
Abstract
Transforming growth factor-β (TGF-β) plays a complex role in skin carcinogenesis, acting as a suppressor early in tumor development but later as a promoter. Smad proteins are important intracellular mediators of TGF-β signaling. To determine the effect of disrupting Smad genes and TGF-β signaling on chemically induced skin carcinogenesis in mice, transgenic mice heterozygous for Smad2 or Smad3 deletions and wild-type controls were treated with topical dimethylbenzanthracene for 7 months. Tumor multiplicity, type, and degree of differentiation were assessed by histopathology and immunohistochemistry. Smad3 ± mice developed significantly fewer tumors than the wild-type group (P < 0.05). This indicated a possible oncogenic function for Smad3 in skin carcinogenesis. Smad2 ± mice formed less-differentiated tumors than their wild-type counterparts, supporting a tumor suppressor role for Smad2. There was a significant difference (P < 0.05) in tumor type between Smad2 ± and Smad3 ± groups, suggesting that Smad2 and Smad3 may regulate different targets.
Keywords
The pathway by which normal epithelial cells give rise to proliferative and invasive tumor cells is complex and involves many genetic and epigenetic changes. Many of these changes have been dissected by studying skin tumor induction in mice. 20 A commonly used regimen for inducing papillomas and skin cancer in mice is the topical application of the carcinogen dimethylbenzanthracene (DMBA) followed by application of one of various promoting agents. Cells with appropriate genetic changes form papillomas; further genetic and epigenetic changes cause these papillomas to undergo malignant conversion to squamous cell carcinomas (SCC) and ultimately to spindle cell carcinomas.
The transforming growth factor-β (TGF-β) superfamily consists of members that function in different developmental and disease processes. Numerous studies have demonstrated the importance of TGF-β signaling in cancer progression and metastasis. TGF-β plays a complex role in skin carcinogenesis. Early in the process of skin tumor formation, TGF-β acts to suppress tumor emergence; 17 indeed, loss of TGF-β activity at this point promotes tumor formation. Paradoxically, later in the process of skin carcinogenesis, tumor cells may produce and secrete high levels of TGF-β, and this TGF-β can act in an autocrine or paracrine manner to enhance tumor growth and malignant conversion. 17 Numerous studies have investigated the effects of TGF-β on epithelial carcinogenesis, but the results of these studies have sometimes been contradictory. Although some studies support the tumor suppressive effects of TGF-β, other studies indicate a tumor-promoting function for TGF-β. Findings in genetically altered mice overexpressing TGF-β1 in the epidermis support a dual role for TGF-β in skin carcinogenesis. Compared with control mice, these mice were more resistant to chemically induced papillomas. However, the papillomas that did develop showed enhanced malignant progression to SCCs and spindle cell carcinomas. 3 Thus, TGF-β appears to have biphasic effects on multistage chemical carcinogenesis of skin, and the precise effect of TGF-β depends on the stage of tumor development under consideration. 3
Smad proteins are an integral part of the TGF-β signaling pathway. 9 After ligand binding, the type-II TGF-β receptor (TβRII) phosphorylates the type-I TGF-β receptor (TβRI), which, in turn, phosphorylates and activates the receptor Smads (Smad2 and Smad3). These receptor Smads bind to a co-Smad (Smad4), and this Smad complex translocates to the nucleus, where the complex regulates transcription of TGF-β–responsive genes. Smad4 has been shown to act as a tumor suppressor gene in colorectal and pancreatic carcinomas. 4 Other data support a tumor suppressor role for Smad2 and Smad3 in colon cancer. 5,21 Complete loss of Smad2, Smad3, and Smad4 protein expression has been observed in chemically induced skin tumors in mice, although the Smad2 and Smad4 genes did not appear to be deleted or to contain mutations. 8 This suggests that Smads may act as tumor suppressor genes in the skin.
