
Abstract
Hemochromatosis is rare in domestic mammals. Five clinical cases and one preclinical case of hemochromatosis were diagnosed in Salers and Salers-cross cattle. Clinical disease developed between 9 and 22 months of age. Animals were healthy until weaning but then lost weight, developed rough hair coats, and lost incisor teeth. In two animals, hemochromatosis was identified by liver biopsy, biochemical evidence of hepatic injury, and/or elevated transferrin saturation values. At necropsy, carcasses were thin, with firm dark brown livers and lymph nodes, soft bones, and brown-colored small bowel. The principal histologic changes were hepatocellular siderosis and periportal, bridging, and perivenular fibrosis. Siderocalcinosis involved collagen, elastin, reticulin, and basement membrane components in liver, lymph nodes, spleen, duodenum, and kidney. Hepatic iron concentrations in clinically affected cattle were 1,500–10,500 μg/g wet weight (reference range for cattle = <300 μg/g). Ultrastructurally, the heaviest intrahepatic deposition was in hepatocytes, which contained large intracytoplasmic siderosomes. Iron deposition in bone was associated with osteopenia. Genetic analysis indicated a common ancestral bull in the pedigrees of five of six affected cattle; no pedigree was available for the remaining animal. Four dams of five affected animals were phenotypically normal and had histologically normal livers. Test mating of four cows to the ancestral bull resulted in a female calf that developed clinicopathologic and histologic evidence of preclinical hemochromatosis by 40 days of age. It was not possible to establish the pattern of inheritance because of the small number of pedigrees from affected cattle.
The Salers breed of cattle is claimed to be one of the oldest in France, taking its name from the town of Salers in Aurillac. The original breed had lyre-shaped horns and characteristic red curly hair coats, although black polled Salers became popular recently. Salers are acclimated to hilly terrain, wide temperature ranges, long winters, and a winter hay diet. Salers were developed as draught oxen and for the production of milk, cheese, and meat. The breed was introduced to North America in 1975. It was promoted as suited to the harsh climate of western states and provinces. The breed's narrow genetic base in North America probably contributed to the emergence in the 1980s and early 1990s of β-mannosidosis, which is transmitted as a homozygous recessive trait. 12,53,60
In 1994, a second familial disease was reported in the breed in the United States. 44 Hemochromatosis was identified in three related cattle in California and Idaho. Affected cattle developed hepatic failure with clinical, chemical, and histologic evidence of iron overload. The authors of the report suggested that a breeding trial was necessary to confirm a suspicion that the disease was inherited. As far as we are aware, there have been no subsequent reports of hemochromatosis in the Salers breed. Hemochromatosis has not been confirmed in other breeds of cattle. A report from New Zealand documented subclinical disease in Aberdeen Angus cattle that was detected at slaughter and was characterized by hepatic siderosis, perilobular hepatic fibrosis, and osteoporosis. 41
Two forms of iron overload are recognized: hemosiderosis and hemochromatosis. Hemosiderosis (secondary iron overload) occurs under various clinical conditions and is characterized by accumulation of iron in the reticuloendothelial system and little or no tissue injury. Conditions and states associated with hemosiderosis include chronic hemolytic disorders, repeated transfusions, changes from natural diet, and metabolic crises such as emaciation. 50,61,75 Hemochromatosis occurs when inappropriately large amounts of iron are absorbed from the gut over an extended period. Unlike hemosiderosis, hemochromatosis is associated with high transferrin saturation values in serum (>60%). 43 Excess iron is stored in parenchymal tissues, particularly liver, and leads eventually to organ failure. Although considered rare when von Recklinghausen first coined the term hemochromatosis in 1889, hereditary hemochromatosis (HH) is now recognized as one of the most common inherited diseases of humans, particularly those of Celtic stock. 2,3,7,72 Two missense mutations of HFE, a protein that complexes with the transferrin receptor, were identified recently in HH patients and are the presumed basis for the disease. 11,28,31,32 Both mutations result in increased absorption and storage of iron by parenchymal cells. There is no published information on whether an HFE-like protein exists in cattle. Few natural or experimental animal models of hemochromatosis exist. Attempts to reproduce hemochromatosis by dietary loading with iron are generally unsuccessful. 45 Forms of iron overload with features of both hemosiderosis and hemochromatosis also exist. 6,54,61
We recently identified five cases of iron overload in Salers and Salers-cross cattle. Here, we supplement original observations of the disease and emphasize lesions in affected cattle at various clinical stages. The disease was reproduced in a sixth calf by mating a presumed female carrier to a founding sire of the American Salers breed.
Materials and Methods
Affected calves, source herds, and experimental breeding
Hemochromatosis was identified in five cattle (calf Nos. 1–5) in three herds (I–III) in the western United States in 1996–1999 (Tables 1, 2). A sixth animal (calf No. 6) was born at the Wyoming State Veterinary Laboratory (WSVL) in 2000 after the dam of an affected calf was bred to the suspected carrier bull (Bull, Fig. 1). The bull was born in France in 1974 and was imported to the United States in 1975. He was in the pedigrees of four of the five affected calves (Nos. 1–4, 6) and of three previously reported affected cattle. 44 No pedigree was forthcoming from the owner of the remaining calf (No. 5).
Clinical and clinicopathologic findings in six female Salers and Salers-cross calves with hemochromatosis.
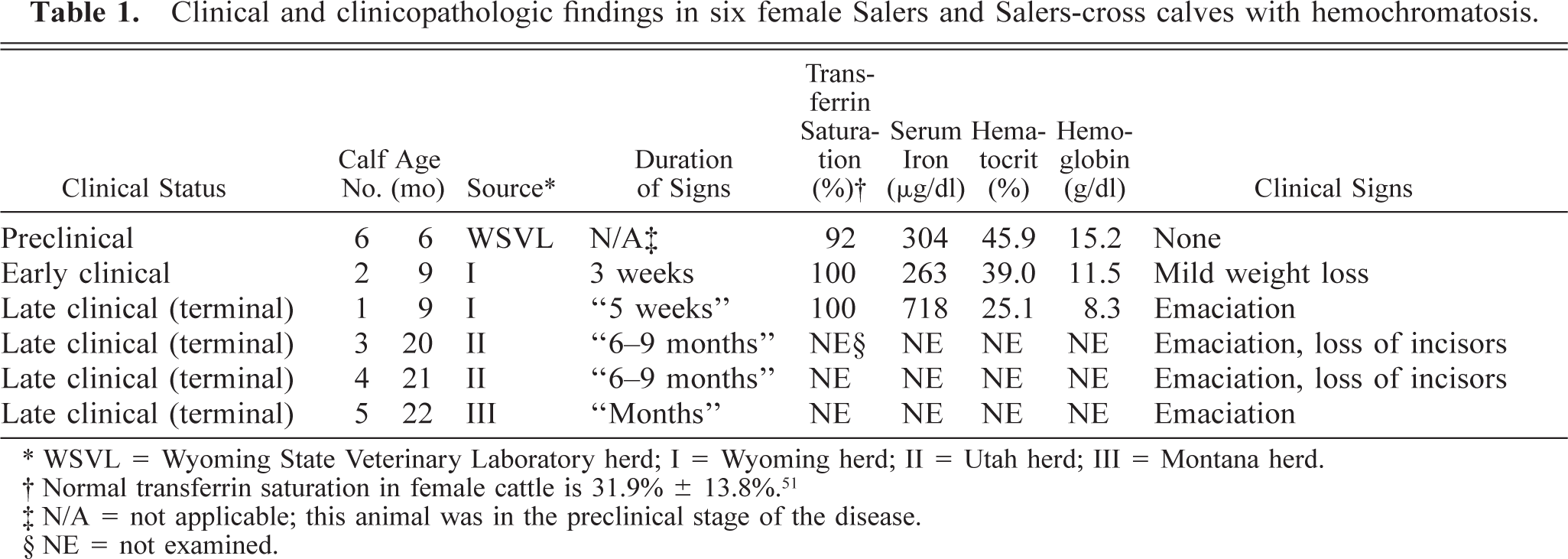
∗ WSVL = Wyoming State Veterinary Laboratory herd; I = Wyoming herd; II = Utah herd; III = Montana herd.
† Normal transferrin saturation in female cattle is 31.9% ± 13.8%. 51
‡ N/A = not applicable; this animal was in the preclinical stage of the disease.
§ NE = not examined.
Lesions in female Salers and Salers-cross calves with hemochromatosis and in their carrier dams.
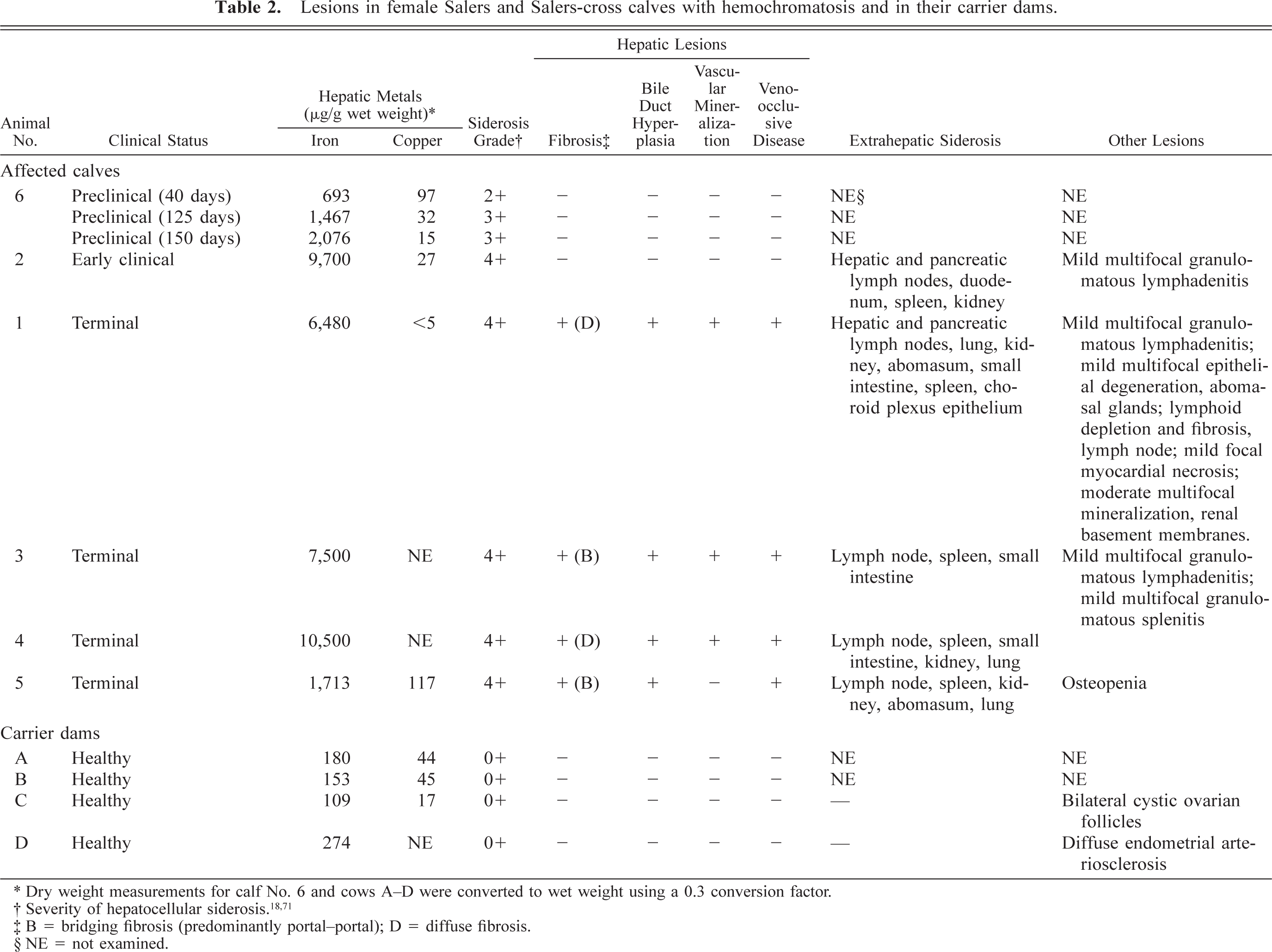
∗ Dry weight measurements for calf No. 6 and cows A–D were converted to wet weight using a 0.3 conversion factor.
‡ B = bridging fibrosis (predominantly portal–portal); D = diffuse fibrosis.
§ NE = not examined.
