
Abstract
A 9-month-old male German Shepherd dog was referred for evaluation of progressive exercise intolerance. Clinical examination revealed a stiff, stilted gait and marked atrophy and hypotonia of skeletal muscle. The dog had raised creatine kinase (181 U/liter), lactate dehydrogenase (510 U/liter), and aspartate aminotransferase (123.6 U/liter) levels, suggesting a muscle disease. Histochemical evaluation of muscle biopsies revealed the presence of subsarcolemmal oxidative activity, reduced nicotinamide adenine dinucleotide, and succinate dehydrogenase, and the absence of cytochrome oxidase activity. Ragged red fibers were demonstrated with Gomori trichrome stain. Ultrastructural examination of the muscle confirmed the presence of subsarcolemmal accumulations of mitochondria and morphologically atypical mitochondria.
Introduction
Mitochondrial disorders are a heterogeneous group of diseases with multisystem presentation. 10,13 Because many metabolic processes take place in the mitochondria, the alteration of adenosine triphosphate production due to diminished activity of the mitochondrial respiratory chain is the causative factor of the majority of these diseases. 5 The respiratory chain is embedded in the inner mitochondrial membrane and is composed of four multi-subunit enzyme complexes encoded by mitochondrial deoxyribonucleic acid (mtDNA) and nuclear DNA. Therefore, mitochondrial diseases may arise from mutations in either mitochondrial or nuclear genomes. Mutations in mtDNA are transmitted by maternal inheritance and are generally heteroplasmic, that is, wild-type and mutant mtDNAs coexist in the same cell. 5,7,9 In mitochondrial disorders, the most severely affected organs are those most dependent on oxidative metabolism, including brain, skeletal, and cardiac muscles; sensory organs; and kidney. 8
Few cases of mitochondrial diseases have been described in veterinary medicine. 2,3,4,11 We report the histochemical and ultrastructural findings of mitochondrial myopathy in a German Shepherd dog.
Case History
A 9-month-old male German Shepherd was referred to the Department of Veterinary Clinical Science–Surgery Section, University of Naples, with a history of exercise intolerance, reluctance to move, and spontaneous pain. Clinical signs had arisen 1 month earlier and had progressively increased in severity.
The dog showed systemic muscle atrophy, mostly in truncal and limb muscles, myalgia, and gait abnormalities (stiff gait, thoracolumbar kyphosis, and bunny-hopping in pelvic limbs).
Orthopedic and neurologic examinations were unremarkable. Blood samples were taken for hematologic and serologic examination.
A diagnosis of skeletal myopathy of unknown etiology was made, and antibiotics (amoxycillin-clavulanic acid, 20 mg/kg bid po) and FANS (aspirin, 25 mg/kg qid po) were given while waiting for results of the analysis. Five days later, the dog showed no improvement.
Routine hematology revealed no abnormalities. Biochemistry showed increased levels of creatine kinase at 37 C (181 U/liter), lactate dehydrogenase (510 U/liter), and aspartate aminotransferase (123.6 U/liter).
Radiographs of coxofemoral and stifle joints were taken, but no abnormalities were found.
Muscle biopsies were taken from the femoral biceps muscle for histopathologic examination.
Materials and Methods
Muscle biopsy specimens from the femoral biceps muscle were frozen in isopentane precooled in liquid nitrogen. Sections were stained by the following histologic and histochemical techniques: hematoxylin and eosin (HE), modified gomori trichrome, oil-red-O, periodic acid–Schiff (PAS), cytochrome oxidase (COX), succinate dehydrogenase (SDH), and reduced nicotinamide adenine dinucleotide tetrazolium reductase (NADH-TR). We used a sample of biceps muscle from a dog euthanatized for a neoplastic disease as a control for histoenzymatic stains.
For transmission electron microscopy, additional muscle samples were fixed in 2.5% glutaraldehyde, postfixed in osmium tetroxide, and embedded in low-viscosity Spurr resin. Ultrathin sections were cut, counterstained with 0.5% uranyl acetate and lead citrate, and examined using a Zeiss Electron Microscope 902.
Results
HE staining showed both single atrophic fibers and fibers with a basophilic rim along the sarcolemma (Fig. 1). Sections stained with modified Gomori trichrome had many “red ragged fibers” (RRF) with subsarcolemmal and intermyofibrillar deposits of reddish granular material (Fig. 2). PAS staining showed an increase in glycogen at the periphery of fibers (Fig. 3), but staining with oil red O was unremarkable. SDH and NADH-TR stains demonstrated concentration of oxidative activity at the periphery of the fibers (Fig. 4). Many muscle fibers and all fiber types failed to stain for COX activity (Fig. 5). Residual subsarcolemmal COX activity was seen in a few fibers. In contrast, normal canine skeletal muscle showed punctate COX staining distributed throughout the cytoplasm of a subpopulation of muscle fibers (Fig. 6).