We investigated the effects of disrupting the TGF-β signaling pathway on the development of DMBA-induced skin tumors in mice by altering the level of expression of Smad genes. Because Smad2 −/− mice die as embryos and Smad3 −/− knockout mice are relatively short lived, we used mice heterozygous for null alleles of Smad2 and Smad3. Our previous work has shown that these mutations decrease the level of Smad2 and Smad3 proteins by roughly 50%. 14 Our work demonstrates that Smad3 heterozygotes exhibit greater resistance to chemically mediated skin carcinogenesis than wild-type or Smad2 ± mice. This would suggest that Smad3 can function in an oncogenic fashion because heterozygous loss is protective against chemically induced cutaneous neoplasms. Interestingly, the tumors that developed in the Smad2 heterozygotes had a less-differentiated phenotype than those found in their wild-type or Smad3 ± counterparts.
Mice were of three genotypes: wild-type, Smad2 ±, or Smad3 ±. Mice were maintained on a mixed genetic background of 50% 129SvEv and 50% Black Swiss, and genotypic analysis of genomic DNA extracted from tail biopsies was performed as described. 16 A breeding scheme (Smad2 ± × Smad3 ±) that produced mice of all three genotypes, such that the mice in any one experimental group were related to mice in the other groups, was used. This was done to minimize the differences in strain background between the different experimental sets. A total of forty-two 8-week-old mice were treated topically with DMBA (Sigma-Aldrich, St. Louis, MO), after removal of dorsal hair by a depilatory, once weekly for 7 months. The mice were initially treated with 200 µl of 80 pM DMBA in acetone for 20 weeks and then with 200 µl of 160 nM DMBA in acetone for 8 weeks. This technique was modified from one reported to induce skin tumors in CD-1 mice. 7 At the end of the study, skin tumors were counted, fixed in formalin, embedded in paraffin, and processed for hematoxylin and eosin (HE) staining and immunohistochemistry.
For immunohistochemistry, sections of paraffin-embedded tumors were dewaxed and subjected to antigen retrieval using Target Retrieval Solution as recommended by the supplier (DakoCytomation, Carpinteria, CA). After endogenous peroxidase activity and nonspecific protein binding were blocked, sections were incubated for 1 hour at room temperature with rabbit primary antibodies against cytokeratins 6, 10, and 14 (Covance, Inc., Princeton, NJ) and desmoplakin (Serotec, Inc., Raleigh, NC), diluted 1:500, 1:1,000, 1:10,000, and 1:1,500, respectively. Immunoreactivity was detected by incubation with biotinylated secondary antibodies, avidin–biotin complex, and diaminobenzidine, as recommended by the supplier of the kit used (Vector, Burlingame, CA). Immunostaining for E-cadherin was performed using a combination of the ARK and CSA staining kits provided by Dako, according to the instructions of the manufacturer. The primary antibody was a mouse monoclonal anti–E-cadherin antibody (BD Biosciences, San Jose, CA) that was biotinylated using the animal research kit as recommended. Staining with components of the catalyzed signal amplification kit was performed as recommended, except that a biotin-blocking step (DakoCytomation, Carpintenia, CA) was added and the buffer used was a high-salt version (0.05 M Tris—pH 7.6, 0.3 M NaCl, 0.05% Tween 20) of the Tris buffer usually used.
Tumor classification was determined based on morphologic features of HE-stained sections. Immunohistochemical staining was also performed to verify the expected focally decreased expression of cytokeratin 10, E-cadherin, and desmoplakin and focally increased levels of cytokeratins 6 and 14 in SCC compared with papillomas. Tumors were evaluated independently by two veterinary pathologists and classified based on growth pattern (endophytic versus exophytic), depth of invasion, inflammatory infiltrate, dermal fibrosis, cellular and nuclear appearance, and immunohistochemical reactivity. Papillomas were exophytic masses that exhibited little cellular atypia and no invasion into the dermis. Papillomas evoked little dermal inflammation or fibrosis and showed strong immunostaining for cytokeratin 10 and cell adhesion molecules (Fig. 1). SCCs were composed of more atypical cells and showed clear evidence of dermal invasion. Thus, SCCs varied from well to moderately differentiated. Compared with well-differentiated SCC (Fig. 2), moderately differentiated SCC demonstrated a more endophytic growth pattern, greater depth of invasion, more intense inflammation and dermal fibrosis, more marked cellular and nuclear atypia, and less intense or extensive immunoreactivity for cytokeratin 10, E-cadherin, and desmoplakin (Fig. 3).
