toxicity. Although intraneuronal inclusions containing alpha-synuclein can be seen in some models, 49, 61, 71 these lack the fibrillar morphology characteristic of Lewy bodies.
c) Alpha synuclein knockout mice
Data from alpha synuclein knockout mice 1 suggest that hereditary Parkinsonism resulting from alpha-synuclein mutations 52 results from a toxic gain-of-function mechanism rather than simple loss of gene function. Alpha synuclein knockout mice 1 are viable and fertile, exhibit intact brain architecture, and possess a normal complement of dopaminergic cell bodies and projection systems. Consistent with the converse findings in alpha-synuclein overexpressing mice, 83 knockout mice display striking resistance to MPTP-induced degeneration of dopaminergic neurons. 17
2) Huntington's disease
Huntington's disease (HD) is an autosomal-dominant, progressive neurodegenerative condition that is caused by expansion of CAG repeats near the 5′ end of the IT15 gene, which encodes a ubiquitously expressed protein of unknown function called huntington. 5, 88 The normal huntington gene allele size is between 6 and 37 CAG repeats. Most Huntington's disease occurs in midlife due to polyglutamine stretches of approximately 50 residues; stretches >57–60 residues trigger symptoms in childhood. 41 Huntington's disease displays selectivity for GABAminergic medium spiny neurons in the striatum—these are selectively vulnerable to a toxic property conferred by the polyglutamine segment. 107 The indirect pathway neurons, which express enkephalin and are enriched in D2 dopamine receptors, are affected earlier than the direct pathway neurons coexpressing substance P and D1 dopamine receptors. 93 Consequently, overactivity of the direct pathway with consequent hyperkinesis characterizes Huntington's disease. Striatal and cerebral cortical neurons contain characteristic neuronal nuclear inclusions (NII). 21, 65, 97 These are rarely present within other subcortical structures, such as the globus pallidus and thalamus, and are generally absent elsewhere in the brain. 21, 65 NII are immunoreactive with antibodies against the N-terminus of huntington (but not the internal segment or C-terminus) and ubiquitin. 97 Similar aggregates are also present in the neuropil, within the dendrites, and have been termed neuropil aggregates or dystrophic neurites. 21 The means by which mutant huntington causes neuropathology is not clear; however, it is thought to arise from the interaction of huntington with a large number of endogenous transcription factors, signaling molecules, and trafficking proteins. 63
In the early stages, Huntington's disease is characterized by absent-mindedness, irritability, depression, and clumsiness. Over time, choreiform movements develop and worsen until the patient is bedridden. Progressive decline in cognitive function and speech occur until severe dementia results in the terminal stages of the disease. 41 The neuropathology of Huntington's disease can be classified into five grades. 104 Grade-0 patients display no gross or microscopic lesions despite Huntington's disease symptomatology. 20 In grades 1–4, shrinkage of the cerebral cortex, white matter, hippocampus, amygdala, and thalamus occurs early, is not progressive, and is caused by loss of both grey and white matter. Grades 1–3 are characterized by progressive atrophy of the caudate nucleus and, to a lesser extent, the putamen and globus pallidus. The most severe grade, 4, is usually found in younger patients and often displays extension of lesions to regions such as the cerebellum not usually affected in adults. 93
Mouse models
Like humans with Huntington's disease, genetically altered mice with striatal pathology display an array of dyskinesias, which are not as clearly defined as those in humans. Most mouse models of Huntington's disease 44, 57, 64, 68, 90, 107, 108 are smaller than their littermates and display progressively worsening foot clasping when suspended by the tail. Most are hypoactive, but in some cases, 57 this can coexist with hyperactivity. Spontaneous abnormalities include stereotypic involuntary movements or tremors, 68 circling, or slow, wide-based or ataxic gait. 44, 90 Gait abnormalities are often quantitated using the painted paw test. Affected mice have a shorter uneven stride. 64, 107, 108 Invariably, these mice display a reduced capacity to remain on a rotarod. 64, 68, 90, 107, 109
A. Transgenic models expressing human huntington gene fragments
Initial transgenic models for Huntington's disease were generated by expressing either the full-length or 5′ fragment of the human huntington gene under direction of a variety of promoters, most of which resulted in widespread expression of the transgene. These models typically display an earlier onset, more severe phenotype than later targeted huntington knock-in models.
