
Abstract
Inflammatory cytokines are suspected to contribute to the pathogenesis of bovine pneumonic pasteurellosis (BPP) through neutrophil recruitment, leukocyte activation, and the induction of a broad array of soluble inflammatory mediators. An in vivo experimental model of BPP was used to characterize the pulmonary expression kinetics of tumor necrosis factor alpha (TNFα), interleukin-1 beta (IL-1β), and interleukin-8 (IL-8) genes and proteins during the acute phase of disease development. Cytokine expression in bronchoalveolar lavage (BAL) fluid, BAL cells, and pneumonic lung parenchyma was quantitated by northern blot analysis, enzyme-linked immunosorbent assay (ELISA), and in situ hybridization at 2, 4, 8, 16, and 24 hours after endobronchial inoculation of Pasteurella (Mannheimia) haemolytica. Expression of TNFα, IL-1β, and IL-8 was significantly increased in the airways and lung lesions of infected calves as compared with mock-infected controls. Although kinetic patterns varied, peak levels of cytokine mRNA occurred within 8 hours postinfection (PI), and peak cytokine concentrations occurred within 16 hours PI. In all samples, IL-8 was expressed to the greatest extent and TNFα was least expressed. Expression of TNFα was restricted to alveolar macrophages. Alveolar and interstitial macrophages produced IL-1β and IL-8 in the first 4 hours; bronchial and bronchiolar epithelial cells were also significant sources of IL-8 during this period. By 8 hours PI, neutrophils were the dominant source of both IL-1β and IL-8. These findings demonstrate a spatial and temporal association between pulmonary expression of inflammatory cytokines and acute lung pathology, supporting the hypothesis that cytokines contribute to inflammatory lung injury in BPP.
Keywords
Bovine pneumonic pasteurellosis (BPP), an acute fibrinous pleuropneumonia caused by Pasteurella (Mannheimia) haemolytica, is a common and economically important disease of North American cattle. One of the hallmark histopathologic features of BPP is extensive infiltration of the lungs by neutrophils. Neutrophil depletion prior to experimental infection with P. haemolytica protects calves from subsequent lung injury, 32,43 indicating that this cell type is directly or indirectly responsible for most of the pulmonary pathology observed in the disease. The mechanisms by which neutrophils mediate lung pathology in BPP remain undefined but are likely to include oxidative injury and structural degradation due to extracellular release of reactive oxygen intermediates and proteolytic lysosomal enzymes. 33 Recognition of the importance of neutrophils in disease pathogenesis prompted us and others to investigate specific factors governing their recruitment and activation within P. haemolytica-infected bovine lung.
The migration and activation of neutrophils in inflamed tissue are regulated by a complex network of interactions among cytokines, leukocytes, vascular endothelium, cellular adhesion molecules, and soluble chemotactic factors. The inflammatory cytokines tumor necrosis factor alpha (TNFα), interleukin-1 beta (IL-1β), and interleukin-8 (IL-8) play a central role in the initiation and orchestration of these interactions. TNFα and IL-1β are pleiotropic early response polypeptides secreted by monocytes and macrophages in response to microbial pathogens and other stimuli. 20 IL-8, a potent chemotactic and activating factor for neutrophils, is a C-X-C chemokine secreted by a variety of immune and nonimmune cell types, including monocytes, macrophages, fibroblasts, epithelial cells, and neutrophils. 3,18
Pulmonary overexpression of inflammatory cytokines is believed to contribute to the pathogenesis of several infectious and inflammatory lung diseases in humans, including adult respiratory distress syndrome, 11,21 cystic fibrosis, 5,17 and pneumoconiosis. 40 This overexpression is also associated with lung pathology in animal models of influenza pneumonia, 24 silicosis, 12 and immune complex alveolitis. 42 Among domestic species, TNFα, IL-1β, and IL-8 are implicated in the pathogenesis of swine influenza, 41 porcine pleuropneumonia caused by Actinobacillus pleuropneumoniae, 2,16 and Corynebacterium pseudotuberculosis-induced pyogranulomas in sheep. 14
A growing body of circumstantial evidence implicates inflammatory cytokines in the pathogenesis of BPP, prompting speculation that it may be possible to treat or prevent the disease through pharmacologic modulation of cytokine expression. Heat-killed P. haemolytica and purified P. haemolytica lipopolysaccharide (LPS) and leukotoxin (Lkt) induce the expression of TNFα, IL-1β, and IL-8 mRNA and proteins by bovine alveolar macrophages in vitro. 19,22,23,36,44,46 In addition, BPP is associated with the pulmonary expression of TNFα, IL-1β, and IL-8 in vivo. 8,45 Because in these studies pulmonary cytokine expression was evaluated 2–4 days after inoculation with P. haemolytica, it is difficult to draw definitive conclusions regarding a causative role for cytokines in disease pathogenesis. A comprehensive understanding of inflammatory cytokine expression during earlier stages of disease development would permit a more accurate assessment of the role of cytokines in disease pathogenesis, and is a necessary prerequisite for the development of therapeutic strategies based upon cytokine inhibition or antagonism.