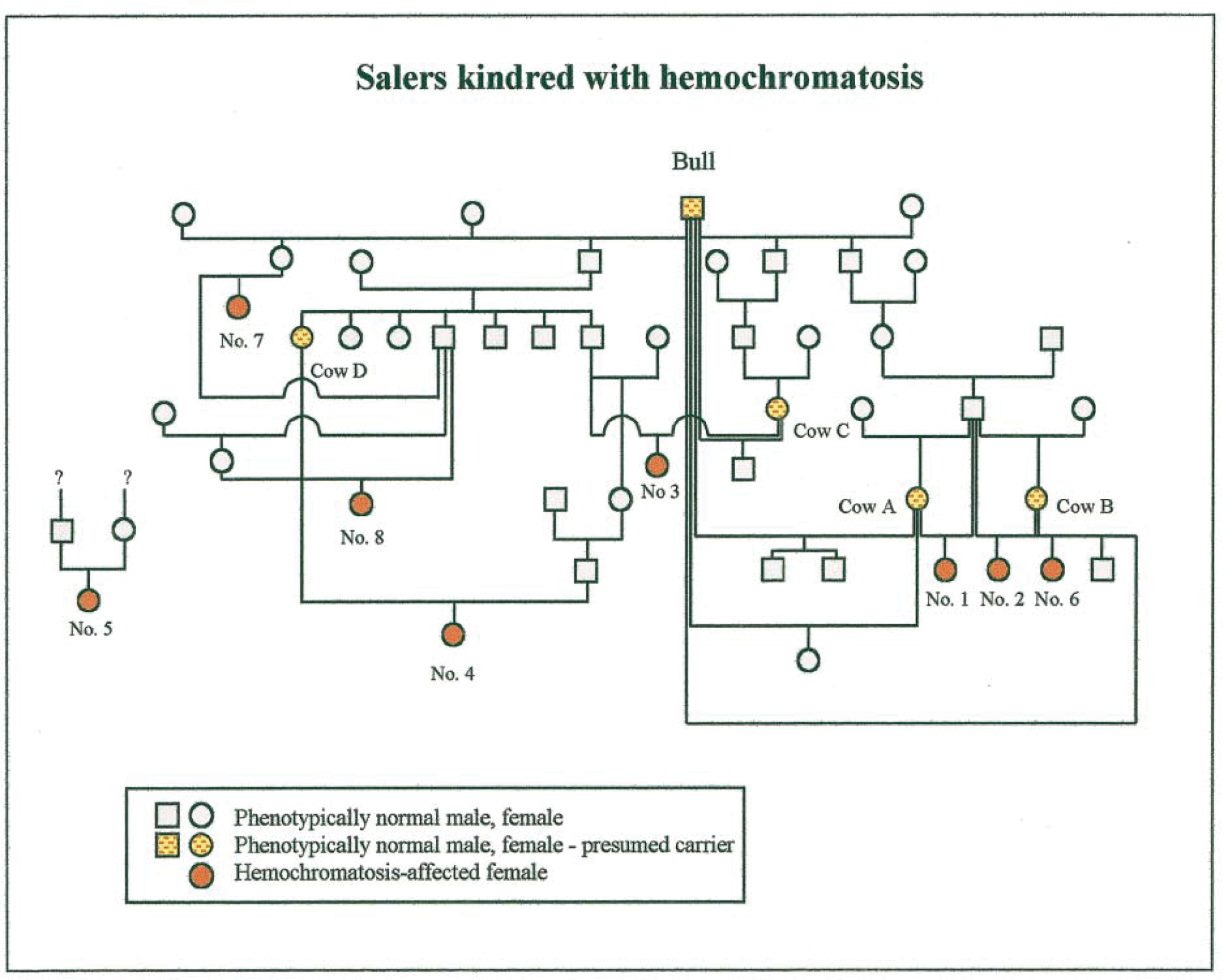
Pedigree of eight Salers and Salers-cross cattle with hemochromatosis. Calf Nos. 1–6 are described here. Animal Nos. 7 and 8 were described previously. 44 No pedigree was available for calf No. 5 or for one of three affected cattle previously reported. 44 In 1998, cows A–D were transferred to the WSVL and bred using semen from the suspect carrier bull (Bull). Cow B produced two calves with hemochromatosis: one in 1996 (calf No. 2) and another in 2000 (calf No. 6).
Herd I was a commercial cow–calf operation in Wyoming. In October 1997, the herd comprised 59 cows, 2 bulls, 3 yearling steers, 1 yearling heifer, and 56 calves. The sire of both affected calves (Nos. 1, 2) was a purebred Salers bull born in 1987. He was introduced into the herd in 1988. The bull was a great-grandson of the Salers sire represented in the earlier pedigree. 44 Animals in the herd were predominantly Salers on a mixed genetic background. The bull was used until 1997, at which time he was sold for slaughter. A line breeding program (father–daughter mating) was used extensively to increase the proportion of Salers genes in the herd. In 1997, a yearling Salers-cross steer lost weight. The animal was segregated from other animals and offered various diets. He lost all incisor teeth and was culled in a state of terminal emaciation. The carcass was not submitted for examination. Subsequently, another Salers-cross calf (No. 1, Fig. 1; Table 1) developed similar clinical signs. This animal, a heifer, was submitted for necropsy examination at 9 months of age. The dam of calf No. 1 was designated cow A (Fig. 1). Following identification of hemochromatosis in the calf, blood samples were obtained from 111 animals in a search for additional cases in the herd. A second affected Salers-cross heifer was thereby identified (calf No. 2). Her dam was designated cow B. Calf No. 2 was submitted for necropsy at 9 months of age.
Herd II was a well-managed purebred Salers breeding herd in Utah. The herd consisted of approximately 30 registered cows, their calves, and several bulls. Two purebred Salers heifers (calf Nos. 3, 4) were born in 1996 to cows (designated C, D, respectively). The heifers developed normally until after weaning at 8 months of age. Over the next 6–9 months, they failed to gain weight, had rough hair coats, and lost all incisor teeth. The dam of calf No. 3 and the sire of calf No. 4 were embryo transfer siblings of the sire of two cattle with hemochromatosis reported previously (animal Nos. 7, 8; Fig. 1). 44 Dietary changes did not arrest progressive weight loss. The emaciated heifers were submitted alive to a slaughterhouse. The owner specifically requested that both carcasses be examined for hemochromatosis.
Herd III was a commercial Salers–Angus herd in Montana. A 22-month-old pregnant Salers-Angus heifer developed signs of weight loss and was presented for necropsy (calf No. 5). A heifer of similar age developed similar signs and died some weeks earlier, but that carcass was not submitted for examination. Pedigree records were not available.
The dams of calf Nos. 1–4 were purchased in 1998 and transferred to the WSVL to determine the mode of inheritance. Cows A–D were test mated using semen from the suspect carrier bull over a 2-year period (1998–2000). Four calves were born alive, and a set of twin calves was stillborn. One of four live-born calves developed hemochromatosis (calf No. 6).
Clinical examination and clinical pathology
Clinical examination of four of six affected animals was minimal because they were presented alive for necropsy or slaughter (calf Nos. 1, 3–5). Serum iron concentration and total iron binding capacity (TIBC) in calf Nos. 1, 2, and 6 were measured using a commercial colorimetric assay (Stanbio Laboratory, San Antonio, TX). Percentage transferrin saturation was calculated using the formula 100 × (serum iron ÷ TIBC). A serum chemistry panel was used to detect evidence of hepatic failure in the same three animals. The panel included total serum protein, serum albumin and globulin, alkaline phosphatase (ALP), alanine aminotransferase (ALT), aspartate aminotransferase (AST), gamma glutamyltransferase, ammonia, and total bilirubin. Calf No. 2 was identified as affected when 8 months old as a result of screening most animals in herd I. Testing was repeated on calf No. 2 on two occasions, when she was 8 and 9 months old. The transferrin saturation of calf No. 6 was tested once or twice each month from birth. Cows A–D were tested intermittently for serum iron and transferrin saturation. Reference ranges for serum iron and transferrin saturation were established using sera from 40 clinically healthy 6-month-old beef calves in the University of Wyoming's herd on summer range. Routine hemograms were performed on calf Nos. 1, 2, and 6 and on cows A–D on one or more occasions.
Pathology
Necropsy examinations were performed on calf Nos. 1–5 following euthanasia or slaughter. Samples of liver of calf No. 6 were collected when she was asymptomatic at 40, 125, and 155 days of age by transcutaneous biopsy through the 10th right intercostal space using a bone marrow punch. Samples were fixed by immersion in 10% buffered neutral formalin, dehydrated, and embedded in paraffin wax. Tissue blocks were sectioned on a microtome, and 5-µm sections were stained with hematoxylin and eosin (HE). Sections of selected tissues were stained to detect iron (Perls' Prussian blue reaction), copper (rubeanic acid stain), and calcium (von Kossa stain), in addition to stains for demonstrating collagen, elastin, and reticulin fibers. 8,77 The extent of siderocalcinosis was assessed by examining adjacent sections from selected tissue blocks following staining for iron and calcium. The severity of hepatocellular siderosis was classified using Perls' stained sections and a 0+ to 4+ grading system 18,71 : 0 = no detectable iron in hepatocytes; 1+ = fine granules in 5–10% of hepatocytes; 2+ = fine granules in 10–50% of hepatocytes; 3+ = fine and large granules in >50% of hepatocytes; 4+ = fine, large, and aggregated granules in virtually all hepatocytes. Published criteria were used to assess the presence and severity of hepatic fibrosis, micronodular cirrhosis, and veno-occlusive disease. 52,57
Samples of formalin-fixed bone from two animals (lumbar vertebra, calf No. 2; rib and skull, calf No. 5) were decalcified and examined by light microscopy. Undecalcified samples from calf No. 5 were ground to a thickness of 100 µm and examined radiographically and with toluidine blue staining. Additional samples of bone were embedded in methylmethacrylate, sectioned at 8 µm, and examined with a light microscope. Samples of rib and skull from a clinically healthy 18-month-old feedlot steer were used as controls.
Samples of liver from three animals were fixed in 4% buffered neutral formaldehyde (calf No. 1) or paraformaldehyde–glutaraldehyde solution (calf Nos. 2, 6), processed, and embedded in epoxy resin. 58 Samples from calf No. 6 were collected by transcutaneous biopsy when she was 150 days old, and samples from calf Nos. 1 and 2 were collected at necropsy. Samples of hepatic lymph node, pancreas, and kidney were collected from calf No. 2. Thick (1µm) sections were stained with methylene blue and examined with a light microscope. Thin sections (approximately 70–90 nm) were stained and examined using an electron microscope.
Hepatic biopsies were obtained from cows A–D and from four calves born to them as the result of test mating and were processed for light microscopic examination using Perls' stain for iron. Following slaughter, samples of the following tissues from cows C and D were sampled for histology: liver, hepatic and mesenteric lymph nodes, gallbladder, duodenum with common bile duct, lung, kidney, heart, ovaries, uterus, and pancreas. Stillborn twin calves born in 1999 to a mating of cow A with the suspect carrier bull were examined postmortem using a standard histologic, microbiologic, and toxicologic diagnostic protocol to establish the cause of perinatal death. 49
Chemical analysis
Samples of liver were collected from calf Nos. 1–5, from cows A–D, and from six calves born to cows A–D following test mating at the WSVL (including calf No. 6 and two stillborn calves). Hepatic concentrations of iron and copper were determined using inductively coupled plasma atomic emission spectroscopy and (after January 2000) inductively coupled plasma atomic mass spectroscopy. Results were reported either as received wet weight samples (liver samples collected at necropsy; calf Nos. 1–5) or on a dry matter basis (liver samples collected by biopsy; calf No. 6 and cows A–D). Concentrations of iron in dried samples was converted to wet weight concentrations by assuming a conversion factor of 0.3. 44 No analysis was performed on the iron content of the diets in herds II or III. A mineral mix offered to cattle in herd I in June 1997 was analyzed for iron and copper. The owner of herd I made available a chemical analysis that was performed of first-cutting grass hay fed to cows and calves in November 1996, which was after calf Nos. 1 and 2 were born.
Pedigree analysis and inbreeding coefficients
Extended pedigrees were obtained and evaluated for five of the six affected calves (Nos. 1–4, 6). Inbreeding coefficients were calculated using the inbreeding procedure of SAS v. 6.12 (SAS Institute, Cary, NC).
Hybridization of zoo blots with human HFE probes
To investigate whether sequences orthologous to the human HFE gene are conserved in evolution and occur in cattle, a radiolabeled probe fragment of the human HFE gene was hybridized to a zoo blot, which is a Southern blot membrane containing genomic DNA from various species. Biologically important sequences, such as coding regions of genes, are under selective pressure; most mutations that change a protein sequence tend to reduce the ability of an organism to survive and reproduce. In contrast, noncoding DNA sequences accumulate mutations relatively rapidly in evolutionary terms and so are not well conserved among species.
Genomic DNA (10 µg each of bovine, porcine, canine, murine, Chinese hamster, and control human DNA) (Clontech, Cambridge, UK) was digested with HindIII overnight at 37 C according to the supplier's instructions (New England Biolabs U.K., Hitchin, UK). The digested DNA was concentrated by ethanol precipitation and resolved by electrophoresis on a 1% (w/v) agarose gel. 69 The DNA was blotted to Hybond-N+ membrane (Amersham Pharmacia Biotech U.K., Little Chalfont, UK) by alkaline capillary transfer. A human DNA probe containing HFE exons 2 and 3 was amplified from human genomic DNA using cDNA primers and served as a positive control. 32 A 1-kb DNA size marker served as a negative control. The membrane was hybridized with the 32P-radiolabeled HFE probe, washed to a final concentration of 0.1× saline sodium citrate, 0.1%(w/v) sodium dodecyl sulfate at 60 C, and exposed to X-ray film. 69
Results
Clinical signs and clinical pathology
Mean (± SD) reference values for serum iron and transferrin saturation for healthy cattle in the university's reference herd were 144 ± 36.01 µg/dl and 35% ± 7.76% (range: 50–217 µg/dl and 20–55%; n = 40 animals).
Clinical signs and clinicopathologic findings are summarized in Table 1. Affected cattle were 9 (calf Nos. 1, 2), 20 (calf No. 3), 21 (calf No. 4), and 22 (calf No. 5) months old when presented for necropsy. All were thin, alert, and ambulatory, with rough hair coats. None of the owners reported lameness or diarrhea as a symptom. Calf No. 1 lost considerable weight in the 5 weeks prior to euthanasia. Calf Nos. 3 and 4 never had an estrous cycle.
Calf No. 2 had the highest serum iron values and transferrin saturation concentration (263 µg/dl and 100%, respectively) of 111 animals in herd I. Mean serum iron and transferrin saturation values in herd I (excluding calf No. 2) were 135 µg/dl and 38% (range: 77–195 µg/dl and 23–71%; n = 110 animals). At that time, calf No. 2 was 8 months old and clinically normal. Shortly thereafter, she lost weight. Serum samples subsequently taken from calf No. 2 on two occasions confirmed that high transferrin saturation values persisted (>90%). The calf's hemogram was unremarkable (11.5 g/ml hemoglobin; 39% hematocrit). The calf lost condition for 3 weeks prior to euthanasia, which was performed to establish whether she was in the early clinical stage of hemochromatosis. Up until euthanasia, there was no biochemical evidence of hepatic failure. Calf No. 1 had clinical evidence of hepatic failure at the time of death: a low albumin/globulin ratio (0.5; reference: 0.6–0.9), elevated total bilirubin (2.17 mg/dl; reference: 0.0–1.6 mg/dl), and elevated serum enzyme activities (ALP = 653 U/liter; ALT = 33 U/liter; AST = 118 U/liter; reference: ALP = 10–149 U/liter; ALT 11–40 U/liter; AST: 0–91 U/liter). 29 The transferrin saturation of calf No. 6 rose during the first 6 months as follows (age in parenthesis): 68.3% (55 days), 95.5% (70 days), 87.4% (92 days), and 92.9% (148 days).