i) The R6 transgenic mouse lines
These models were generated by using exon 1 of the human huntington gene containing 115–156 CAG repeat units driven by huntington promoter sequences in the 5′UTR. 68 Clinically, the mice develop a progressive neurologic phenotype at 2–3 months of age. This includes a resting tremor, stereotypic involuntary movements, hind-limb clasping, choreiform movements (rapid abrupt shuddering of the trunk), mild dysmetria, handling-associated seizures, and abnormal vocalization. The brain of these mice is cytoarchitecturally normal but demonstrates uniform diminution due to reduced size of all central nervous system (CNS) structures. These mice develop pronounced neuronal intranuclear inclusions, containing the proteins huntington and ubiquitin, prior to developing a neurological phenotype. 18
ii) N-terminal huntington driven by mouse prion promoter
This model uses the prion promoter to drive expression of the N-terminal fragment (171 amino acids) of huntington with 82 (juvenile onset HD), 44 (adult onset HD), or 18 (normal) glutamines in virtually every neuron of the CNS. 90 Mice expressing huntington with 82 glutamine repeats (N171–82Q) develop behavioral abnormalities, including loss of coordination, tremors, hypokinesis, hind-limb clasping, abnormal gait, and inability to stay on a rotarod, before dying prematurely. In mice exhibiting these abnormalities, diffuse nuclear labeling, intranuclear inclusions, and neuritic aggregates, all immunoreactive with an antibody to the N-terminus (amino acids 1–17) of huntington (AP194), were found in multiple populations of neurons. 90 Although slightly smaller, brains of these mice are grossly normal, with no sign of abnormal development.
iii) N-terminal huntington driven by rat neuron-specific enolase promoter
Broad CNS expression of the 5′ region of human huntington with 18, 46, and 100 CAG repeats was obtained using the rat neuron-specific enolase promoter. 57 Clinically, mice with 46 and 100 repeats display a spectrum of neurologic signs, including poor rotarod performance, hind-limb clasping, hyperactive stereotypic behavior, hypoactivity, wide-based gait, or slow gait with tremulousness. Neuropathology of these is characterized by cortical cytoplasmic huntington protein accumulation and dysmorphic dendrites. Although Huntington's disease is considered primarily a disease of striatal degeneration, evidence suggests that cortical changes are as important in creating the neurologic phenotype.
iv) Huntington's disease yeast artificial chromosome (YAC) transgenic
YACs containing human genomic DNA spanning the full-length gene, including all regulatory elements, were used 44 to express huntington with either 46 (to replicate adult-onset HD) or 72 (juvenile onset) CAG repeats. Both normal and mutant human huntington are expressed in the same tissues as the endogenous mouse protein, with highest levels seen in brain and testes. The YAC72 mouse exhibits the most severe clinical signs, including circling, choreiform movements of the head and neck, as well as gait ataxia and foot clasping. In the striatum, neuronal intranuclear inclusion immunoreactive for N-terminal huntington is detected only in medium spiny neurons, accompanied by striatal degeneration.