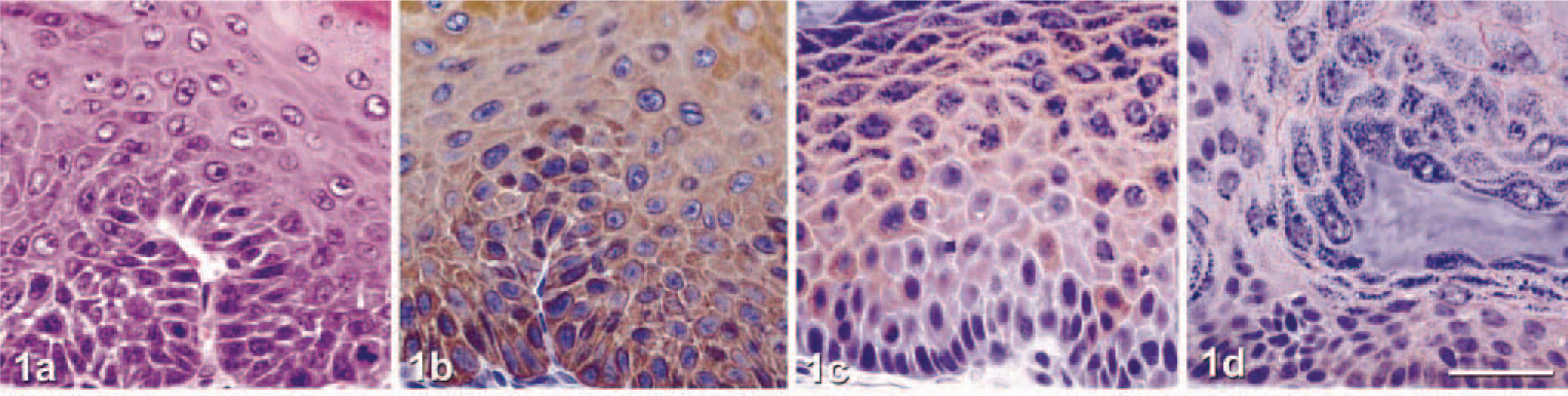
Chemically induced cutaneous papilloma. Wild-type SvEv/Black Swiss mouse.
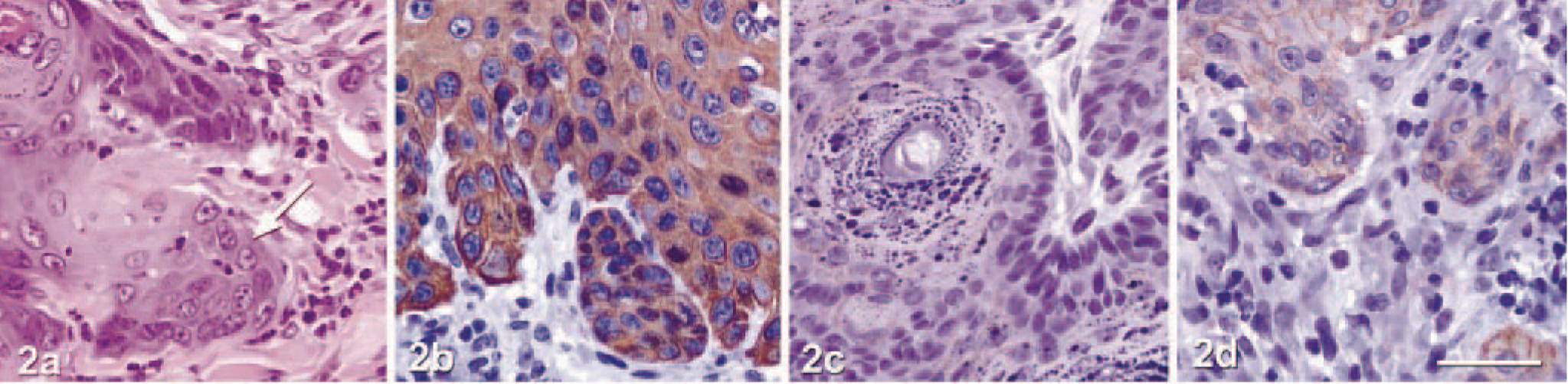
Chemically induced well-differentiated SCC. Wild-type SvEv/Black Swiss mouse.