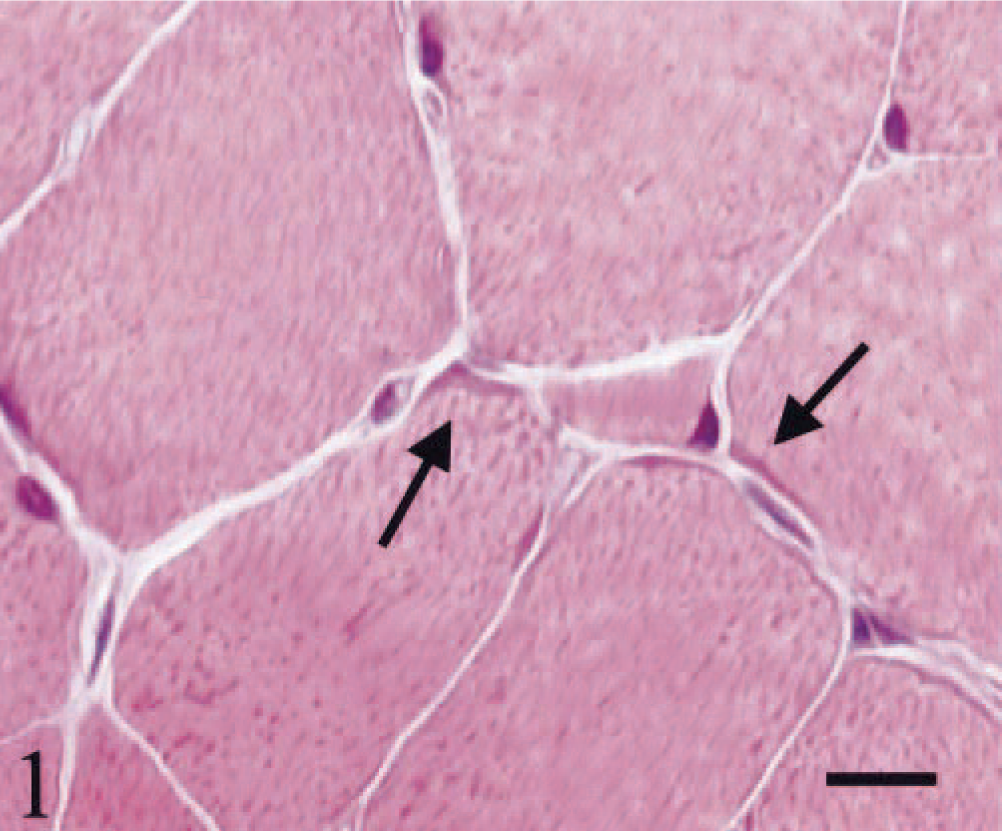
Skeletal muscle; dog. Atrophy of single-muscle fibers and fibers with a basophilic rim (arrows) along the sarcolemma. HE stain. Bar = 22 µm.
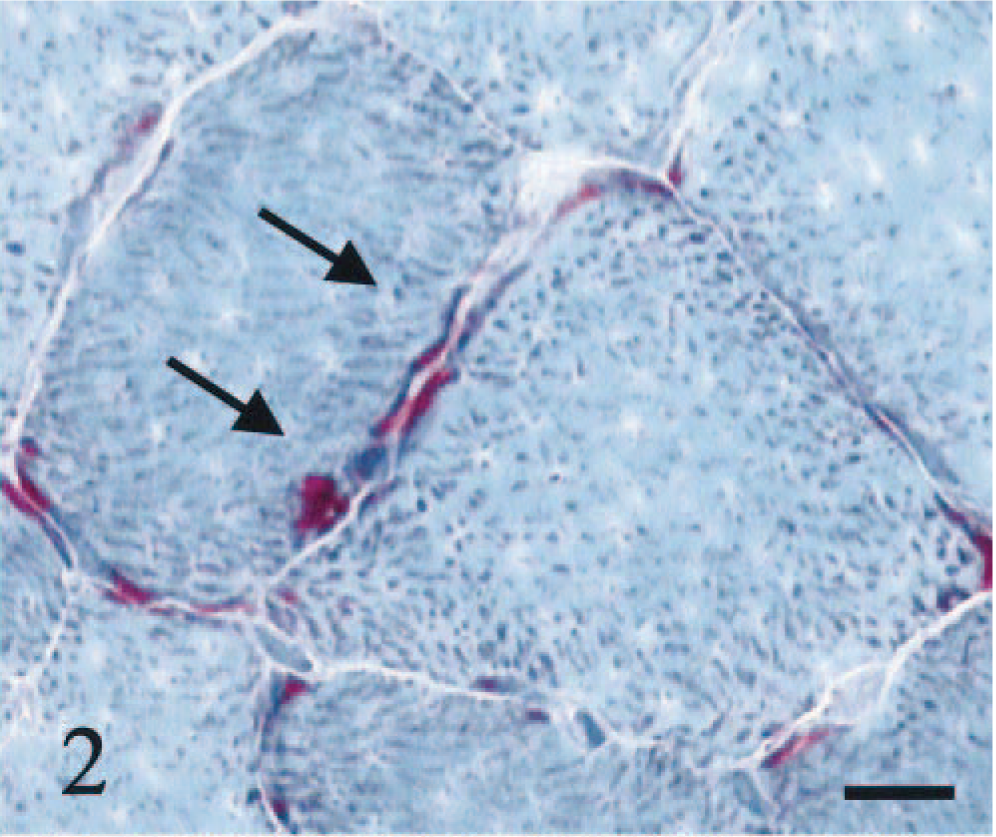
Skeletal muscle; dog. RRF with subsarcolemmal and intermyofibrillar reddish deposits of granular material (arrows). Modified Gomori trichrome stain. Bar = 22 µm.