Experimental breeding and pedigree analysis
Cows were test bred by artificial insemination using semen of the suspect carrier bull over two breeding seasons (1998–2000). At the end of the first year, cows A, B, and C gave birth to full-term calves. Cow A delivered twin full-term stillborn male calves; neither had clinical or histologic evidence of hemochromatosis. Infectious viral and bacterial agents were absent. Hepatic iron concentrations were in the normal range (105 and 175 µg/g, respectively; reference range for fetal bovine liver: 40–400 µg/g). 64 The presumed cause of death of the twins was dystocia, as indicated by abundant intrapulmonary placental debris in one calf. Bull calves born to cows B and C did not exhibit clinical or histologic signs of hemochromatosis during the first 18 months of life. Serum iron concentrations and ferritin saturation never exceeded 210 µg/dl iron and 45% saturation. Hepatic biopsies taken at 18 months of age revealed normal hepatic tissues, and no lesions were found subsequently in any tissue when both animals were slaughtered for human consumption. Cow D was culled because of infertility. Suspect carrier cows A–C were rebred to the suspect carrier bull. Cow A gave birth to a healthy male calf in 2000. He developed normally and did not show clinical, morphologic, or biochemical evidence of hemochromatosis. Cow B, a Salers–Hereford–Galloway cross, delivered a full-term female calf (No. 6, Fig. 1). Chemical and morphologic signs of hemochromatosis were present in this calf at 40 days of age. Dam C was culled because of infertility.
Evaluation of the extended pedigrees of five affected animals showed that although some had multiple relatives on both sides of the pedigree, in all cases line breeding to the suspected carrier bull was apparent. Inbreeding coefficients for individual animals ranged from 3.37% to 25.06%. Although high inbreeding levels increase the probability that a recessive gene passes through both the sire and dam of an animal, low inbreeding coefficients do not preclude that possibility. Based on this analysis, it was not possible to establish with certainty that hemochromatosis was inherited as an autosomal recessive trait.
Pathology
Gross and light microscopic lesions
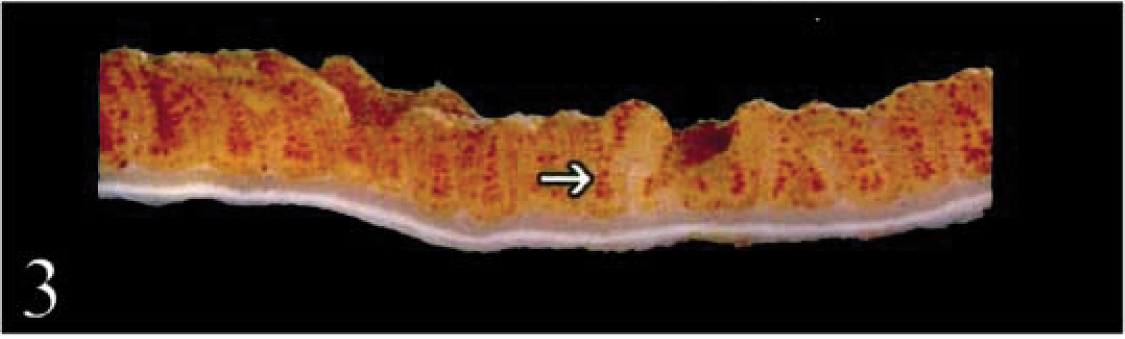
Representative gross and microscopic lesions in liver and other tissues are illustrated in Figs. 2–13 (Table 2). Findings at necropsy were similar. Terminally ill animals were thin, with muscular atrophy and little body fat. Two carcasses (calf Nos. 3, 4; Fig. 1) were jaundiced. Livers were dark brown in five animals (calf Nos. 1–5) and firm in three (calf Nos. 1, 3, 4). The liver of calf No. 1 had arborizing white fibrous bands throughout. The mucosa of a 1-m segment of proximal duodenum, including the area surrounding the intramural bile duct, was brown (Fig. 2). On cut section, the duodenal mucosa contained <1-mm dark brown foci (Fig. 3). Hepatic and pancreatic lymph nodes were large with brown discoloration, particularly in the medullary areas (Fig. 4). Skull bones of calf No. 5 were soft, gray, and pliable.
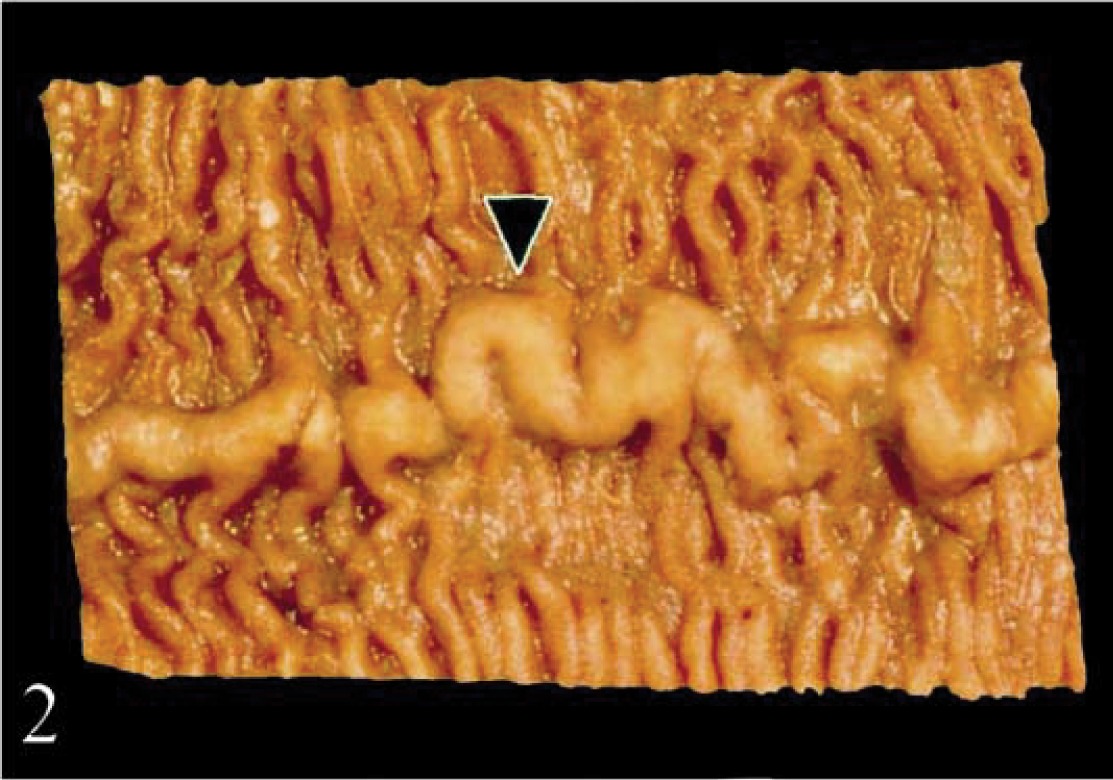
Duodenum; calf No. 2. The mucosa is discolored brown. The raised ridge (arrowhead) corresponds histologically to gut-associated lymphoid tissue.
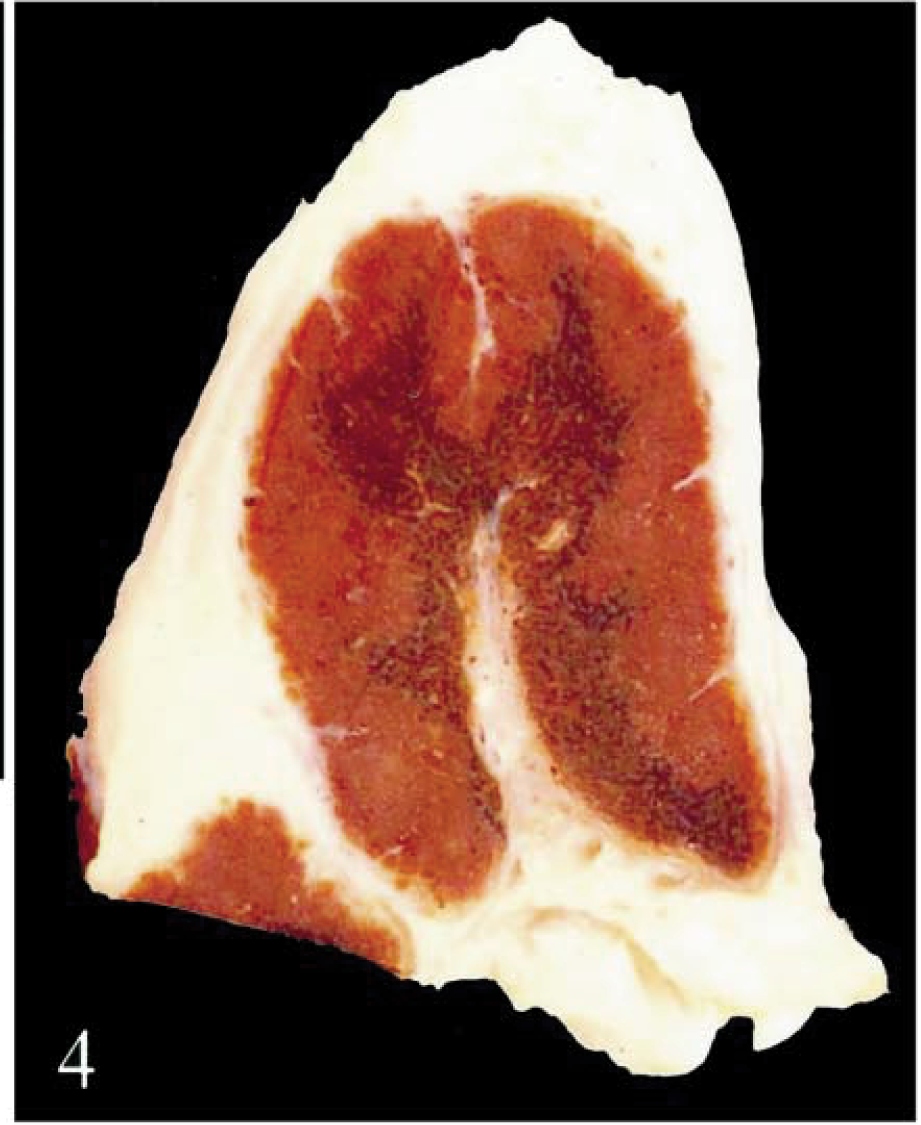
Hepatic lymph node; calf No. 2. Dark brown discoloration is evident in the medulla.
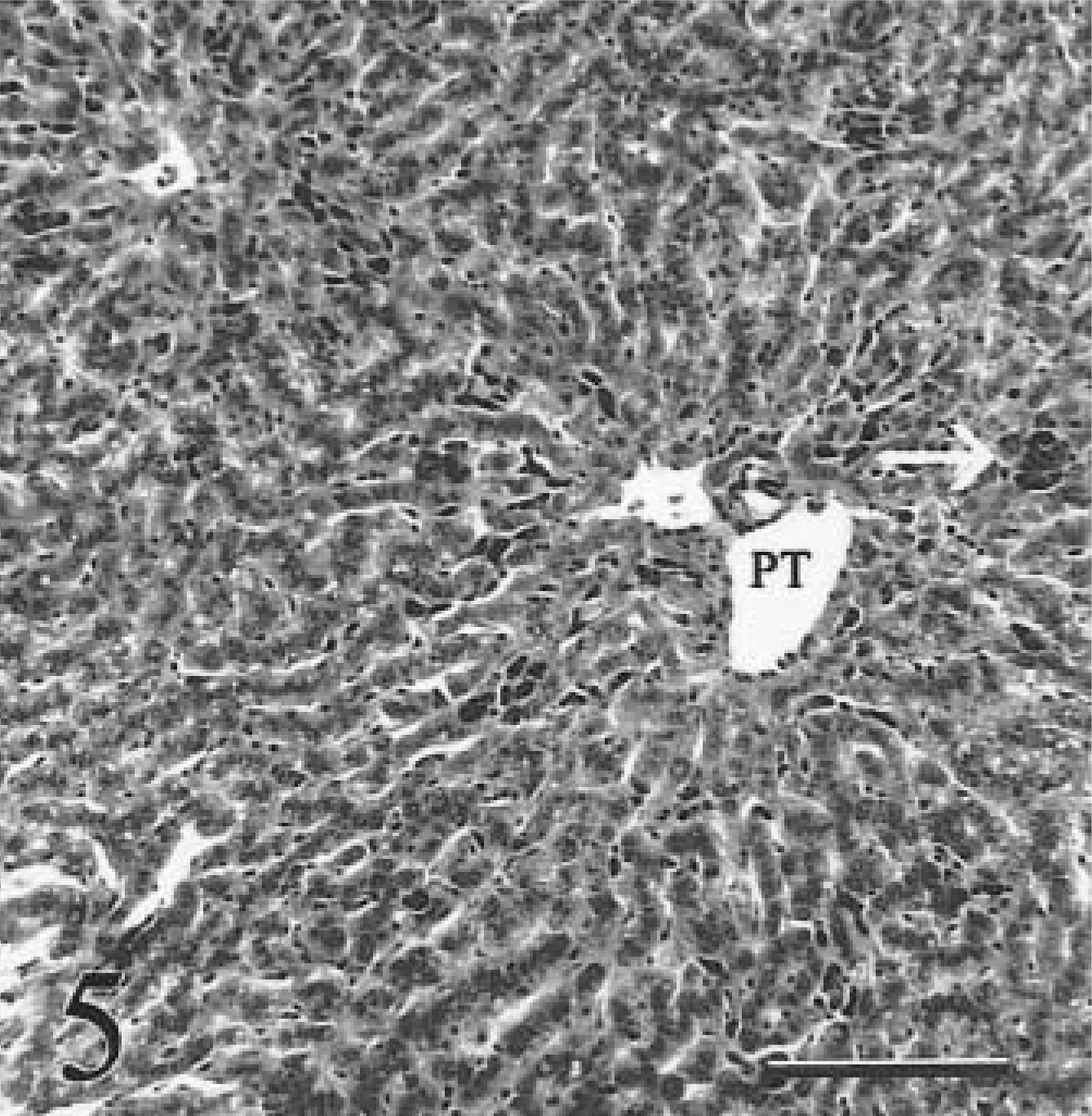
Liver; calf No. 2. Iron is present in hepatocytes and in aggregates of Kupffer cells (arrow) adjacent to portal triad (PT). There is no detectable fibrosis in this calf with early clinical hemochromatosis. HE. Bar = 100 µm.