B. Targeted models containing the endogenous mouse Hdh gene
i) Huntington knock-in mice
The murine wild-type Hdh gene contains 7 CAG N-terminal repeats. Expanded polyglutamine repeats of 50, 92, 111, and 150 repeats were inserted in the endogenous mouse Hdh gene, with consequent expression of mutant proteins in a distribution most similar to that seen in human patients. 64, 107, 108 Expanded repeats of lengths causing adult onset (50 repeats) do not rapidly provoke abnormalities, suggesting that the mouse's short lifespan precludes the accumulation of disease-causing pathology seen in humans. 108 Longer stretches of 92 and 111 CAG repeats induce cytoplasmic accumulation followed by nuclear translocation of mutant huntington protein to striatal neurons. 107 The larger the repeat, the earlier this occurs. No behavioral abnormalities were noted in mice with 92 and 111 repeats. 107 Double immunolabeling studies using calbindin D antibody to identify medium spiny neurons confirmed that nuclear relocation in the striatum targeted this cell population. 107 Insertion of 150 CAG repeats 64 resulted in earlier onset neuropathology accompanied by behavioral and gait abnormalities.
3) Additional mouse models with striatal pathology
Both spontaneous mutants (the weaver mouse) 94 and mice genetically altered for genes originally not suspected to result in striatal pathology have been described. 27, 70
a) The weaver mouse
The weaver (wv) mouse is a spontaneous autosomal recessive mutant with a predominantly cerebellar phenotype caused by failure of granular cells to migrate normally and their subsequent death. 94 In addition, wv is also dopamine deficient in the forebrain. This deficiency is profound in the dorsal striatum, whereas the accumbens nucleus, ventral caudate-putamen, and medial olfactory bulb are much less severely affected. 67, 86 Although no human homologue of the weaver mouse exists, the combined cerebellar-striatal phenotype is reminiscent of hereditary striatonigral and cerebello-olivary degeneration in the Kerry Blue Terrier. 19, 74 Although cerebellar abiotrophy prevails in these dogs, they also demonstrate marked degeneration of the caudate nuclei and the substantia nigra.
b) Genetically altered mice
Mice in which the transcription factors Creb1 and Crem are disrupted in the postnatal forebrain show progressive neurodegeneration in the hippocampus and in the dorsolateral striatum. 70 The striatal phenotype is reminiscent of Huntington disease and is consistent with the postulated role of CREB-mediated signaling in polyglutamine-triggered diseases. 80, 115 Ablation of the murine ataxia-telangiectasia gene (Atm) results in adult-onset loss of dopaminergic neurons in the A9 and A10 areas of the mesencephalon. 27 These lesions are accompanied by locomotor abnormalities manifested as stride-length asymmetry, which could be corrected by peripheral application of the dopaminergic precursor L-dopa. 28
II. Mouse models of dopaminergic neurotransmission
Aspects of dopaminergic synthesis, secretion, and receptor activity have been explored by ablating genes controlling these functions. The mice resulting from these experiments have no human counterparts. In addition, their phenotype is locomotor, behavioral, or vegetative rather than neuropathologic. Consequently, they have been used to explore the role of dopaminergic neurotransmission in motivational 22, 113 or hyperactivity behaviors 2, 6 as well as gait. 48
1) Dopamine synthesis, metabolism, and transport
Dopamine is synthesized from the amino acid tyrosine
85,
101
within the neuronal cytoplasm. Tyrosine is transported into the neuron via membrane-bound amino acid carrier and is converted to
Mouse models
a) Tyrosine hydroxylase deficient mice
Ablation of TH 53, 114 eliminates synthesis of both dopamine and norepinephrine. The majority of TH−/− embryos die between embryonic days 11.5 and 15.5 and display cardiac dilation and bradycardia. Those that are born fail to breathe. Administration of L-DOPA to pregnant females results in survival and birth of TH−/− pups, 114 indicating that catecholamines are essential for mouse fetal development. To isolate the role of dopamine alone, Zhou and Palmiter 113 restored norepinephrine synthesis in sympathetic neurons by crossing TH−/− mice with transgenic mice expressing TH under control of the dopamine-beta hydroxylase promoter. Dopamine-deficient pups were born at the normal Mendelian frequency but became hypoactive and aphagic a few weeks after birth, indicating that dopamine is essential for movement and feeding but that the neural networks that utilize it can develop in its absence. The postnatal syndrome in dopamine-deficient mice can be alleviated by administration of L-DOPA. 96, 113 An alternate pathway for catecholamine synthesis was identified by Rios et al., 84 who detected catecholamines in sympathetic nuclei, adrenal medulla, and brain of pigmented, but not albino, TH−/− mice. They concluded that tyrosinase in melanocytes provides an alternate pathway for the synthesis of L-DOPA and catecholamines.