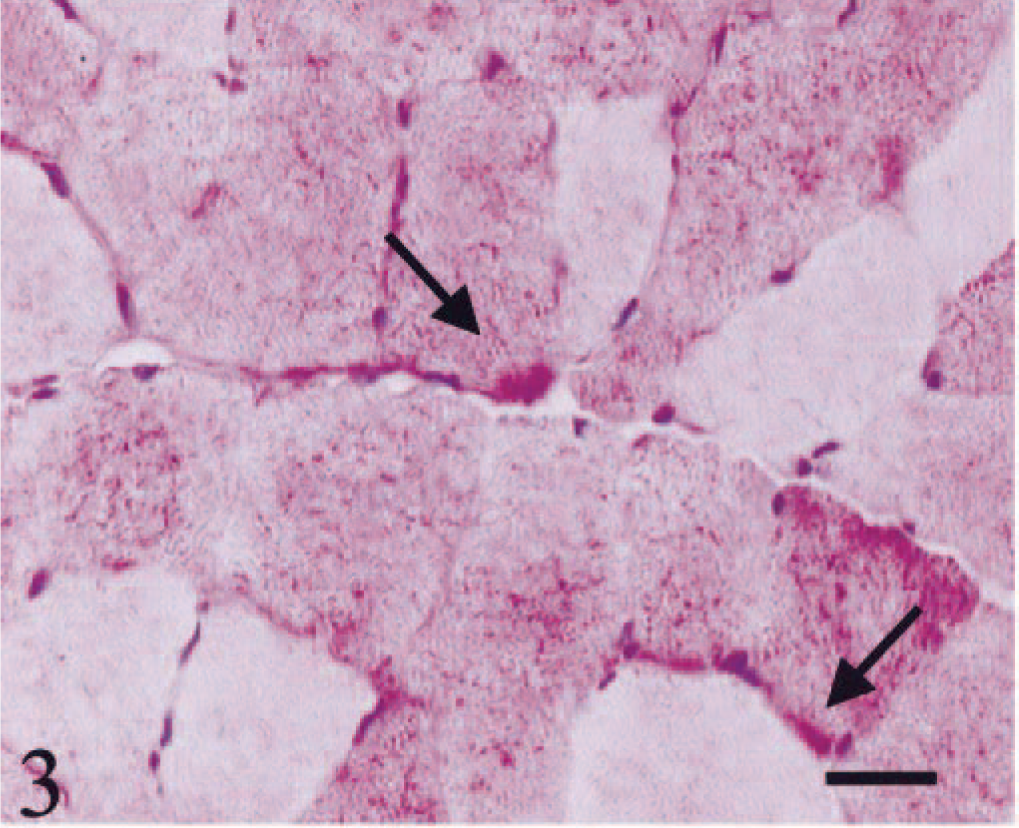
Skeletal muscle; dog. Increase in glycogen at the periphery of the fibers (arrows). Bar = 40 µm.
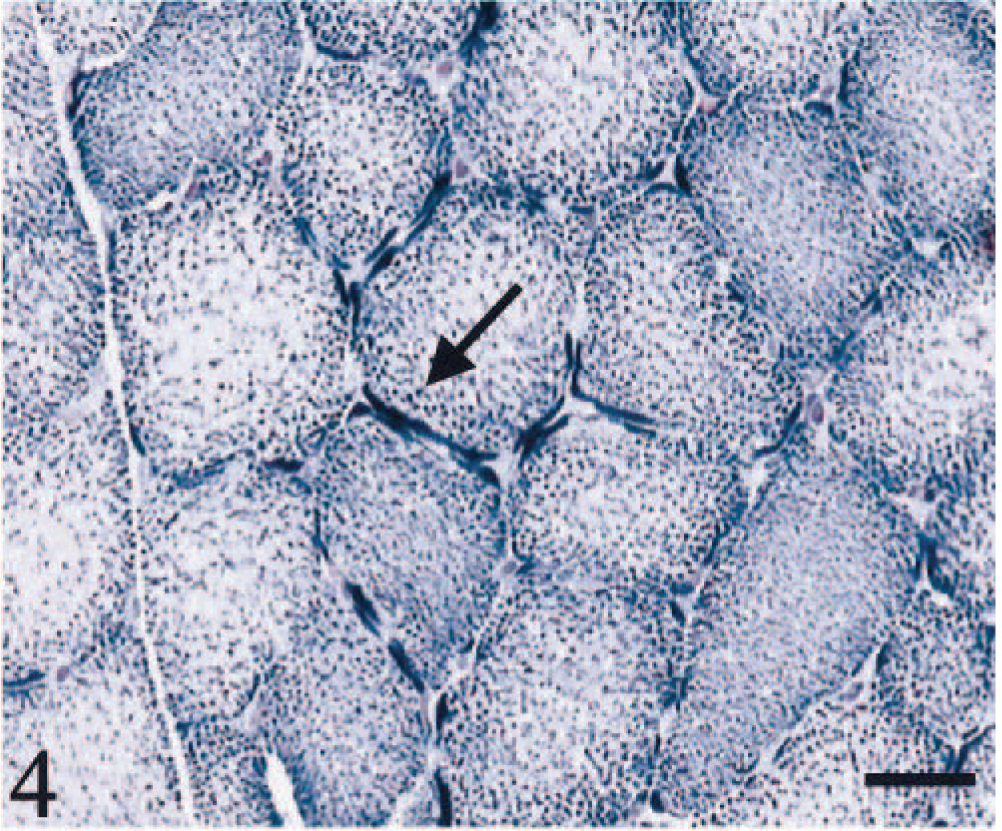
Skeletal muscle; dog. Concentration of oxidative activity at the periphery of the fibers (arrow). SDH stain. Bar = 40 µm.