The objectives of this study were to 1) characterize the kinetics of pulmonary TNFα, IL-1β, and IL-8 gene and protein expression in the first 24 hours of experimental BPP, 2) compare patterns of cytokine expression in airways with those in lung lesions, and 3) identify major cellular sources of these cytokines within affected lung. Northern analysis was used to quantitate the expression of cytokine mRNA in bronchoalveolar lavage (BAL) cells and diseased lung parenchyma. Immunoreactive cytokines in BAL fluid and lung tissue extracts were measured by enzyme-linked immunosorbent assay (ELISA). In situ hybridization was used to localize the expression of cytokine mRNA within lung tissues; numbers of positively stained cells were enumerated by quantitative morphometric techniques, and stained cells were identified by cell type on the basis of cell morphology and location.
Materials and Methods
Preparation of bacterial inoculum
Pasteurella (Mannheimia) haemolytica A1 strain 12296, a field isolate recovered from a yearling calf with fatal pneumonic pasteurellosis, was propagated in phenol red–free RPMI 1640 medium (BioWhittaker, Walkersville, MD) supplemented with 2 mM
Animals and induction of experimental disease
Eighteen healthy male Holstein calves between 4 and 7 weeks of age were purchased from the University of Minnesota Department of Animal Science. The calves were weaned at birth and raised in individual hutches and were free of detectable serum antibodies against P. haemolytica surface antigens and Lkt as determined by ELISA using previously described methods. 34 Pneumonic pasteurellosis was induced in 15 calves using a well-characterized, reproducible experimental model developed in our laboratory. 1 Calves were sedated with xylazine (0.1 mg/kg intravenously) and positioned in sternal recumbency for the passage of a sterile fiberoptic bronchoscope into the left caudal lung lobe. With the tip of the endoscope wedged in a large bronchus, 5 ml (5 × 109 cfu) of logarithmic phase P. haemolytica was deposited into the airway, followed by 30 ml of sterile LPS-free phosphate-buffered saline (PBS), pH 7.4. Groups of three infected calves, randomly selected on the basis of birth order, were euthanatized at each of the following times postinfection (PI): 2, 4, 8, 16, and 24 hours. At necropsy, focally extensive regions of hemorrhage, interlobular edema, and consolidation consistent with pneumonic pasteurellosis were present in the left caudal lung lobe of all inoculated calves. Bacteriologic cultures of pulmonary lesions yielded pure colonies of P. haemolytica. Three control calves received mock infections in which an equal volume of sterile culture medium was substituted for bacteria. These animals were euthanatized at 24 hours PI and exhibited no gross pulmonary pathology at necropsy.