b) Vesicular monoamine transporter 2 knockout mice
Vesicular monoamine transporter 2 (VMAT2) is the predominant dopamine transporter expressed in the brain. 98, 101 VMAT2−/− mice 106 die within a few days after birth, manifesting severely impaired dopamine storage and vesicular release. Heterozygous VMAT2 knockouts are viable into adult life but display VMAT2 levels one half that of wild-type values, accompanied by smaller changes in monoaminergic markers, heart rate, and blood pressure. 106
c) Dopamine transporter knockout mice
Ablation of the DAT results in increased levels of dopamine into extracellular space and enhanced dopaminergic neurotransmission. Deletion of the DAT 6 results in increased dopaminergic tone, rostral pituitary hypoplasia, dwarfism, and an inability to lactate, thus revealing an unexpected and important role for dopamine in the control of developmental events in the pituitary gland. Behaviorally, DAT knockout mice demonstrate marked hyperactivity characterized by increased locomotion, rearing, and stereotypic behaviors. These behaviors are completely reversed by dopamine antagonists. 32
2) Dopamine receptors
The central actions of dopamine are mediated by 5 distinct dopamine-receptor classes that are expressed on dopamine synthesizing neurons and exert their effects via activation of heterotrimeric guanine nucleotide regulatory proteins. 37, 73 The D1 dopamine receptor is the most widely distributed central dopamine receptor. It is present in the cortex and rostral olfactory nuclei, accumbens nuclei, and amygdaloid nuclei. 101 Both D1 and D2 dopamine receptor types are expressed by medium spiny neurons, which constitute over 90% of the striatal population. 33 Medium spiny neurons can be divided into two primary populations determined by whether they utilize substance P or enkephalin. These two populations also possess distinct dopamine-receptor types. Striatal neurons projecting directly to the medial globus pallidus express the D1 dopamine receptor and the neuropeptides substance P and dynorphin. Striatal neurons giving rise to the indirect pathway (projecting to the medial globus pallidus via the lateral globus pallidus and subthalamic nucleus) express the D2 dopamine receptor and enkephalin. 22, 37, 101 Ablation of D1 or D2 receptors results in phenotypes that reflect an imbalance between the direct and indirect pathways. 7, 22
Mouse models
a) D1A dopamine receptor knockout mouse
D1A−/− mutants are small and exhibit postnatal growth retardation. 22, 110 These findings are consistent with the role of dopamine in motivational and reward behaviors mediated by the ventral striatum. Neurologically, D1A−/− mice exhibit normal coordination and locomotion, but display conflicting results for tests of exploratory activity in novel environments. 22, 110 Decreased rearing in an open field was identified by Drago et al., 22 suggesting reduced exploratory phenotype, while increased activity based on interruption rate of a photocell beam was reported by Xu et al. 110 Results such as these highlight the potential of different test protocols to affect results of behavioral testing. 50 Using a double transgenic Cre-lox strategy, Drago et al. 24 expressed diphtheria toxin gene in D1A-positive cells. This resulted in death of all D1A-positive neurons and a much more severe motor phenotype. Double mutant mice display bradykinesia (a Parkinsonian phenotype), dystonia, and myoclonia (more consistent with signs seen in Huntington's disease). In addition, these mutants, like their predecessors, 110 are hypophagic and usually die in the first 2 postnatal weeks.