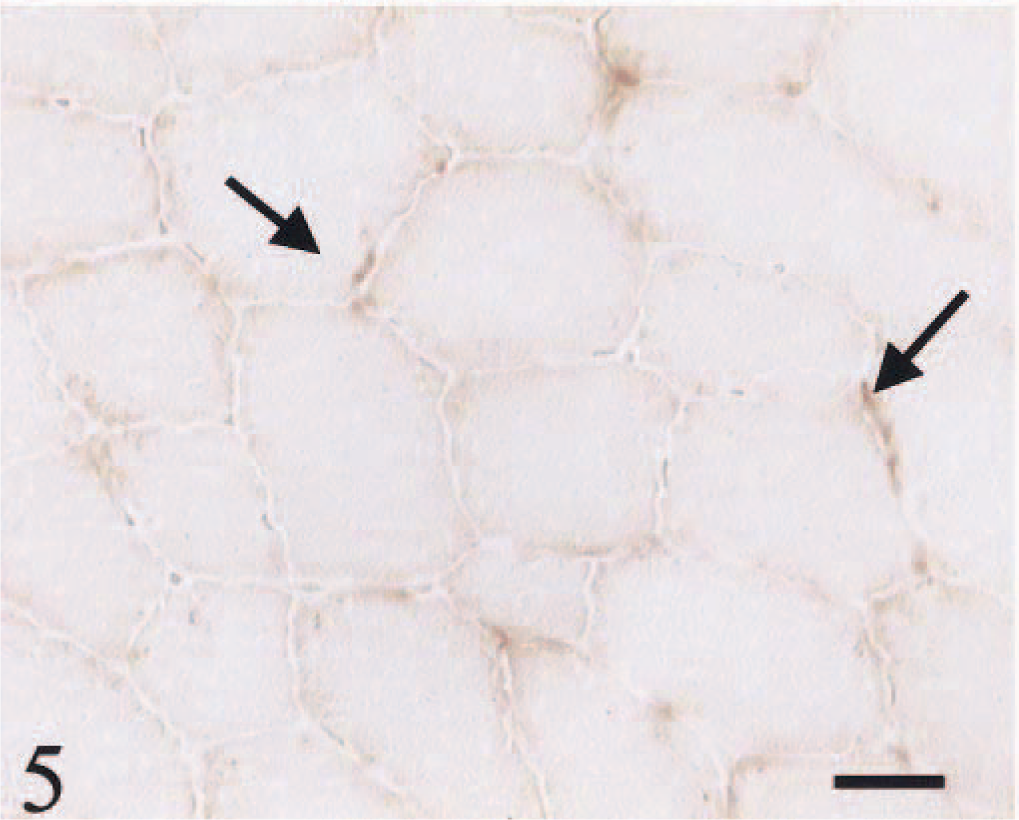
Skeletal muscle; dog. High proportions of muscle fibers and all fiber types failed to react for COX. Residual COX activity is observable in a few fibers (arrows). COX stain. Bar = 40 µm.