BAL and lung tissues
BAL fluid was collected from the right caudal lung lobe of all calves immediately prior to infection or mock infection and from the left caudal lung lobe at necropsy. Using a sterile fiberoptic endoscope, 60 ml of LPS-free PBS was infused into a large bronchus and immediately retrieved by gentle suction. This process was repeated with three additional 60-ml aliquots of PBS. Samples were pooled and centrifuged for 10 minutes at 400 × g (4 C) to separate fluid and cellular components. The supernatant was centrifuged again for 30 minutes at 15,000 × g (4 C) and stored at −80 C for quantitation of cytokines and urea. BAL cells were washed once with LPS-free PBS and lysed in 4 M guanidinium isothiocyanate containing 8 mM sodium citrate, 0.5% sodium lauroyl sarcosinate, and 8% (v/v) β-mercaptoethanol. Cell lysates were stored at −80 C for RNA extraction and northern blot analysis.
Nonlavaged lung tissues were collected from the left caudal lung lobe of all calves. Samples were taken from the margins of gross pneumonic lesions in infected calves and from grossly normal lung in control calves. Tissue samples for in situ hybridization were fixed in neutral buffered formalin for 24 hours, stored at −20 C in 70% ethanol for 12–24 hours, processed, and embedded in paraffin by standard methods. Tissues for RNA extraction and northern analysis were snap-frozen in liquid nitrogen, homogenized in lysis buffer (4 M guanidinium isothiocyanate, 0.5% sodium lauroyl sarcosinate, 8 mM sodium citrate, and 8% [v/v] β-mercaptoethanol), and stored at −80 C. Tissue extracts were prepared by homogenizing 1 g of fresh lung tissue per 3 ml of PBS containing 0.05% Tween-20 and recovering the supernatant after centrifugation for 10 minutes at 15,000 × g (4 C). Extracts were stored at −80 C for quantitation of cytokines by ELISA.
Plasmids
Bovine TNFα, IL-1β, and IL-8 cDNAs (488, 474, and 230 base pairs [bp] in length, respectively) were cloned and sequenced in our laboratory, 19,44 ligated into pcDNA3 (Invitrogen, Carlsbad, CA), and transformed into Escherichia coli DH5α. A 1,250-bp human glyceraldehyde phosphate dehydrogenase (GAPDH) cDNA in pBluescript KS+ (Stratagene, La Jolla, CA) was the generous gift of Dr. M. Murtaugh (University of Minnesota, St. Paul, MN). All plasmids were purified by alkaline lysis using a commercial kit (Qiagen, Valencia, CA) according to the manufacturer's instructions.
RNA extraction and northern blot analysis
All solutions were treated with 0.1% diethylpyrocarbonate (DEPC) and glassware was baked overnight at 350 C before use. Total cellular RNA was extracted from BAL cells and lung tissues using the acid guanidinium thiocyanate and phenol–chloroform extraction method. 9 Ten micrograms of RNA from each sample was electrophoretically fractionated in a 1.2% agarose gel containing 6.5% formaldehyde, transferred to a neutral nylon membrane (Schleicher and Schuell, Keene, NH), and covalently linked to the membrane by ultraviolet illumination. Membranes were prehybridized at 45 C for 2 hours in solution containing 50% formamide, 5× saline sodium citrate (SSC), 5× Denhardt's solution, 1% sodium dodecyl sulfate (SDS), and 0.1 mg/ml yeast tRNA.
Gel-purified TNFα, IL-1β, IL-8, and GAPDH cDNA plasmid inserts were labeled with [α-32P]dCTP by DNase/DNA polymerase I nick translation, and unincorporated [α-32P]dCTP was removed using Elutip-d affinity columns (Schleicher and Schuell). Labeled probe was added to prehybridization buffer at 2 × 106 cpm/ml, and membranes were hybridized overnight at 45 C. Blots were washed to a stringency of 0.1× SSC/0.1% SDS at 50 C. Autoradiographs were prepared by exposing membranes to Kodak X-OMAT AR x-ray film (Eastman Kodak, Rochester, NY) with an intensifying screen for 1–3 days at −80 C. Phosphor screen autoradiographs were prepared (Phosphorimager SF, Molecular Dynamics, Sunnyvale, CA), and relative levels of cytokine-specific mRNA were quantified by densitometric analysis using ImageQuant software (Molecular Dynamics). Data were normalized to the expression of GAPDH mRNA. For each cytokine, values for infected calves were presented relative to mean normalized expression in mock-infected control calves.