Liver; calf No. 5. Terminally affected heifer with moderate bridging fibrosis extending from portal areas (arrows). Mild lymphocytic-histiocytic portal hepatitis is present. There is a zonal gradient of iron accumulation, with large granular deposits of iron evident in periportal macrophages and hepatocytes. CV = central vein. HE. Bar = 100 µm.
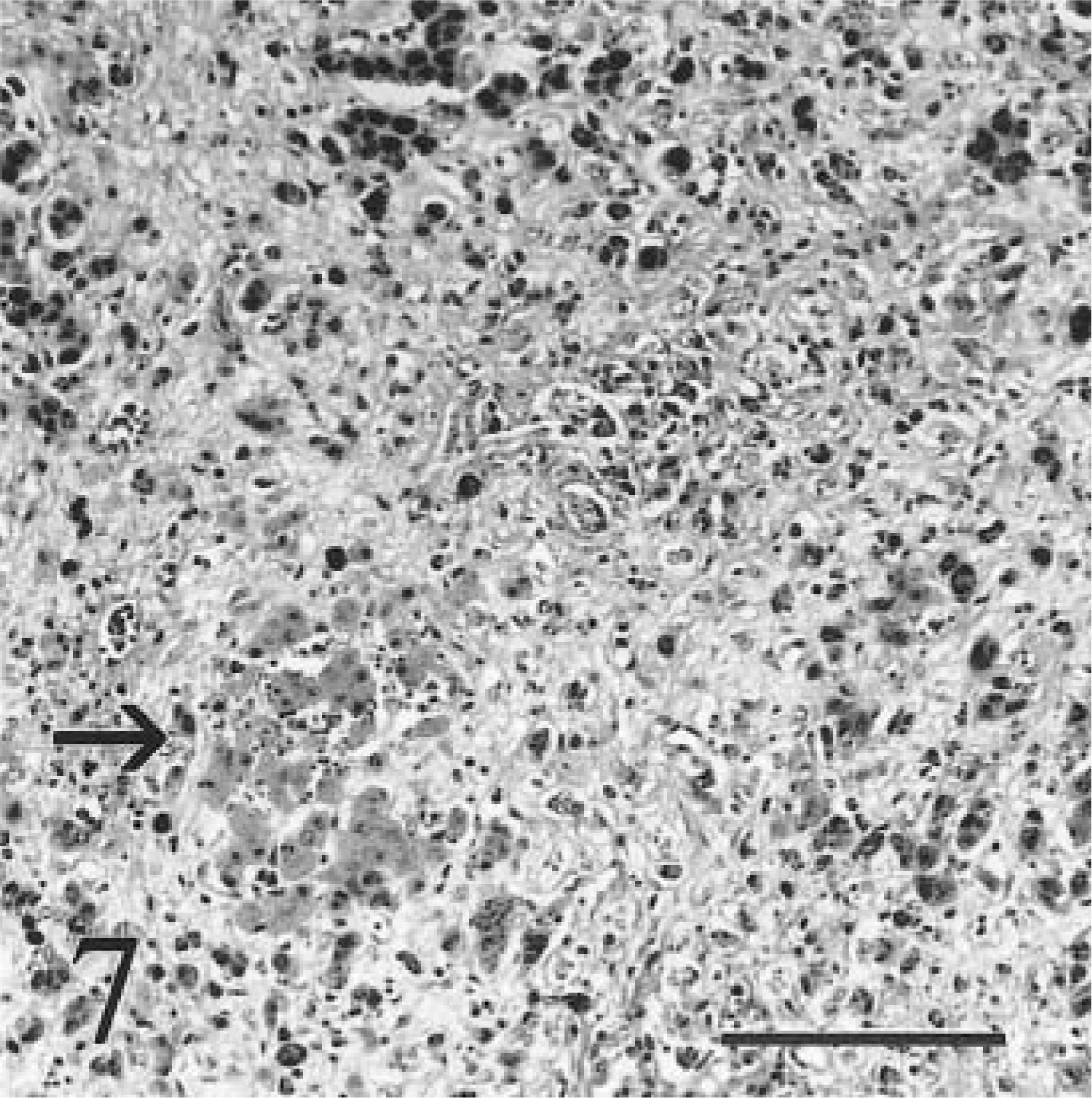
Liver; calf No. 5. Terminally affected heifer with severe diffuse fibrosis, loss of hepatocytes, and loss of zonal organization. A small aggregate of hepatocytes is more lightly stained because of less intracytoplasmic iron (arrow); such aggregates may represent hyperplasia. HE. Bar = 100 µm.
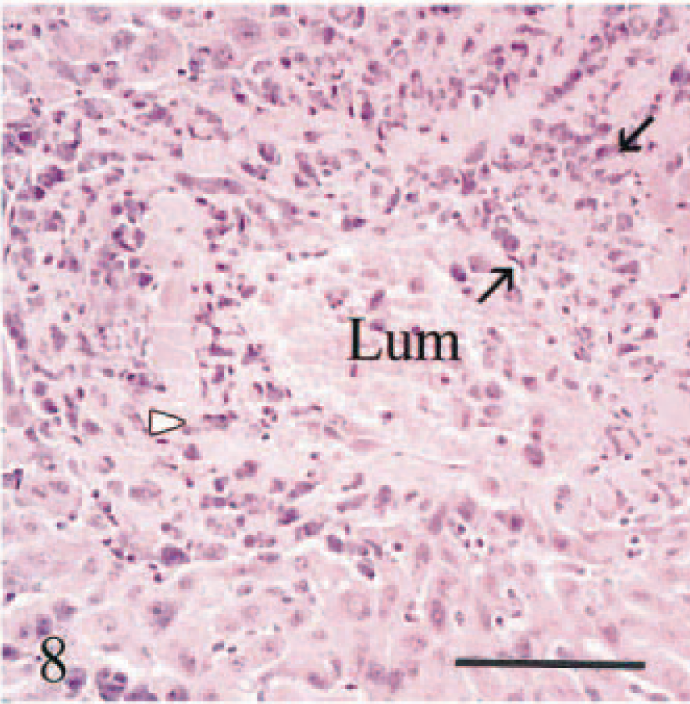
Liver; calf No. 5. Eccentric intimal fibrosis (between two arrows) in a sublobular vein, resulting in reduced vascular diameter (veno-occlusive disease). Iron-laden macrophages (arrowhead) are in thickened intima. Lum = Vascular lumen. HE. Bar = 50 µm.
Duodenum; calf No. 3. Hemosiderin-laden macrophages accumulate in lamina propria at the tip of a villus sectioned horizontally. Hemosiderin is present in a line of cells directly beneath epithelial membrane (arrows). No discernable iron is present in villous enterocytes. Perls' stain. Bar = 25 µm.
Duodenum; calf No. 4. Diffuse intracytoplasmic positive staining for iron in cryptal duodenal enterocytes. Small granules of hemosiderin are in apical cytoplasm (arrow). Perls' stain. Bar = 25 µm.
Duodenum; calf No. 1. Iron deposition involving elastic lamina, endothelium, and perivascular macrophages of small artery in duodenal lamina propria. Perls' stain. Bar = 25 µm.
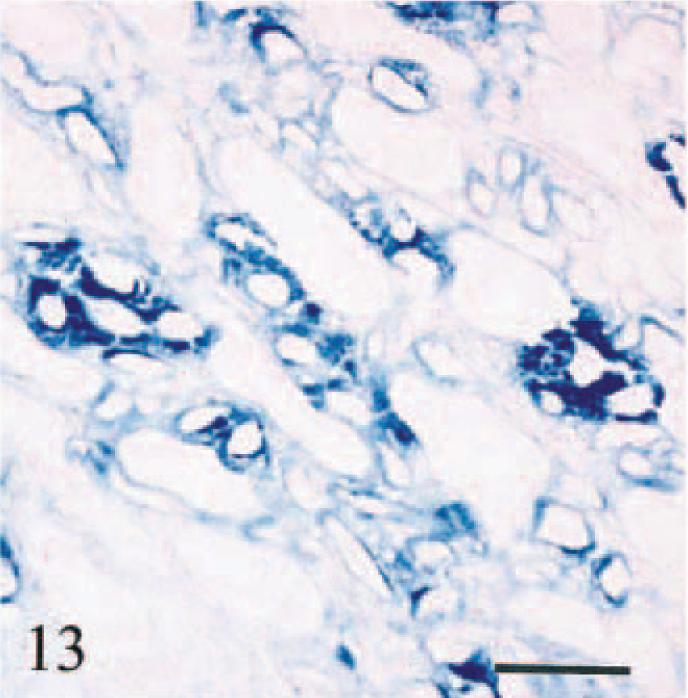
Kidney; calf No. 1. Iron deposition on vascular and tubular basement membranes and, to a lesser degree, interstitium of renal medulla adjacent to renal calyx. Perls' stain. Bar = 100 µm.
Calf Nos. 1–6 had histologic evidence of excessive iron accumulation in hepatocytes. Three progressive stages of iron overload were evident histologically: a preclinical stage (calf No. 6), an early clinical stage (calf No. 2), and a terminal stage (calf Nos. 1, 3–5) (Table 1).
In the preclinical stages of the disease, there was grade 2+ iron accumulation in hepatocytes at 40 days of age, progressing to grade 3+ at 125 and 155 days of age. By contrast, normal cattle have little or no iron in hepatocytes (data not shown). Iron in hepatocytes at 40 days affected primarily centrilobular and midzonal areas (acinar zones 2 and 3). Detectable iron occurred as diffuse, nongranular cytoplasmic staining (blush reactivity) of ferritin–iron complexes 11 and as granules (hemosiderin). Iron granules were concentrated around bile canaliculi. By 125 and 155 days, all hepatic zones including periportal areas (acinar zone 1) were affected equally. There was no evidence of hepatic fibrosis, iron deposition in Kupffer cells, or mineralization of hepatic vessels.
During the early clinical stage of the disease, there was grade 4+ iron accumulation in hepatocytes. All acinar zones were affected equally. Fibrosis was absent (Fig. 5). Kupffer cells contained large amounts of hemosiderin and formed hyperplastic foci. Modest numbers of iron-laden portal macrophages were present.
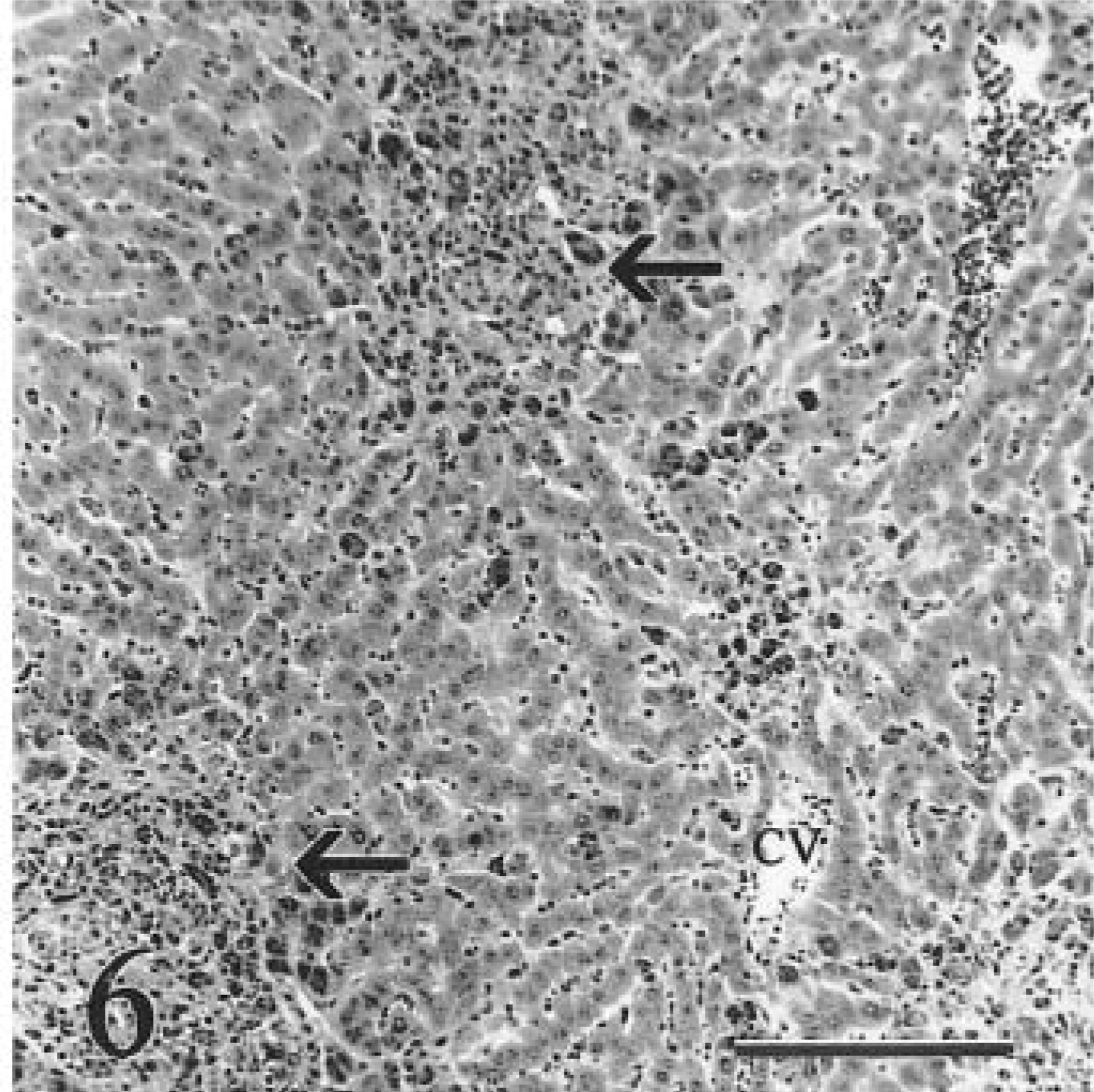
In the advanced stage of the disease, terminally ill animals had moderate to severe diffuse hepatic fibrosis and 4+ iron accumulation in hepatocytes. A zonal gradient was evident in one of four terminally affected animals, with the highest concentration of hemosiderin in periportal areas (calf No. 5, Fig. 6). Heavy accumulation of hemosiderin in Kupffer cells and portal macrophages resulted in the formation of siderotic nodules. There was marked reduction of hepatocellular mass and disorganization of remaining hepatic plates. Sparse foci of hepatocytes that were larger and contained less hemosiderin than hepatocytes in surrounding parenchyma were interpreted as hyperplastic; 52 however, discreet hyperplastic nodules were absent (Fig. 7). Moderate to marked periportal fibrosis resulted in portal-based bridging and centrilobular fibrosis. Moderate bile duct hyperplasia was accompanied by mild lymphocytic-histiocytic portal hepatitis. There was subendothelial fibroplasia and partial to complete vascular occlusion in sublobular and central veins (Fig. 8). Some occluded veins were recanalized. Bile duct epithelium was negative for iron. There was heavy encrustation by iron of the inner and outer elastic lamina of arteries. No copper was detectable histologically in calf Nos. 1–6.