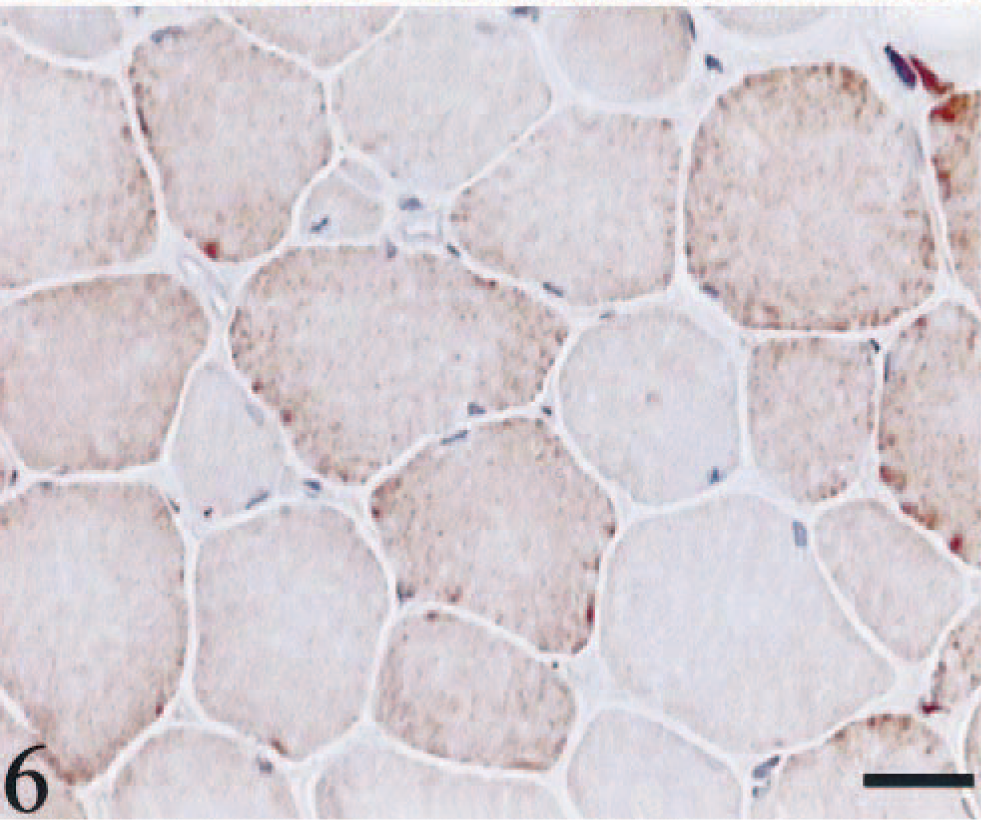
Skeletal muscle; dog. Positive control that shows the normal distribution of COX activity. COX stain. Bar = 40 µm.
Ultrastructural examination confirmed the presence of abnormal, large and giant mitochondria with bizarre shapes (e.g., “golf-club-shaped mitochondrion”) in a subsarcolemmal position and near the nucleus (Figs. 7, Figs. 8). These mitochondria contained abundant matrix and showed either disintegrating cristae or irregularly undulating cristae (sinuous cristae). Many