TNFα ELISA
A capture ELISA was developed to quantitate immunoreactive TNFα in BAL fluid and lung extracts. Mouse monoclonal antibody 2C4 ascites and rabbit anti-TNFα antiserum were generously provided by Dr. T. H. Elsasser (USDA-ARS, Beltsville, MD), and purified recombinant bovine (rb)TNFα for use as a standard was generously provided by Dr. Dale Godson (Veterinary Infectious Disease Organization, Saskatoon, SK, Canada). All samples, standards, and detection antibodies were diluted in PBS containing 10% (v/v) bovine serum albumin blocking concentrate (Kirkegaard & Perry, Gaithersburg, MD). Unless otherwise indicated, all reactions were conducted in a volume of 100 µl, and plates were incubated at 37 C for 1 hour on a platform shaker. After each step, plates were washed five times with PBS containing 0.01% Tween-20. Monoclonal antibody 2C4 ascites diluted 1:1,000 in coating buffer (15 mM sodium carbonate, 35 mM sodium bicarbonate, and 3 mM sodium azide, pH 9.6) was adsorbed to 96-well ELISA plates (Costar Corp., Cambridge, MA) overnight at room temperature. After blocking nonspecific protein binding sites, samples and standards were added to plates. Samples were assayed in duplicate at twofold dilutions from neat to 1:8. Rabbit anti-bovine TNFα antiserum diluted 1:2,000 in blocking buffer was used for primary detection of bound cytokine, followed by secondary detection with horseradish peroxidase (HRP)-labeled polyclonal goat anti-rabbit IgG (Kirkegaard & Perry) at a dilution of 1:6,000. The color substrate tetramethylbenzidine (Kirkegaard & Perry) was added, and plates were incubated for 10 minutes at room temperature. The reaction was stopped with 100 µl of 1 M phosphoric acid, and absorbance was measured at 450 nm. For each plate, a standard curve was constructed using duplicate threefold dilutions of rbTNFα. Sample dilutions yielding absorbance readings in the linear region of the standard curve were used to calculate cytokine concentrations by interpolation using SOFTmax PRO software (Molecular Devices, Sunnyvale, CA).
IL-1β ELISA
A capture ELISA was developed to quantitate immunoreactive IL-1β in BAL fluid and lung extracts. Mouse monoclonal antibody SA22 specific for bovine IL–1β 25 was produced from hybridoma CRL-2052 (American Type Culture Collection, Manassas, VA) and purified by standard methods. 15 Purified rbIL-1β for use as a standard was generously provided by Dr. Kathleen Heaney (Fort Dodge Animal Health, Princeton, NJ). Samples were assayed in duplicate at twofold dilutions from neat to 1:8. Methods were as for the TNFα ELISA with the following exceptions. All samples, standards, and detection antibodies were diluted in PBS containing 10% (v/v) milk blocking concentrate (Kirkegaard & Perry). Purified monoclonal antibody SA22 in coating buffer (5 µg/ml) was adsorbed to plates overnight at room temperature. Rabbit anti-ovine IL-1β antiserum (Chemicon, Temecula, CA) diluted 1:1,000 was used for primary detection, and HRP-labeled goat anti-rabbit IgG diluted 1:4,000 was used for secondary detection. Color development was allowed to proceed for 30 minutes before the reaction was stopped with 1 M phosphoric acid, and absorbance was measured at 450 nm.
IL-8 ELISA
Purified rbIL-8 and mouse monoclonal antibody 170.13 specific for bovine IL-8 were produced and characterized in our laboratory. A cDNA encoding the mature bovine IL-8 protein 19 was subcloned into the pET15b expression vector (Novagen, Madison, WI), transformed into E. coli BL21(λDE3)pLysS cells (Novagen), and expressed according to the manufacturer's recommendations. Histidine-tagged rbIL-8 was expressed, affinity purified on a Ni2+-resin column (Novagen), and concentrated by dialysis against polyethylene glycol. Purified rbIL-8 was used to produce hybridomas and monoclonal antibodies by standard methods in collaboration with Immunochemistry Technologies (Bloomington, MN). 15 Monoclonal antibody 170.13 (IgG1) recognized rbIL-8 and recombinant human IL-8 in western blots and neutralized the neutrophil chemotactic activity of rbIL-8 in vitro.