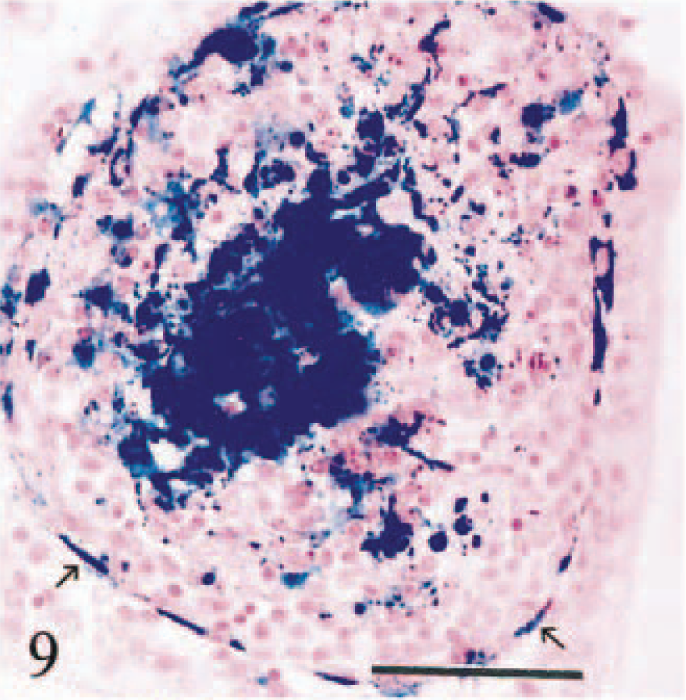
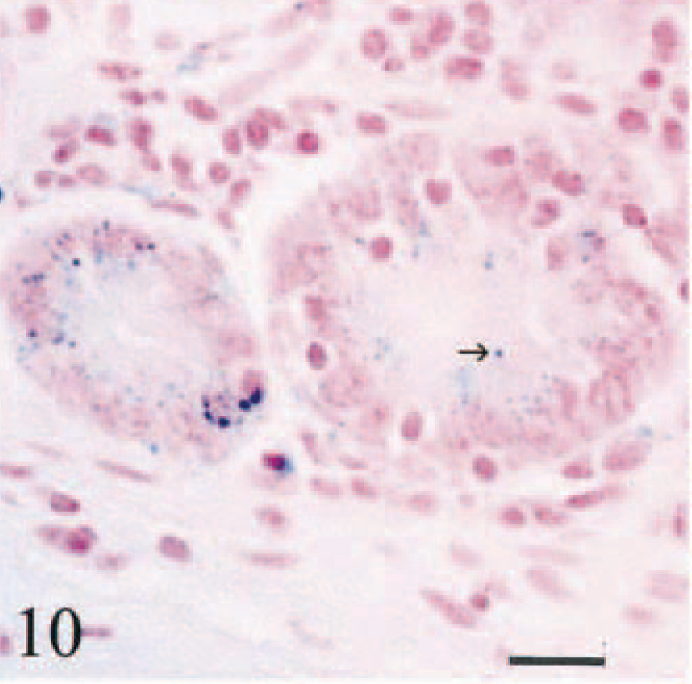
In descending order of severity, siderosis and siderocalcinosis were evident in hepatic and pancreatic lymph nodes, duodenum, kidneys, abomasum, bones, spleen, lung, and choroid plexus epithelium. Hemosiderin in duodenal macrophages was present at the tips of villi and in lamina propria beneath crypts. Iron in villi was present in macrophages surrounding lacteal vessels and in cells immediately beneath villous enterocytes (Fig. 9). Small hemosiderin granules were present in the apical cytoplasm of villous and cryptal duodenal enterocytes (Fig. 10) and in parietal cells in the deep half of abomasal glands. Hemosiderin in large bowel and small intestine distal to affected duodenum was scant and restricted to scattered siderophages in lamina propria.
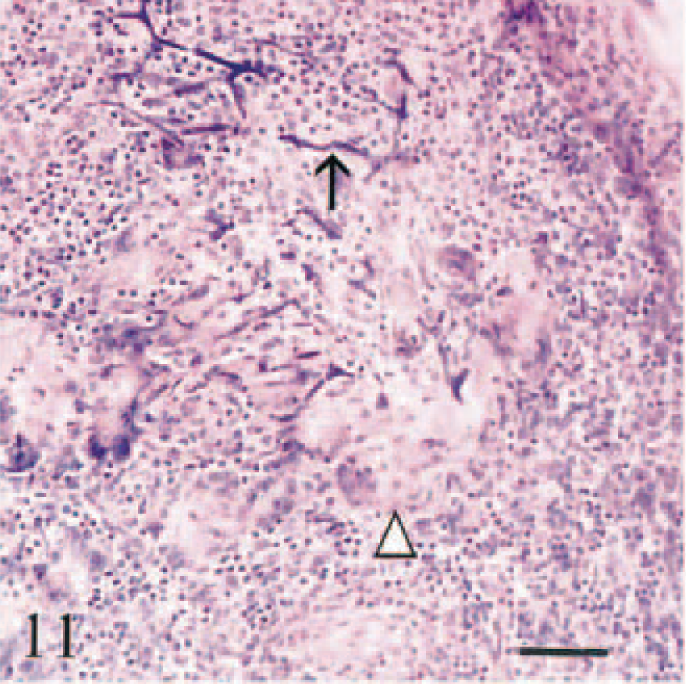
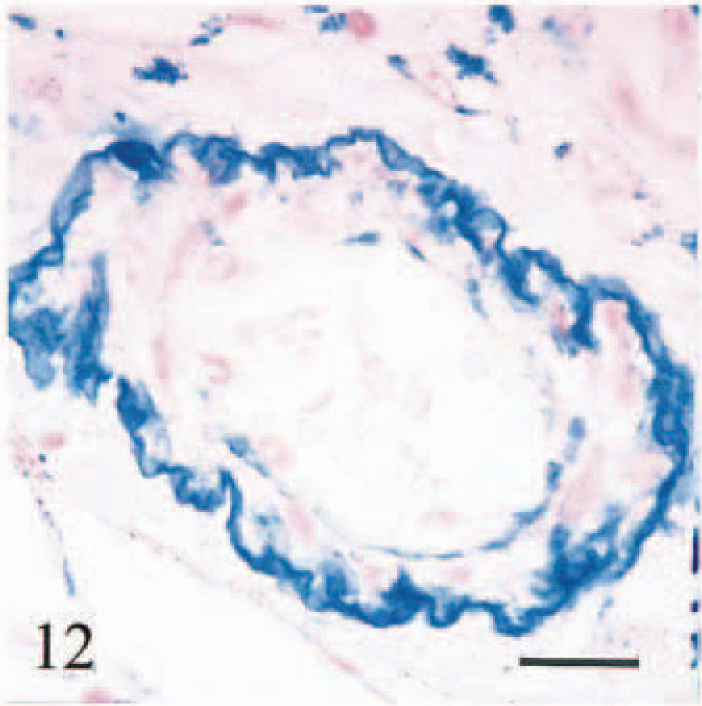
There was heavy accumulation of iron in macrophages in hepatic and pancreatic lymph nodes, accompanied by lymphoid depletion, fibrosis, and partial effacement of sinusoids. Plaques of iron and calcium encrusted hepatic and splenic capsules, splenic trabeculae, and reticulin at the margins of lymphoid follicles. Three calves (Nos. 1–3) had multifocal granulomatous lymphadenitis associated with siderocalcinosis of hyalinized fibers in hepatic and pancreatic lymph nodes and/or spleen (Fig. 11). Iron was accumulated on the walls of arterioles and small-caliber arteries (<130 µm diameter), on venous valves and elastic laminae of arteries, and in vascular endothelium (Fig. 12). This accumulation was most pronounced in vessels in duodenum, hepatic and pancreatic lymph nodes, liver, and spleen.
Renal siderocalcinosis occurred in small discrete zones at the tips of renal papillary tissue. Iron encrustation of vascular and tubular basement membranes and interstitial connective tissue (Fig. 13) was colocalized with calcium deposits. A discrete band of intracytoplasmic hemosiderin was present in renal tubular epithelium at the corticomedullary junction of four terminally ill animals (calf Nos. 1, 3–5). Calf No. 1 had mild multifocal myocardial necrosis. There were no detectable lesions or iron deposition in brain (apart from choroid plexus epithelium), spinal cord, pituitary gland, pancreas, and joints. No hemosiderin was detected in cardiac myocytes.
The iron content in bone ash samples of cranium and two ribs from calf No. 5 ranged from 168 to 704 mg/kg (reference steer: 11–20 mg/kg). In undecalcified sections, iron staining was seen diffusely in the mineralized matrix and at the surface. No staining was seen in decalcified sections, indicating iron was associated with the mineral phase. Histologically, changes in bone were those of osteopenia, with decreased cortical and trabecular bone. Widened osteoid seams and areas of decreased mineralization were consistent with a mineralization defect. Periosteal bone formation in the rib and a layer of poorly mineralized porotic bone was present in outer cortex surface. No abnormalities were detected microscopically in bone from calf No. 2.
Cows A–D had grade 0+ liver samples. Hepatic lesions were absent and iron concentrations were in the normal range (Table 2). Cow D had severe bilateral cystic follicular disease. No abnormalities were found in tissues of cow C, apart from moderate diffuse endometrial arteriosclerosis.
Ultrastructure
Hepatic lesions in three animals (calf Nos. 1, 2, 6) were consistent with previously published reports of the fine structure of HH. 45–47 In the preclinical stage (calf No. 6; 150 days old), electron-dense granules (200–900 nm) consistent with hemosiderin were accumulated in hepatocytes at all levels of hepatic lobules (Fig. 14), whereas hemosiderin in Kupffer cells was minimal. Larger granules were membrane bound. Hepatocytes were otherwise unremarkable, many normal primary lysosomes were present, and fibrosis was absent. Fine granules up to 7 nm in diameter (ferritin) were dispersed throughout the cytoplasm. Canaliculi contained aggregates of hemosiderin (Fig. 15).
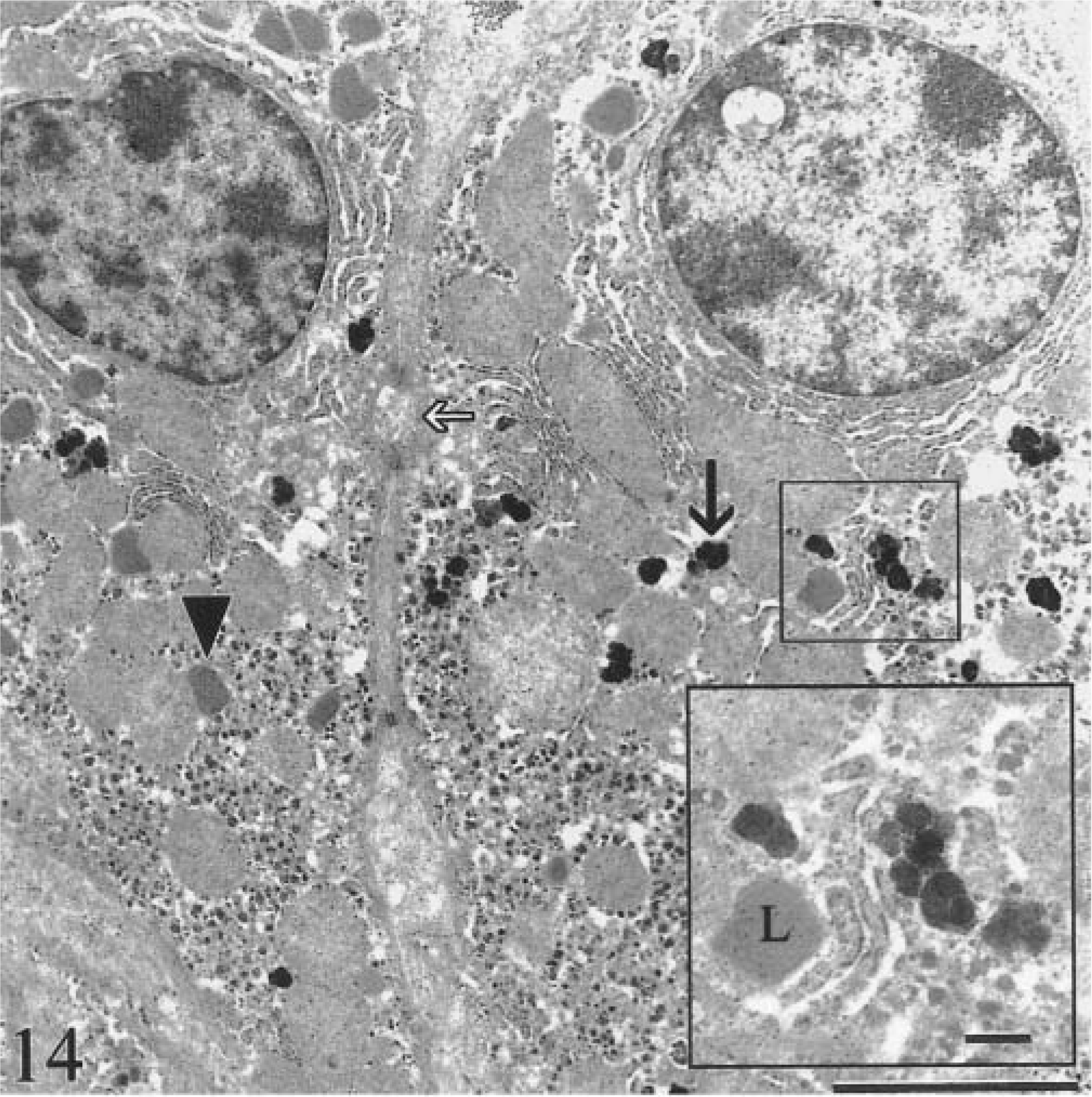
Transmission electron micrograph. Liver; calf No. 6. Iron accumulation in two hepatocytes in preclinical stage of the disease. Aggregates of hemosiderin (black arrow) are dispersed throughout the cytoplasm. Many normal lysosomes are present (arrowhead). Canaliculi (white arrow) contain aggregates of hemosiderin. Hepatocytes are otherwise unremarkable. Bar = 2.5 µm. Inset: Higher magnification of hemosiderin aggregates. L = lysosome. Bar = 500 nm.