swollen mitochondria were observed. The mitochondrial matrix appeared vacuolated, sometimes containing osmiophilic, electron-dense bodies (Fig. 9). In a few mitochondria, breaks in the wall were evident.
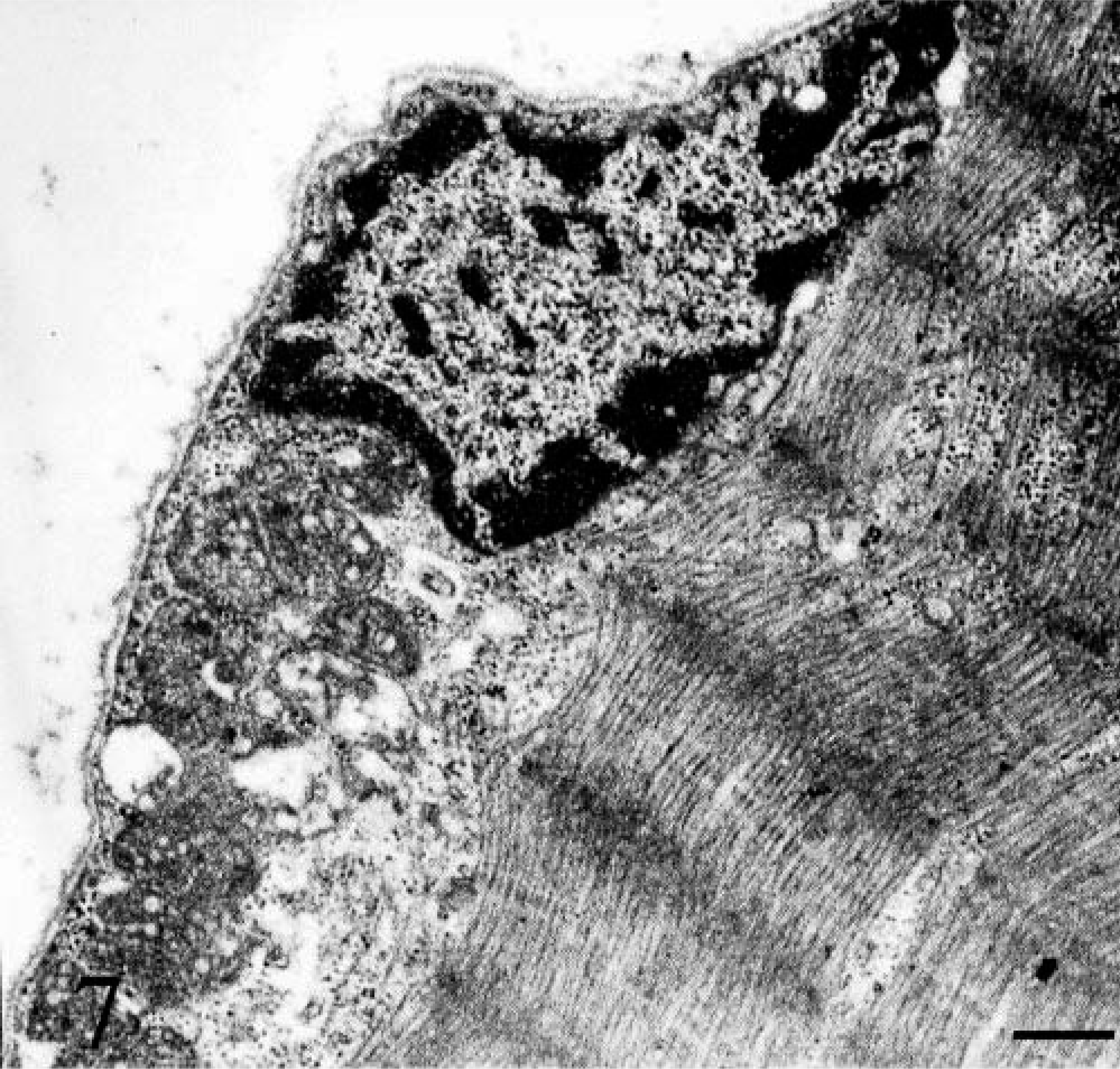
Skeletal muscle; dog. Electron micrograph. Abnormal and large mitochondria at the subsarcolemmal position and near the nucleus. Bar = 2.5 µm.
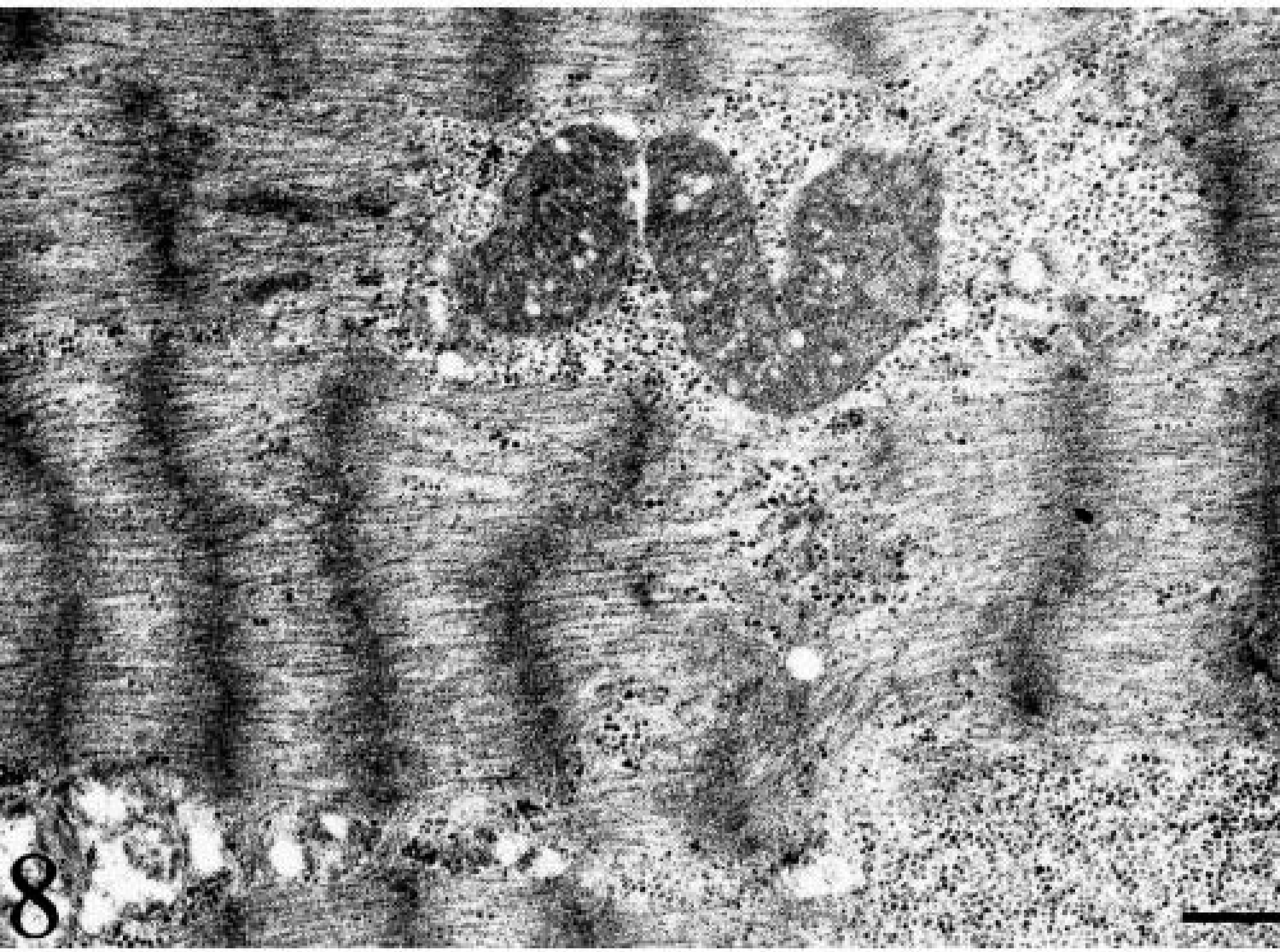
Electron micrograph. Skeletal muscle; dog. A giant golf-club–shaped mitochondrion. Bar = 6 µm.