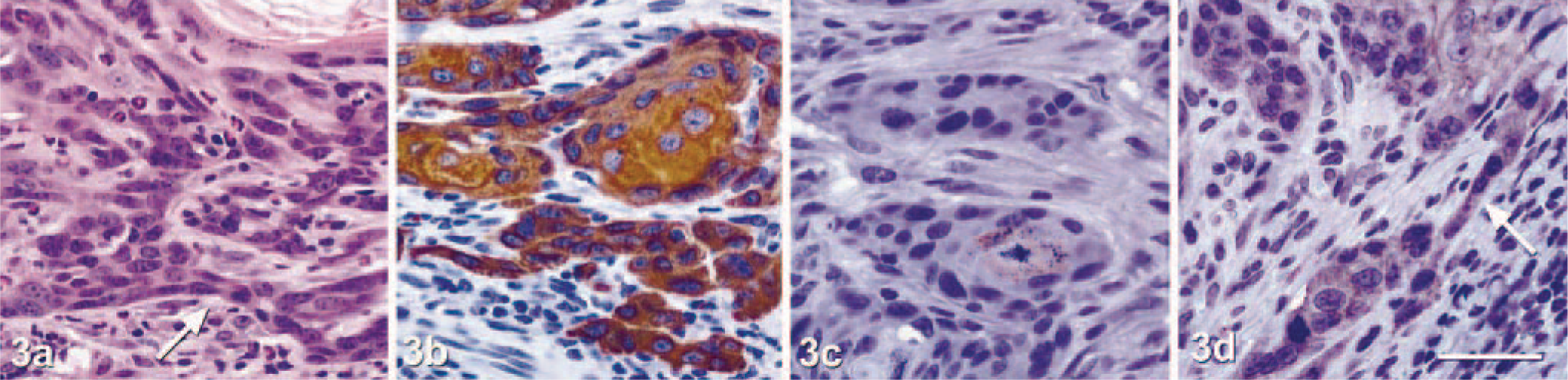
Chemically induced moderately differentiated SCC. Smad 2± SvEv/Black Swiss mouse.
Within the wild-type group, seven of 18 mice (38.9%) developed a total of nine tumors (one mouse had three tumors and six mice had one tumor each); three of the tumors were papillomas and six were SCCs (three well differentiated and three moderately differentiated). Within the Smad2 ± group, three of nine mice (33.3%) developed a total of five tumors (two mice developed two tumors each), all of which were moderately differentiated SCCs. Within the Smad3 ± group, one of 15 mice (6.7%) developed two papillomas. Tumor incidence (number of mice per group with tumors) was compared by Fisher exact test, both overall and by conducting pairwise comparisons of the groups.