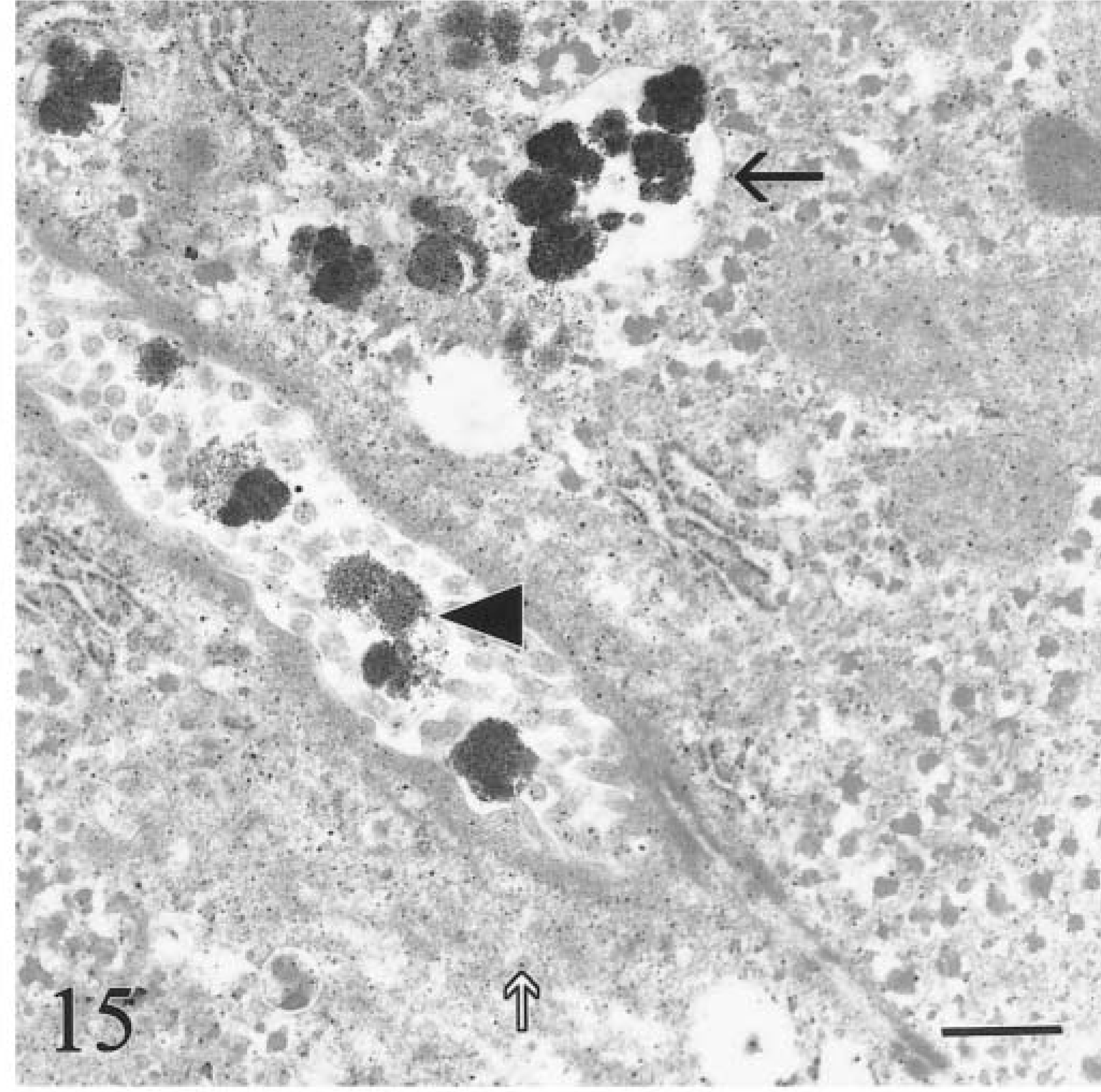
Transmission electron micrograph. Liver; calf No. 6. Membrane-bound siderosomes containing hemosiderin in cytoplasm of hepatocyte (black arrow). Similar aggregates, but devoid of a membrane, are in bile canaliculus (arrowhead). Fine granular particles (white arrow) of ferritin-like material are throughout the cytoplasm. Bar = 0.5 µm.
In the clinical stage (calf Nos. 1, 2; both 9 months old), hemosiderin aggregates in hepatocytes were larger (0.6–3.5 µm), more heterogeneous, and incorporated into phagolysosomes (siderosomes). Normal primary lysosomes were rare, and individual degenerate hepatocytes were present (Fig. 16). Neither animal had intracanalicular hemosiderin. Intercellular matrix of lymph nodes and liver contained round, electron-dense, 50–300-nm granules consistent with iron (Fig. 17). Many granules were deposited along collagen fibrils (Fig. 18). Granulomas in lymph nodes were associated with large, laminated, electron-dense concretions up to 5.5 µm in diameter that were interpreted as calcified aggregates (Fig. 19). Similar material occurred in the cytoplasm of giant cells. There was no iron deposition in exocrine or endocrine components of the pancreas. Scant intracytoplasmic iron was observed in mesangial cells of glomerular tufts.
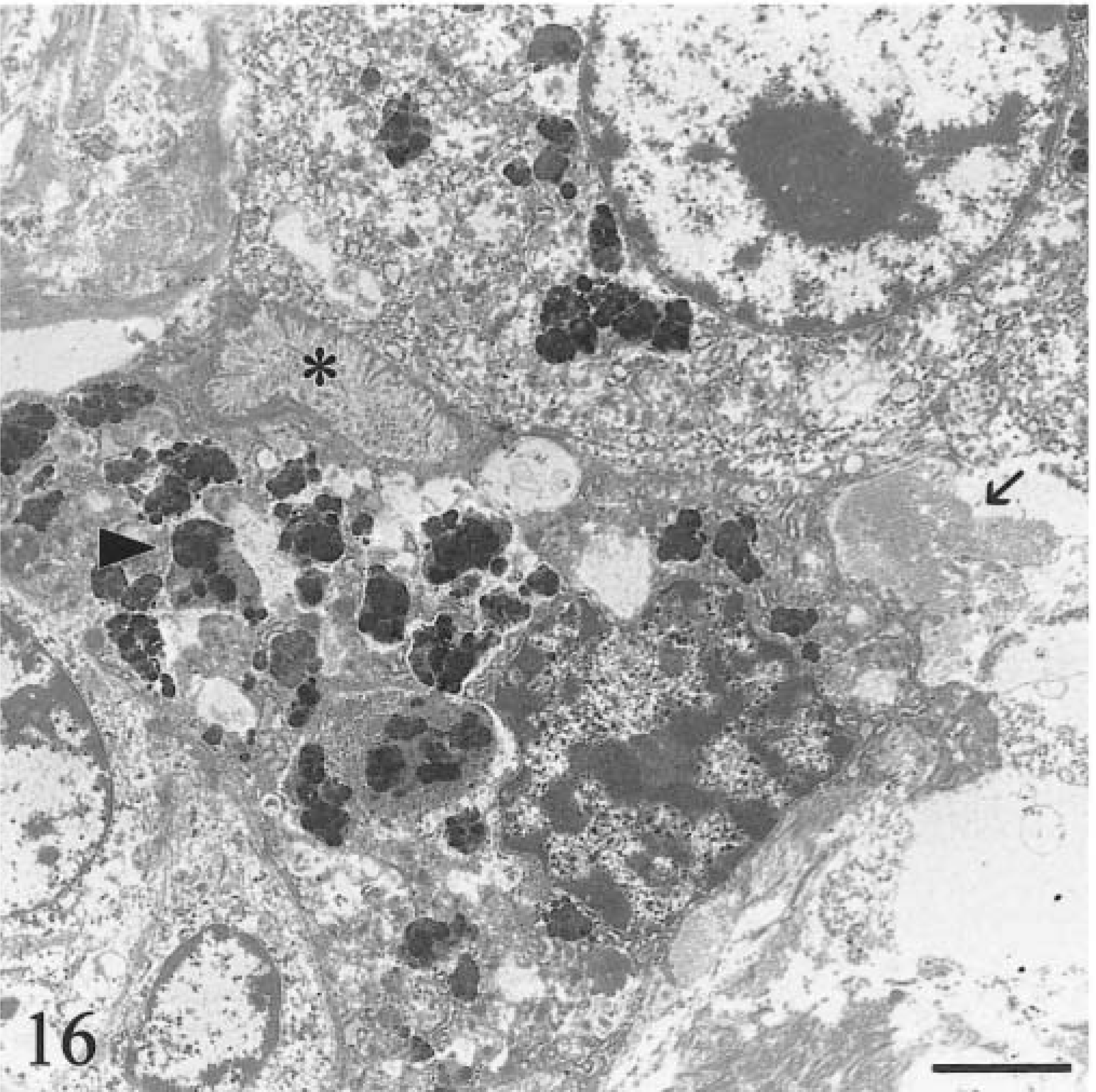
Transmission electron micrograph. Liver; calf No. 1. Acute degeneration in hepatocyte containing large siderosomes. Siderosomes (arrowhead) are larger and more heterogeneous than in the preclinical stage (see Fig. 14). Primary lysosomes are no longer evident. There is collagen deposition (arrow). Canaliculus (asterisk) contains no hemosiderin. Bar = 2.5 µm
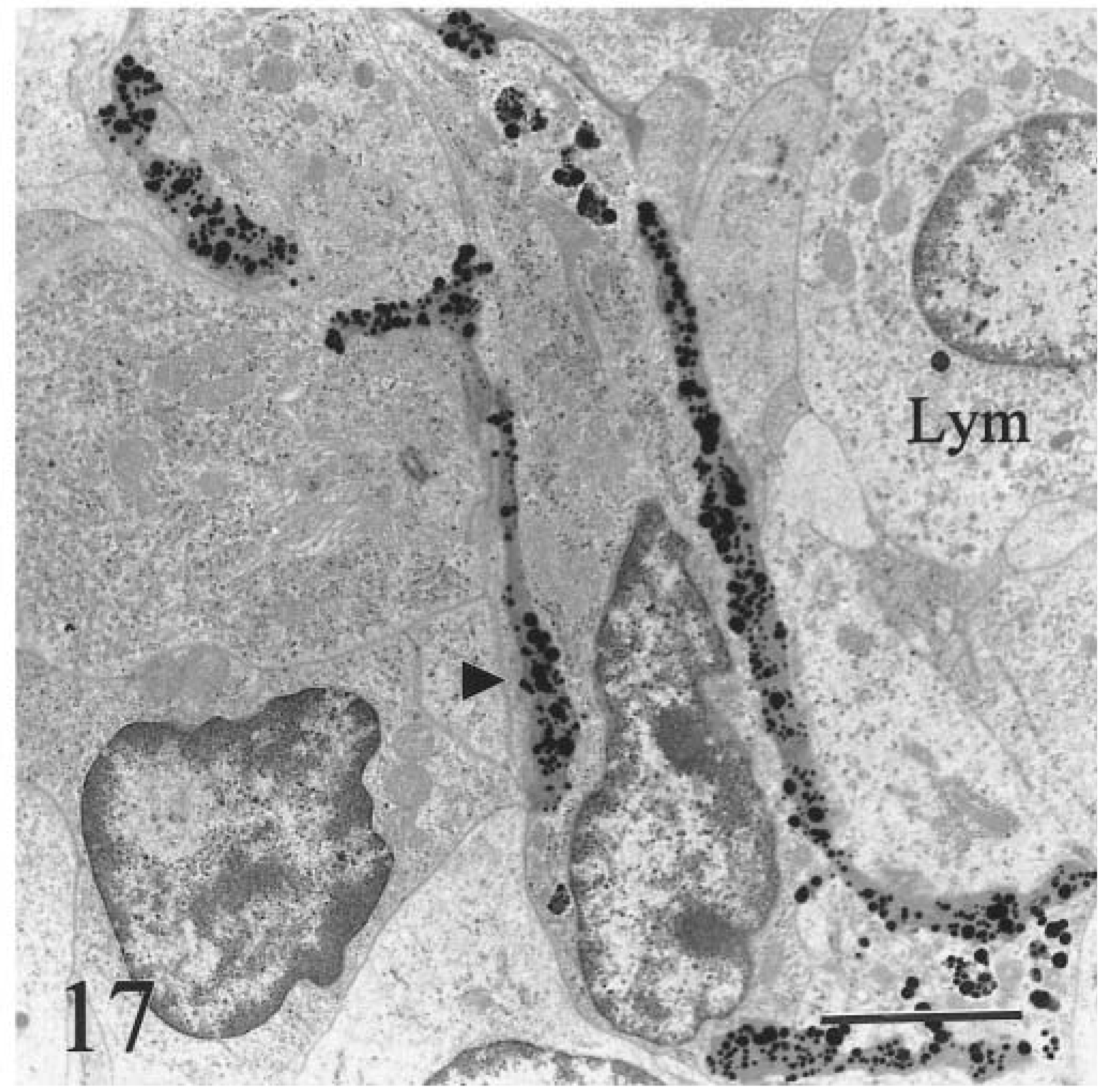
Transmission electron micrograph. Hepatic lymph node; calf No. 2. Electron-dense granules of iron (arrowhead) encrust intercellular matrix in cortex of lymph node. Lymphocytes (Lym) contain little intracytoplasmic iron. Bar = 2.5 µm.