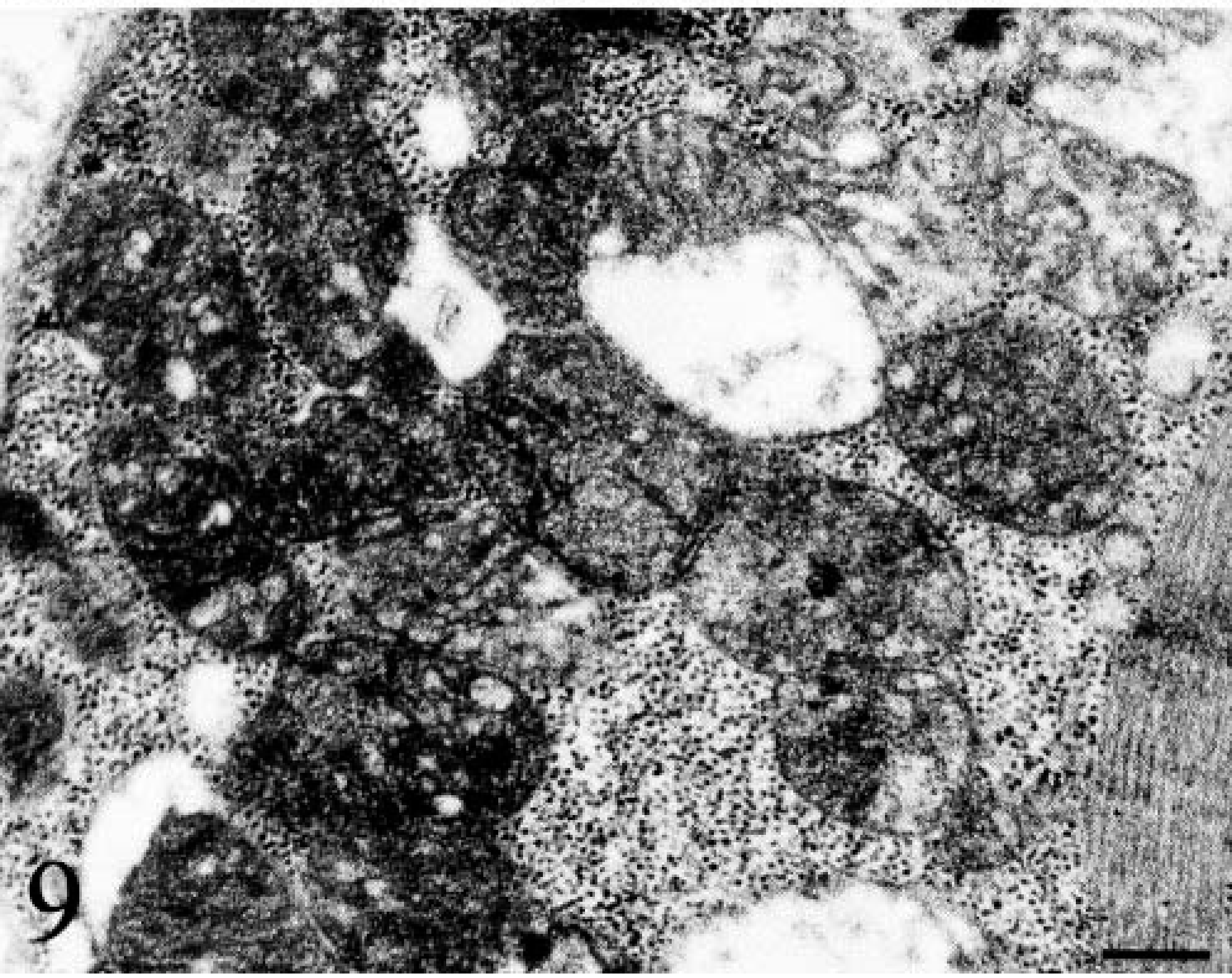
Electron micrograph. Skeletal muscle; dog. Swollen mitochondria with cavitation of the matrix and disruption of the cristae. Bar = 8 µm.
Muscle mitochondria were embedded in a sarcoplasm containing a large amount of glycogen, as suggested by PAS staining.