Surprisingly, Smad3 ±mice were significantly less likely to develop tumors than control mice (6.7% versus 38.9% of mice with tumors, P < 0.05). Because heterozygous loss of the gene led to a marked decrease in tumor formation, it is possible that Smad3 functions as an oncogene in the normal mouse. Although the role of Smad3 in epithelial carcinogenesis has not been well established, Smads are generally thought of as tumor suppressors. 13 Indeed, Smad3 protein has been shown to be lost in chemically induced skin carcinomas. 8 In contrast, analysis of archived head and neck SCCs for Smad3 protein by immunohistochemistry demonstrated comparable nuclear staining in both normal and neoplastic cells. 10 Similarly contradictory results have been obtained in colon cancer studies. Although one laboratory reported high rates of metastatic colon cancer in Smad3-deficient mice, 20 others have reported no colon cancer at all in these mice. 18
The manner in which Smad3 functions as an oncogene in the skin remains to be determined, although there are numerous possibilities. Heterozygous loss of Smad3 facilitates wound healing by enhancing keratinocyte proliferation and protects against cutaneous injury by ionizing radiation through the disruption of immune functions and loss of fibrotic responses. 1,6 Smad3 is also known to control the expression of a number of dermal extracellular matrix (ECM) components, including matrix metalloproteinase I, 19 collagens 1A2, 3A1, 6A1, 6A3, 1A1, and 5A2, and TIMP-112. It may be that modulation of ECM components in Smad3 heterozygous mice leads to an environment less permissive to the development of cutaneous tumors. It is further possible that heterozygous loss of Smad3 is antioncogenic, whereas loss of heterozygosity of Smad3 enhances tumorigenesis. Such antipodal effects in gene dosage are not unknown in TGF-β signaling, and examples were discussed earlier regarding skin tumorigenesis.
The difference in tumor type between Smad2 ± and Smad3 ± mice was statistically significant (P < 0.05). Only two papillomas arose in Smad3 ± mice, whereas multiple moderately differentiated SCC occurred in Smad2 ± mice. It is interesting that there was a difference in roles between the two Smads in our model because there is 95% homology between Smad2 and Smad3, and they act through the same ligands and receptors. 11 We previously hypothesized that despite their similarities, Smad2 and Smad3 regulate different targets and that this leads to the phenotypic differences seen experimentally when the genes are disrupted. 15 This hypothesis is supported by the results of this study.
The skin tumor incidence in Smad2 ± mice (33.3%) did not differ significantly from the incidence in control mice (38.9%). Interestingly, Smad2 ± mice formed exclusively moderately differentiated SCC, whereas wild-type mice formed papillomas, well-differentiated SCCs, and moderately differentiated SCCs. This difference in distribution of tumor types was not statistically significant, suggesting that heterozygous Smad2 loss does not affect the development of skin cancers. Nevertheless, these results do not rule out a function of Smad2 as a tumor suppressor gene because the number of Smad2 ± mice available for the studies was low, and differences in tumor incidence might have emerged in studies using a larger cohort of experimental animals. In addition, potent modifiers of TGF-β function in different inbred mouse strains may affect tumor susceptibility. 2 For example, Smad3-deficient hybrid 129/Sv × C57BL/6 mice developed colon cancer more slowly than inbred mice, and tumors were smaller and less aggressive. 21 These results clearly demonstrated that strain-specific genetic modifying factors could influence experimental results in studies of Smad-deficient mice. Previously reported studies investigating the role of Smads and TGF-β in skin carcinogenesis used various inbred mouse strains, including ICR 8 and NIH/Olac strains. 3 In contrast, we used outbred mice (129SvEv crossed with NIH Black Swiss). Given the pivotal nature of background-specific modifiers, it is possible that an effect of Smad2 loss on tumor formation was overlooked, and it will be interesting to test the effects of strain background on tumor susceptibility in both Smad2 ± and Smad3 ± mice. It has been shown in other studies that Smad2 is a tumor suppressor gene because loss of Smad2 function has been reported in colorectal cancer 5 and head and neck SCCs in humans. 10 It is therefore reasonable to think that this might also be the case in the skin.
Footnotes
Acknowledgements
This study was funded by National Cancer Institute (CA 77911, T. J. Rosol; CA 83766, S. H. Tannehill-Gregg) and National Center for Research Resources (RR00168, T. J. Rosol) grants. We thank Kathy Hopwood and Christie Newland for animal support, Tim Vojt for technological support, Mary Ross for help in immunohistochemical studies, and members of the Weinstein and Rosol laboratories for critical review of the manuscript.