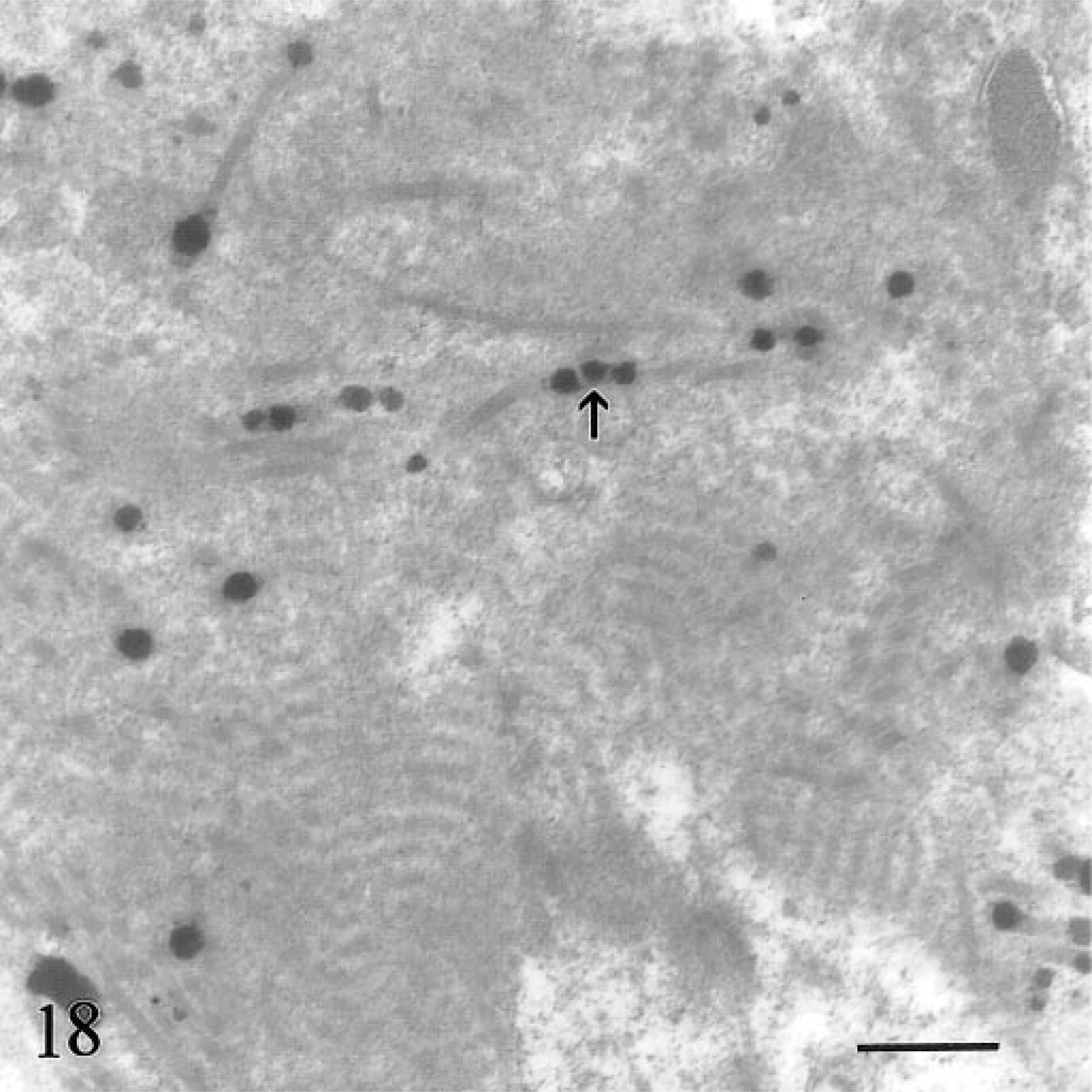
Transmission electron micrograph. Hepatic lymph node; calf No. 2. Higher magnification of iron deposition in intercellular matrix. Some granules decorate collagen fibrils (arrow). Bar = 0.5 µm.
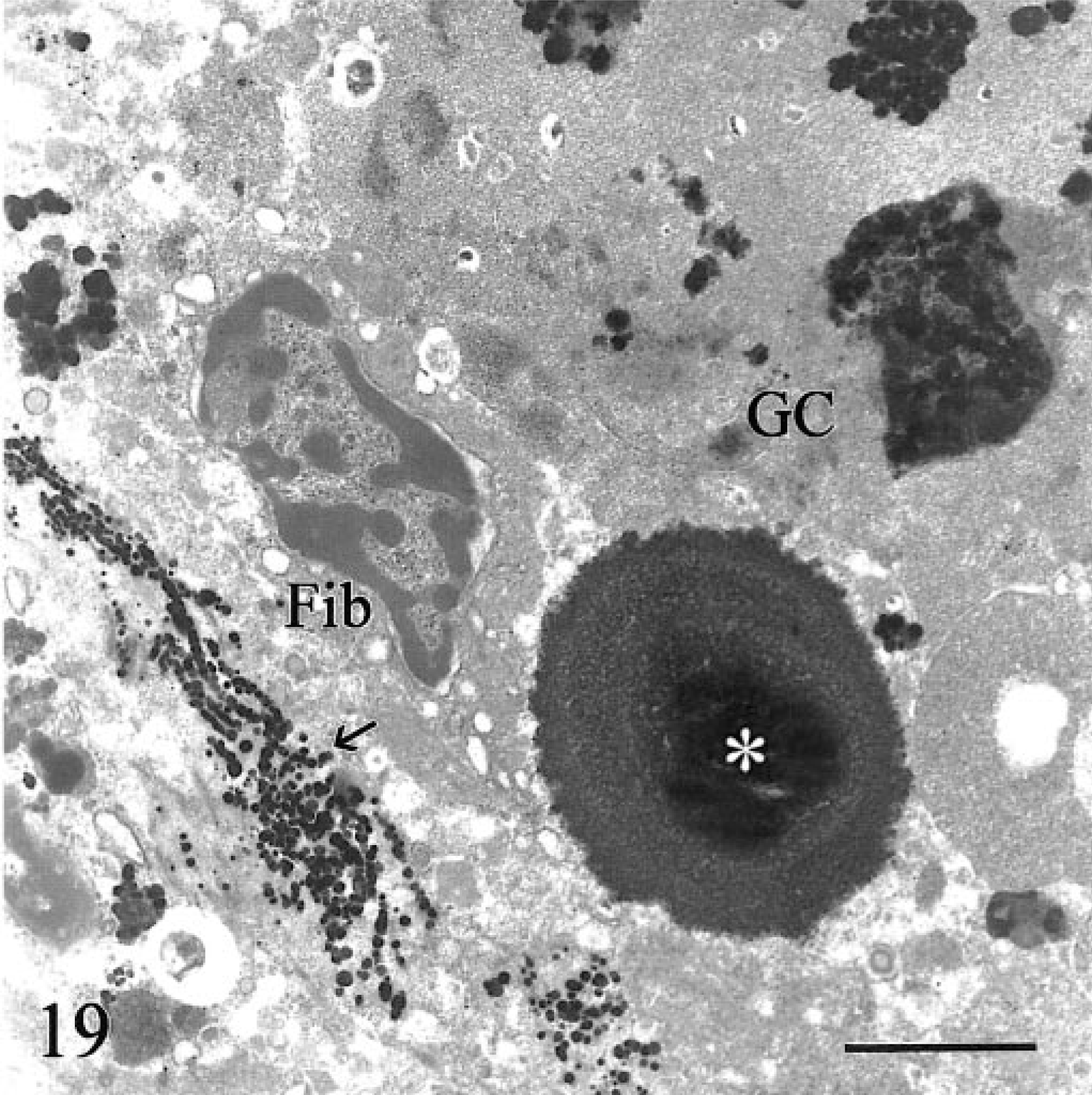
Transmission electron micrograph. Hepatic lymph node; calf No. 1. A large extracellular laminated mineralized deposit (asterisk) is partly invested by a foreign body giant cell (GC) that contains electron-dense intracytoplasmic inclusions. Linear arrays of iron granules decorate extracellular fibers (arrow). Fib = fibrocyte. Bar = 2.5 µm.
Chemical analysis
Hepatic iron concentrations in clinically affected animals ranged from 1,713 (calf No. 5) to 10,500 µg/g wet weight (reference concentration: <300 µg/g) (Table 2). 44 Calf No. 6 had a hepatic iron concentration above the reference range for cattle when first examined at 40 days of age (493 µg/g wet weight). The concentration of hepatic iron rose to 1,467 and 2,076 µg/g wet weight at 125 and 150 days, respectively (Table 2). The concentration of iron in liver for cows A–D (109–274 µg/g wet weight) was in the normal range for cattle. Two grass hay samples from the premises of herd I collected in November 1996 had an iron content of 75.4 and 41.9 µg/g (as received basis) and 87.9 and 49.0 µg/g (dry matter basis) (AgSource Soil and Forage Laboratory, Bonduel, WI). The mineral mix offered to cattle at the time calf No. 2 was identified on the ranch contained 4,377 µg/g iron, 1,902 µg/g copper, and 1,499 µg/g zinc.
HFE in cattle
A zoo blot containing DNA from various species was hybridized with a radiolabelled probe fragment of human genomic DNA containing exons 2 and 3 of the HFE gene. Cross-hybridizing bands were detected in bovine, porcine, canine, and murine DNA and were absent in hamster DNA (Fig. 20). In bovine DNA, two clear bands were detected: a strongly hybridizing band at 6.5 kb and a weakly hybridizing band at 3.5 kb, indicating HFE-related sequence in bovine DNA.
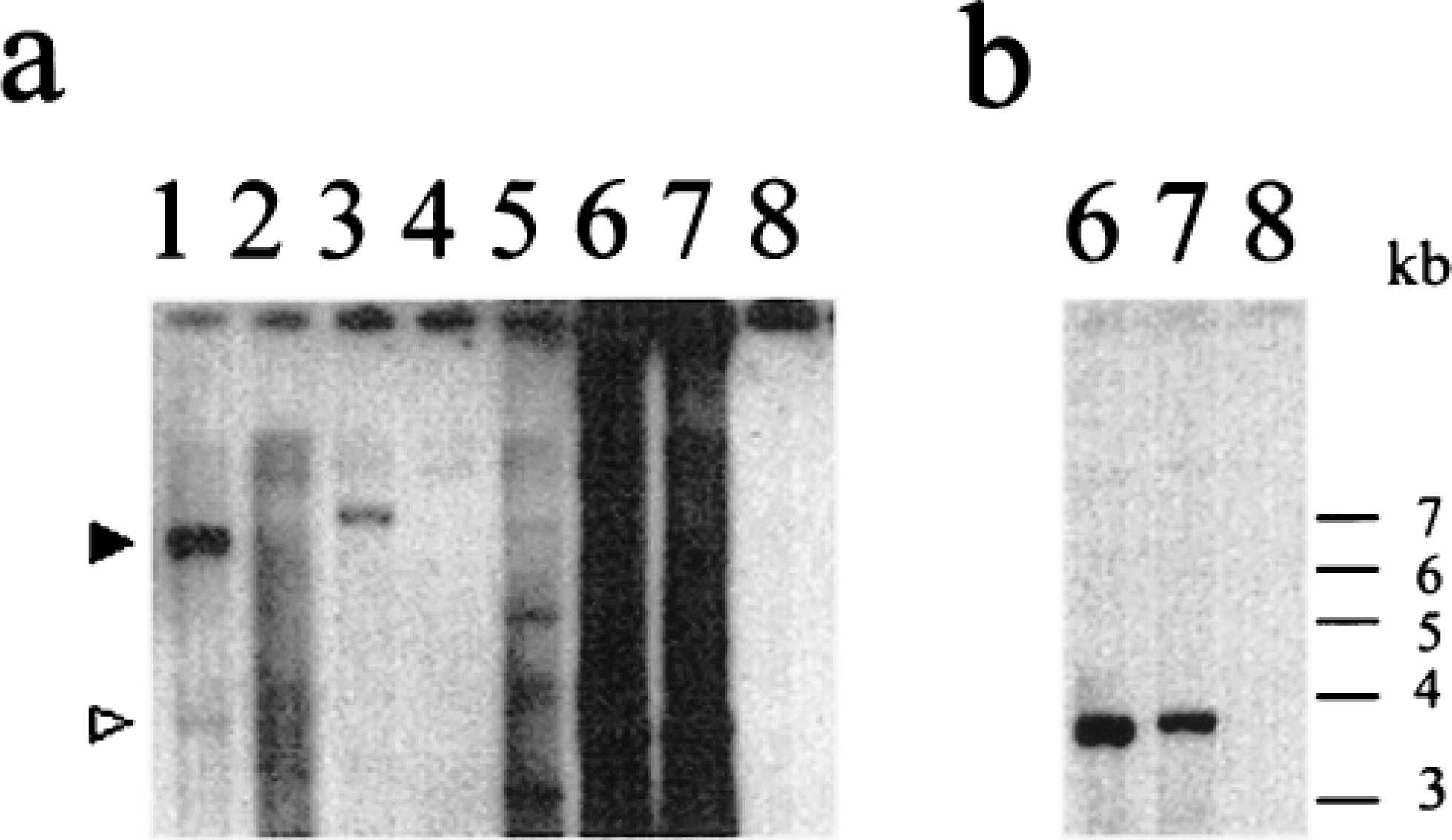
Zoo blot containing genomic DNA from various species, digested with the restriction enzyme HindIII and hybridized with a 32P-labeled genomic DNA probe containing human HFE exons 2 and 3. The lanes contain DNA as follows: 1, cow; 2, hamster; 3, pig; 4, dog; 5, mouse; 6, human individual A; 7, human individual B; 8, 1-kb size marker. Fig. 20a Autoradiograph after moderate stringency washing (0.5× saline sodium citrate [SSC], 0.1% [w/v] sodium dodecyl sulfate [SDS] at 55 C for 30 minutes) and overnight exposure. Specific bands that hybridized across species were detected in bovine, porcine, canine, and murine DNA but were not clearly demonstrated in hamster DNA. In bovine DNA, a strongly hybridizing band at 6.5 kb (solid arrowhead) and a weakly hybridizing band at 3.5 kb (open arrowhead) were seen. The hybridization to human genomic DNA in lanes 6 and 7 (positive control) comprised a specific band at approximately 3.5 kb due to HFE hybridization and a nonspecific component due to the presence of human-specific repeated sequences in noncoding DNA (lane smear). There was no nonspecific hybridization to the 1-kb marker DNA in lane 8 (negative control). Fig. 20b Autoradiograph after high stringency washing (0.1× SSC, 0.1% [w/v] SDS at 65 C for 30 minutes) and overnight exposure to reveal the human HFE signal from human individuals A (lane 6) and B (lane 7).