b) Dopamine D2 receptor knockout
Mice lacking D2 dopamine receptors are smaller, with reduced postnatal growth and reduced fertility. 7, 48 Motor signs appear between PN 30 and 45, consistent with maturation of the D2 receptor system. 48 They exhibit postural abnormalities, including a hunched posture, paw flattening, and sprawling of hind limbs, that are more pronounced when voluntary movement is initiated. Absence of D2 receptors results in animals that are akinetic and bradykinetic in behavioral tests and which show significantly reduced spontaneous movements. This phenotype presents analogies with symptoms characteristic of Parkinson's disease. 7, 23
c) Dopamine D3 receptor knockout
Homozygous mice lacking D3 receptors display increased locomotor activity and rearing behavior. 2 Those heterozygous for the D3 receptor mutation show similar, albeit less pronounced, behavioral alterations, suggesting that D3 receptors play an inhibitory role in the control of certain behaviors. 2
d) Double D1/D3 receptor knockout
Combined deletion of both receptors abolishes the exploratory hyperactivity of D3 dopamine-receptor mutant mice and further attenuates the low exploratory phenotype of some D1 receptor knockout mice, 22, 50 suggesting that dopamine D1 and D3 receptors may interact in an opposing or synergistic fashion.
e) Double D2/D3 receptor knockout
D2/D3 double mutants develop motor phenotypes that, although qualitatively similar to those seen in D2 single mutants, are significantly more severe and persist into adulthood. 48 These results suggest that D3 receptors compensate for some of the lacking D2 receptor functions and that these functional properties of D3 receptors, detected in mice with a D2 mutant genetic background, remain masked when the abundant D2 receptor is expressed. 48
f) Dopamine D4 receptor knockout
The human dopamine D4 receptor (D4R) has received considerable attention because of its polymorphic nature and possible role in the phenomenon of novelty seeking. 81 While knockout mouse data support the role of the D4 dopamine receptor in modulating responses to novelty, a recent analysis of the studies linking D4 dopamine receptor variants to novelty seeking, alcoholism, drug abuse, and attention deficit hyperactivity disorder determined that many of these studies had methodologic concerns. 81 D4 dopamine receptor knockout mice display reduced novelty-induced activity. 25
g) Dopamine D5 receptor knockout mice
D5 dopamine receptor-deficient mice are viable and fertile but develop hypertension and exhibit significantly elevated blood pressure (BP) by 3 months of age. 46 Behavioral changes are minimal. 47
Summary
The intent of this review is to provide an overview of the anatomy and physiology of the dopaminergic system as well as the mouse mutants that describe major categories of dopaminergic function and dysfunction. The requirements of each study (mouse model used, extent of pathologic examination, or choices of behavioral testing paradigms) are heavily dependent on the research question. Human movement disorders, such as Huntington's and Parkinson's diseases, have been quite comprehensively replicated using mouse models that develop lesions in the relevant anatomical areas. The specific neuropathology in these models is relatively subtle and ranges from loss of specific cell groups, neurodegeneration, and accumulation of intracytoplasmic or intranuclear inclusions. Genetically altered mice with striatal pathology display an array of dyskinesias, which are not as clearly defined as those in humans. Easily noted changes include alterations in body weight, posture, gait, and spontaneous activities. Motor abnormalities can be quantitated using tests of motor function, such as the rotarod, balance-beam test, vertical pole test, and painted paw test. Aspects of dopaminergic synthesis, secretion, and receptor activity have been explored by ablating genes controlling these functions. The mice resulting from these experiments have no human counterparts. In addition, their phenotype is locomotor, behavioral, or vegetative rather than neuropathologic. Consequently, they have been used to explore the role of dopaminergic neurotransmission in motivational or hyperactivity behaviors as well as gait.
Footnotes
Acknowledgements
The author gratefully acknowledges Dr Alexander deLahunta for his review of the anatomical portion of this manuscript.