Discussion
DNA analysis has allowed better understanding of the molecular bases of mitochondrial diseases that result in myopathies or encephalomyopathies. 8 Mitochondrial disorders associated with mutations involving mtDNA can have several unique genetic characteristics, including 1) maternal inheritance, 2) heteroplasmy due to the high mutation rate of mtDNA and random segregation of mitochondria during cell division, 3) threshold expression of phenotype, and 4) variable clinical phenotype resulting from differences in defects and the extent to which a tissue relies on mitochondrial energy production. 7,8 Mitochondrial disorders due to mtDNA mutations are not always transmitted from an affected mother. Some cases may be sporadic. Sporadic cases probably result from mtDNA deletions during embryogenesis, which is favored in development because deleted DNA reproduces more rapidly than its normal counterpart. Because most mtDNA mutations either alter or eliminate transfer ribonucleic acid (tRNA) genes, thus altering biosynthesis of most mitochondrial (mt)-encoded proteins, the diversity of the clinical and biochemical consequences is puzzling. The pleiotropic effects of these diseases suggest that additional factors may be important in determining the clinical and biochemical phenotypes. 8
The diagnosis of mitochondrial myopathy is based on clinical presentation and on the following criteria: evidence of mutations or deletions in mtDNA, deficiency of respiratory chain enzymes in fibroblasts or muscle, characteristic changes such as RRF or decreased SDH or COX staining in muscle biopsy, evidence of abnormal mitochondrial structure, and a family member with a proven mitochondrial disease. Unfortunately, we had no data on the lineage of the animal in the present case, but the owner knew no other similarly affected relative.
Few cases of mitochondrial myopathies 2,3,11 have been reported in domestic animals. Altered cytochrome c oxidase activity and reduced mt messenger RNA (mRNA) was demonstrated, by enzyme assay, in fibroblasts and skeletal muscle of an old English sheepdog. 4 Some authors suggested that mt dysfunction may be the causative factor of exertional metabolic myopathy with lactic acidosis in dogs. 4,11
The muscle biopsy changes, although nonspecific, often provide the initial diagnostic clue and prompt further investigations to discover the underlying defects. The morphologic hallmark of mitochondrial myopathies, due either to nuclear or mtDNA mutation involving the respiratory chain, is the presence of RRF. They are irregular in outline and show reddish subsarcolemmal and intermyofibrillar granular deposits with the modified Gomori trichrome stain. The sarcoplasm appears fragmented; changes resemble artifactual cracking. This appearance represents the accumulation of mitochondria beneath the sarcolemma and between myofibrils. However, RRF also can occur in adult acid maltase deficiency and as a nonspecific isolated finding in various myopathies. In normal human muscles, their frequency increases with age, reaching a maximum of about 0.4% by the eighth decade. This probably reflects the age-related accumulation of abnormal mitochondria with DNA damage. 1
RRF are most commonly seen with primary mutations in mtDNA, usually in the transfer RNA genes, and rarely with mutations in nuclear DNA. 3
SDH, which indicates abnormally increased numbers of mitochondria, has been frequently used as a qualitative histochemical marker for RRF in skeletal muscles. SDH is the complex II of the respiratory chain, embedded in the inner mitochondrial membrane, and is specifically involved in the oxidation of succinate. It carries electrons from flavin adenine dinucleotide (reduced form) to coenzyme Q. SDH is the only enzyme in the respiratory chain complex that is entirely encoded by the nuclear genome. 12 Either partial deficiency of SDH activity or increased SDH activity may be seen in mitochondrial myopathy. 12
In our case, a peculiar finding was the failure of muscle fibers to stain histochemically for COX activity. COX-negative fibers are invariably present in human patients with progressive external ophthalmoplegia, Kearns-Sayre syndrome, and benign reversible form of cytochrome c oxidase deficiency, but they often occur in other mitochondrial encephalomyopathies and in patients with myopathy alone. Recent studies showed that mitochondria with deleted mtDNA are almost exclusively concentrated in segments of the muscle fibers that lack COX activity. 8 These studies also have shown that most deleted mtDNAs are transcriptionally active, but the overabundant mt mRNAs, which are mainly transcribed from partially deleted genomes, are not translated into proteins. 8 Translational failure in ragged red, COX-negative fiber segments has been attributed to lack of indispensable tRNA genes eliminated by the deletions or depletion of wild-type mtDNA or interference with expression of near-normal levels of wild-type mtDNA by an overabundance of abnormally short molecules. 8
COX-negative muscle fibers outnumbered RRF, indicating a more widespread distribution of abnormal mitochondria than was expected on the basis of the frequency of the RRF.
This finding was confirmed in our case by ultrastructural examination, which showed large numbers of structurally abnormal muscle mitochondria. Ultrastructural changes in mitochondria are nonspecific and are not correlated with type of mtDNA mutation, site of respiratory chain blockade, or clinical phenotype. 6 Nevertheless, electron microscopic examination of muscle biopsy specimens is a useful screening method to select specimens for further biochemical analysis and to confirm the light microscopic diagnosis.
Footnotes
Acknowledgements
We thank Mr. R. Ilsami for technical assistance.