Discussion
Iron is a potent cellular poison, yet it is essential for life. 76 The liver is a major storage depot for iron, containing one third of total body reserves in healthy animals. Unlike the relatively high tolerance of macrophages for iron, many parenchymal tissues have limited capacity to store iron in nontoxic forms. 38 Absorption of iron in most mammalian species is tightly regulated because of the absence of a natural pathway for eliminating iron following enteric absorption. 52 The only way that substantial quantities of iron can be lost following absorption is by blood loss. 38 Normal regulatory mechanisms for iron uptake are incompletely understood. Significant recent advances in the understanding of iron homeostasis were made in part through identifying the genetic basis for the most common form of human hemochromatosis and through the use of animal models. 33,34,78
An important aspect of the disease in Salers cattle is that it is one of the few spontaneous forms of familial hemochromatosis in a domesticated species. One in 200 humans is homozygous for HH, involving an estimated 1.5 million Americans. 9 Another 32 million Americans carry the trait. 79 Chronic uncontrolled HH is associated with hepatomegaly, micronodular cirrhosis, cardiomyopathy, arthritis, gastric and duodenal siderosis, osteopenia, abnormal cutaneous pigmentation, hepatocellular carcinoma, diabetes, and hypogonadism. 1,9,17,20,30,55,72,74,79 The clinical signs of HH are probably due to abnormally high uptake of dietary iron in the duodenum. 20 Three inherited forms of human hemochromatosis are now recognized. The most common is HFE-related hemochromatosis (hemochromatosis type 1), where the majority of patients are homozygous for the C282Y mutation of the HFE gene on chromosome 6. 31,32 A second missense of HFE (H63D) may contribute to iron loading in HH patients with the C282Y mutation. 2 Mutations in HFE other than C282Y and H63D also occur. 4 Separate genes are implicated in juvenile hemochromatosis (hemochromatosis type 2), which has been mapped to chromosome 1, and in a disease affecting Italian families wherein there is a homozygous nonsense mutation in the gene encoding transferrin receptor 2 (TFR2) on chromosome 7. 13–16,67 Few spontaneous animal models exist to facilitate study of the mechanisms leading to iron accumulation and associated clinical complications, particularly in the extended preclinical phase. 25 Dietary models of iron overload exist in laboratory species, but they generally lack key clinical and morphologic features of HH. 40,45,55 Genetically engineered models of HH were recently developed in rodents. 25,34,68,80 Chronic dietary iron overload occurs in some mammalian species fed iron-rich forage or incorrectly formulated diets or as a seasonal phenomenon, but this dietary overload has been reported only once in cattle. 6,37,41,54 Iron overload in avian species such as mynah birds (Gracula sp.) has features of hemochromatosis. 39 Uptake of dietary iron in birds is more loosely regulated than that in mammalian species, and hepatic siderosis occurs in a variety of physiologic states and diseases. 5,21,59 The pertinence of birds as models for HH is questionable.
Iron-induced lysosomal injury and peroxidation by free radicals are two major mechanisms responsible for hepatocellular necrosis in hemochromatosis and for sequelae such as fibrosis, bile duct hyperplasia, veno-occlusive disease, and hepatic neoplasia. 10,76 Clinical signs and lesions in the six cattle in this report were consistent with iron overload and hepatic failure due to hemochromatosis. We presume that iron overload in Salers cattle is due to poorly regulated and excessive absorption of iron from the gastrointestinal tract, probably via duodenum or abomasum. 1,51 Although duodenal siderosis in affected cattle might have been due to reuptake of iron following biliary excretion, in normal mammals little iron is lost via bile. 22,23,51,75 Iron in the duodenum was found predominantly in macrophages in the lamina propria, which does not exclude the possibility that in hemochromatosis of Salers cattle the primary abnormality involves iron-absorbing enterocytes or parietal cells. 31,38 We excluded a primary dietary overload of iron because the disease occurred in multiple herds, including one at a research facility, only a few individuals in herds were affected, and the iron content of hay for herd I met National Research Council dietary guidelines for adult and neonatal cattle (50 and 100 µg/g, respectively). 75
The disease described herein closely matches that in the one previous report of bovine hemochromatosis, with the exception that none of the affected cattle in the present series had cirrhosis. 44 Secondary iron overload occurs in diseases other than primary hemochromatosis, such as the iron-loading anemias, viral hepatitis, cryptogenic cirrhosis, porphyria cutanea tarda, aceruloplasminemia, and congenital atransferrinemia. 10,38,45 Serum ceruloplasmin concentrations in 111 animals in herd I, including calf No. 2, were within the normal reference range for cattle (Dr. S. Aust, personal communication). The presence of high serum transferrin saturation and the absence of neurologic lesions exclude aceruloplasminemia as the basis for the disease. 56,61 None of the calves was anemic, thereby ruling out chronic anemia and atransferrinemia. The earlier demonstration that phlebotomy induced a clinical response in affected Salers cattle further excludes chronic anemia as the basis for the disease. 44
Ultrastuctural features of the disease match those in previous reports of HH and various animal models of iron-loading disorders. During the early compensated stage of hemochromatosis (calf No. 6), iron accumulated primarily in hepatocytes without evidence of damage in organelles. The presence of hemosiderin granules in bile canaliculi indicates that some excretion of iron may occur via bile early in the clinical course. Intracanalicular iron has been observed ultrastructurally in iron-loaded baboons and hypotransferrinemic mice but not in mice, gerbils, or human HH patients. 45–47 As the disease progresses (represented ultrastructurally by calf Nos. 1, 2), the number and internal complexity of iron-laden secondary lysosomes increases. A striking feature in affected cattle was the extent to which siderocalcinosis occurred in the extracellular matrix. Calciphylaxis is the term used to describe calcification occurring in response to deposition of toxic compounds, including iron, in tissues. 65 Although calciphylaxis is considered protective, the granulomatous reaction in lymph nodes appeared to be driven in part by the presence of aggregates of calcium salts.
The extent of hemochromatosis in Salers cattle in the United States is difficult to establish. This report brings to nine the number of histologically confirmed cases. To date, the disease has been confirmed in five states: Wyoming (2 cases), Utah (2), California (2), Montana (1), and Idaho (1). The insidious, nonspecific wasting nature of the disease makes it likely that producers and veterinarians will overlook hemochromatosis unless specific tests for iron overload are performed. Hemochromatosis may occur in bloodlines outside of the United States; the suspected carrier bull was born to parents in France. Reproduction of the disease by breeding a cow to the original suspected carrier bull strongly supports the suggestion by House et al. 44 that hemochromatosis in Salers cattle is genetic in origin. Unfortunately, we were unable to characterize the form of inheritance. Conclusive determination of the mode of inheritance for genetic conditions depends on access to either a sufficiently large pedigree of affected animals or sufficient carrier animals to conduct test mating to achieve an acceptable level of statistical probability. Neither condition has yet been met. The predominance of females (eight of nine histologically confirmed cases) is interesting; HFE-related hemochromatosis in humans occurs predominantly in men, and many C282Y homozygous women are free of clinical disease. 24 One explanation for the predominance of females in affected cattle is that bull calves are more likely than heifers to be sold and lost to follow-up before clinical disease is evident. New owners are less likely to recognize and investigate a familial pattern for nondescript ill thrift in recently purchased stock, particularly in nonbreeding animals such as steers.
Confirmation of hemochromatosis is straightforward. This disease should be included as a differential diagnosis when Salers or Salers-cross cattle of ages 9 months to 2 years lose weight, shed incisor teeth, and have biochemical evidence of hepatic failure. Persistently high serum transferrin saturation values support the diagnosis. Published ranges for transferrin saturation in healthy cattle are 21.0–57.5%. 51 In the present study, none of 40 yearling cattle in a university herd had saturation values above 53.5%. Serum transferrin saturation values of >50% suggest a diagnosis of hemochromatosis in humans, although higher values have been suggested. 16,52,79 A cutoff of >60% may be more appropriate for cattle suspected to have the disease. High serum transferrin saturation values should be documented in animals on more than one occasion to minimize the confounding effects of normal daily fluctuations. 38,51 Bovine hemochromatosis can be confirmed by chemical and histologic examination of subsequent hepatic biopsy. In decompensated stages of the disease, the amount of hepatic iron may decline as iron-loaded hepatocytes are replaced by fibrous tissue containing less iron.
Clinical disease supervened after a clinically silent period of 6–9 months. The earliest sample examined from an affected calf indicated that excessive iron was present by 40 days of age. The rate of progression of the disease is probably determined in part by the concentration of dietary iron, which may explain the variability in onset and rate of progression among the six affected cattle. Clinical progression of HH in humans and in HFE-deficient mice is also variable, predicated on dietary iron, physiologic and pathologic blood loss, and unidentified modifying genetic factors. 80 Approximately one in three humans homozygous for the C282Y mutation shows no clinical or biochemical expression. 24,62,63 The penetrance of the genotype in Salers cattle remains to be established, including the possibility that disease in some animals is clinically silent for their lifetime. Hemachromatosis in humans is thought to have become common due to a selective advantage conferred on heterozygotes who have increased capacity to absorb dietary iron. 34 Analogous selection pressure may have favored emergence of a similar trait in Salers cattle. Perhaps the breed originated in an area of France where dietary iron in native forage was marginal or low.
Osteopenia was confirmed histologically in one animal in the present study. Osteopenia is a feature of HH, and its occurrence is positively correlated with the development of hepatic cirrhosis and hypogonadism. 27,73 The finding of poorly mineralized matrix and osteopenia was consistent with a mineralization defect such as osteomalacia. Osteomalacia is not a recognized feature of human hemochromatosis. Interference with vitamin D metabolism in the liver may occur in human hepatic osteodystrophy, but osteomalacia is absent. 26,27 The segmental nature of the mineralization defect indicates that other transient factors may be involved in hemochromatosis of Salers cattle or it may have been peculiar to the one animal that was studied. Two affected animals lost incisor teeth, a feature noted in the first report of the disease. 44 The most likely basis for this loss is loss of alveolar bone and osteopenia. Changes in bone are probably a late development; vertebral tissue from a calf in the early stages of hemochromatosis was histologically normal.
The disease in Salers cattle is similar but not identical to HFE-related hemochromatosis (Table 3). Similarities include a long preclinical period, heavy accumulation of pericanalicular siderosomes in hepatocytes followed by accumulation of iron in Kupffer cells and in other tissues, absence of estrous cycles, hemosiderin accumulation in gastric glands and small intestine, granulomatous lymphadenitis, and progression to cirrhosis and hepatic failure. 11,38,45,72
Comparison of human HFE hemochromatosis and Salers cattle hemochromatosis.

† Based on present report and three cases previously published. 3
‡ Sheldon 72 reported “haemofuscin” but not iron in hepatic vessels, hepatic capsule, and connective tissue cells.
The bovine form of hemochromatosis differs from HFE-related HH in several respects. In affected cattle, differences include greater amounts of iron in duodenal and gastric mucosa, more extensive mineralization of collagen, elastin, and reticulin, earlier onset, more rapid clinical progression, presence of renal lesions, minimal cardiac lesions, and females rather than males being more commonly identified. 1,19,20,24,36,72 Five of the six Salers cattle had a panacinar rather than a portal-to-centrilobular (acinar zones 1–3) gradient of iron accumulation typical of HH. 11,52,72 These differences may be due to species-determined differences in normal iron metabolism or may reflect a fundamentally different pathogenesis for iron overload. The disease in Salers cattle has features of early onset aggressive human HH described in Italian and Canadian families, which lack a recognized HFE mutation. 13,15,48,66 The major difference from juvenile HH in humans is that hepatic rather than cardiac failure dominates the clinical picture in Salers cattle.
Additional studies may establish whether hemochromatosis cases in humans and in Salers cattle share a similar pathogenesis due to a common mutation affecting proteins involved in iron metabolism. The role of HFE represents an attractive target for study, given preliminary evidence presented here that an HFE-like gene exists in normal cattle. HFE orthologues have been reported in mice and rats (GenBank accession AJ001517). 42,70 We are investigating bovine HFE as a candidate gene for bovine hemochromatosis, in addition to testing positional candidate genes and markers linked to human non-HFE related hemochromatosis. Other candidate genes are those for transferrin (Tf), transferrin receptors (Tfr), ferritins, iron regulatory proteins (IRP-1 and IRP-2), iron responsive elements (IRE), and the Nramp2 gene. As Sheldon 72 stated when he speculated that hemochromatosis was an inherited inborn error of iron metabolism, justification for his hypotheses lay “in the acquisition of any new knowledge entailed by their destruction.” Elucidation of the genetic basis for bovine hemochromatosis should extend our knowledge about iron metabolism in cattle and humans and the mechanisms associated with subcellular injury early in the clinical course. It may also permit the development of a diagnostic DNA test for bovine carriers of the disease.
Footnotes
Acknowledgements
This study is dedicated to the memory of Dr. Rue Jensen (24 October 1911–28 November 2000). Dr Jensen worked as a veterinary diagnostic pathologist in Colorado and Wyoming. This work was made possible by active cooperation of the owners of herds I and II, who generously shared pedigree information. Dr. John House provided pedigree information for two of three previously reported cases. We appreciate the assistance of Dr. Lynn Woodard, Cody Molle, Paula Jaeger, Carol Hearne, Roger Simeon, Angie McGuire, Dr. Steve Lucas, and Dr. John Koger. We thank Dr. M. Keven Jackson (Utah State University), who examined calf Nos. 3 and 4, and personnel at the Texas Veterinary Diagnostic Laboratory for permission to use chemical analytical data for calf Nos. 3 and 4. We are grateful to Dr. S. Aust (Utah State University) for examining ceruloplasmin concentrations in herd I. This study was supported by the University of Wyoming Agricultural Experiment Station.