A capture ELISA was developed to quantitate immunoreactive IL-8 in BAL fluid and lung extracts. Methods were as for the TNFα ELISA with the following exceptions. Samples and standards were diluted in PBS containing 10% (v/v) milk blocking concentrate. Purified monoclonal antibody 170.13 in coating buffer (5 µg/ml) was adsorbed to plates overnight at room temperature. BAL fluids were assayed in duplicate at twofold dilutions from neat to 1:8, and lung extracts were assayed in duplicate at 10-fold dilutions from neat to 1:1,000. Rabbit anti-ovine IL-8 antiserum (Chemicon) diluted 1:4,000 in blocking buffer containing 5% normal mouse serum was used for primary detection, and HRP-labeled goat anti-rabbit IgG diluted 1:6,000 in blocking buffer was used for secondary detection. Color development was allowed to proceed for 20 minutes before the reaction was stopped with 1 M phosphoric acid, and absorbance was measured at 450 nm.
Correction for variable dilution of BAL samples
Urea was used as an endogenous marker of dilution to calculate the extent to which epithelial lining fluid (ELF) was diluted during the BAL procedure in each calf. 26 Blood for quantitation of serum urea nitrogen was drawn from the jugular vein of all calves immediately prior to euthanasia. Urea concentration in serum and BAL fluid was determined by enzymatic methods using a commercial kit (Sigma, St. Louis, MO) according to the manufacturer's instructions. Measured cytokine concentrations in BAL fluid were corrected for dilution and expressed as cytokine concentration per milliliter of ELF.
In situ hybridization
All solutions were treated with 0.1% DEPC, and glassware was baked overnight at 350 C before use. Plasmids containing TNFα, IL-1β, and IL-8 cDNA inserts were linearized by restriction digestion, and sense and antisense digoxigenin (DIG)-labeled RNA probes were synthesized by in vitro transcription using a commercial kit (Boehringer Mannheim, Indianapolis, IN) according to the manufacturer's instructions. Labeled probes were ethanol precipitated and dissolved in ultrapure water. Probe concentration was determined by dot blot hybridization and subsequent immunologic detection using components of the DIG Nucleic Acid Detection Kit (Boehringer Mannheim).
Sections of paraffin-embedded lung tissue (5 µm) were cut, transferred to Superfrost Plus slides (Fisher Scientific, Pittsburgh, PA), deparaffinized in xylene, and rehydrated through a series of graded ethanol baths to ultrapure water. Except where otherwise indicated, steps preceding hybridization were carried out at room temperature, and slides were rinsed with ultrapure water between steps. Tissue sections were hydrolyzed in 0.2 N HCl for 20 minutes, incubated in 0.15 M triethanolamine (pH 7.4) for 15 minutes, and transferred to 0.3% (v/v) Triton-X in PBS for 5 minutes. Tissues were permeabilized with 25 µg/ml proteinase K (Boehringer Mannheim) for 20 minutes at 37 C, acetylated with 0.25% (v/v) acetic anhydride in 0.1 M triethanolamine (pH 8.0), and incubated in 2× SSC for 10 minutes at 70 C. Sections were then dehydrated through a series of graded ethanol baths, air dried, and covered with 60 µl of hybridization buffer (50% deionized formamide, 7% dextran sulfate, 1× Denhardt's solution, 0.6 M NaCl, 0.05% SDS, 20 mM HEPES, 1 mM ethylenediaminetetraacetic acid, 1 mg/ml poly[A], and 250 µg/ml yeast tRNA) containing 180 ng of DIG-labeled antisense riboprobe specific for TNFα, IL-1β, or IL-8. Siliconized glass cover slips were applied and sealed with rubber cement, and hybridization was conducted overnight at 43 C. Negative control sections were hybridized with buffer containing an equal concentration of the corresponding sense riboprobe or with buffer containing no added riboprobe.
Nonspecifically bound riboprobe was removed by enzymatic digestion and stringency washes as follows: 2× SSC for 30 minutes at 42 C, 50% formamide/2× SSC for 20 minutes at 52 C, 25 µg/ml ribonuclease A (Sigma) in enzyme buffer (10 mM Tris-HCl, pH 8.0, 0.5 M NaCl) for 30 minutes at 37 C, 1× SSC for 15 minutes at 37 C, and 0.1× SSC for 15 minutes at 37 C. Tissue sections were blocked for 30 minutes with Tris-buffered saline (0.1 M Tris-HCl, pH 7.4, 0.15 M NaCl) containing 3% normal sheep serum and then covered with sheep anti-DIG/alkaline phosphatase conjugate (Boehringer Mannheim) diluted 1:500 in blocking buffer and incubated for 1 hour at room temperature. After washing three times for 5 minutes each in Tris-buffered saline (TBS), slides were incubated for 15 minutes in TBS containing 4 mM levamisole to inactivate endogenous alkaline phosphatases. The color substrates nitroblue tetrazolium and 5-bromo-4-chloro-indoyl phosphate were applied to sections, and slides were incubated in the dark for 18 hours at room temperature. After a final rinse in ultrapure water, tissues were counterstained with nuclear fast red (Vector Laboratories, Burlingame, CA) and mounted with aqueous medium.
Morphometric analysis
Cells staining positive for cytokine mRNA in lung tissue were quantified using a combination of light microscopy and digital image analysis. For each calf, images of a minimum of three microscopic fields from each of at least three tissue sections (a minimum of nine fields/animal) were electronically captured using an Eclipse E800 microscope (Nikon, Melville, NY) equipped with a CoolCam 2000 digital video camera (Cool Camera Company, Atlanta, GA). Nonoverlapping fields were selected at random along a line defining the long axis of the tissue section. Positively stained cells were identified by light microscopy. Image-Pro Plus image analysis software (Media Cybernetics, Silver Spring, MD) was used to mark and count stained cells in digital images and to calculate the surface area of tissue fields analyzed. Results were expressed as the number of positively staining cells per square millimeter of tissue.
Cells staining positive for cytokine mRNA in lung tissue were classified according to cell type. Fifty stained cells in each of at least three light microscopic fields per tissue section (three sections/animal) were identified by cell type on the basis of cell morphology and histologic location. Fields were selected as for morphometric analysis; additional nonoverlapping fields were examined as needed to type a minimum of 450 cells/animal. Data from all calves in a given group were combined, and results were expressed in terms of the percentage of total staining cells.
Statistical analysis
All values were expressed as the mean ± SEM. Data from all groups were analyzed using the Kruskal–Wallis test, and individual groups were compared with the Mann–Whitney U-test. Differences were considered significant when P < 0.05.
Results
Kinetics of cytokine mRNA expression in BAL cells
Northern analysis was used to characterize inflammatory cytokine gene expression within airways during the first 24 hours of experimental pneumonic pasteurellosis. Steady-state levels of TNFα, IL-1β, and IL-8 mRNA in BAL cells collected at 2, 4, 8, 16, and 24 hours PI were compared with those in cells from mock-infected control calves (Fig. 1). For all three cytokines, postinfection mRNA levels were significantly higher than those of controls (P < 0.05), and maximal levels occurred within 4 hours of disease onset. Peak levels of TNFα mRNA occurred at 2 hours PI, and peak levels of IL-1β and IL-8 mRNA occurred at 4 hours PI. At maximum expression, levels of IL-1β and IL-8 mRNA were roughly twofold greater than those of TNFα mRNA. Expression of TNFα and IL-1β mRNA declined to control levels by 8 hours PI, but IL-8 mRNA levels were significantly increased throughout the 24-hour study period (P < 0.05). To rule out the possibility that experimental results were influenced by preexisting differences in levels of gene expression between the groups of calves euthanatized at various time points, preinfection BAL cells collected from the right lung of all calves were subjected to identical northern analysis. Results showed no significant differences in preinfection cytokine mRNA expression among groups (data not shown).
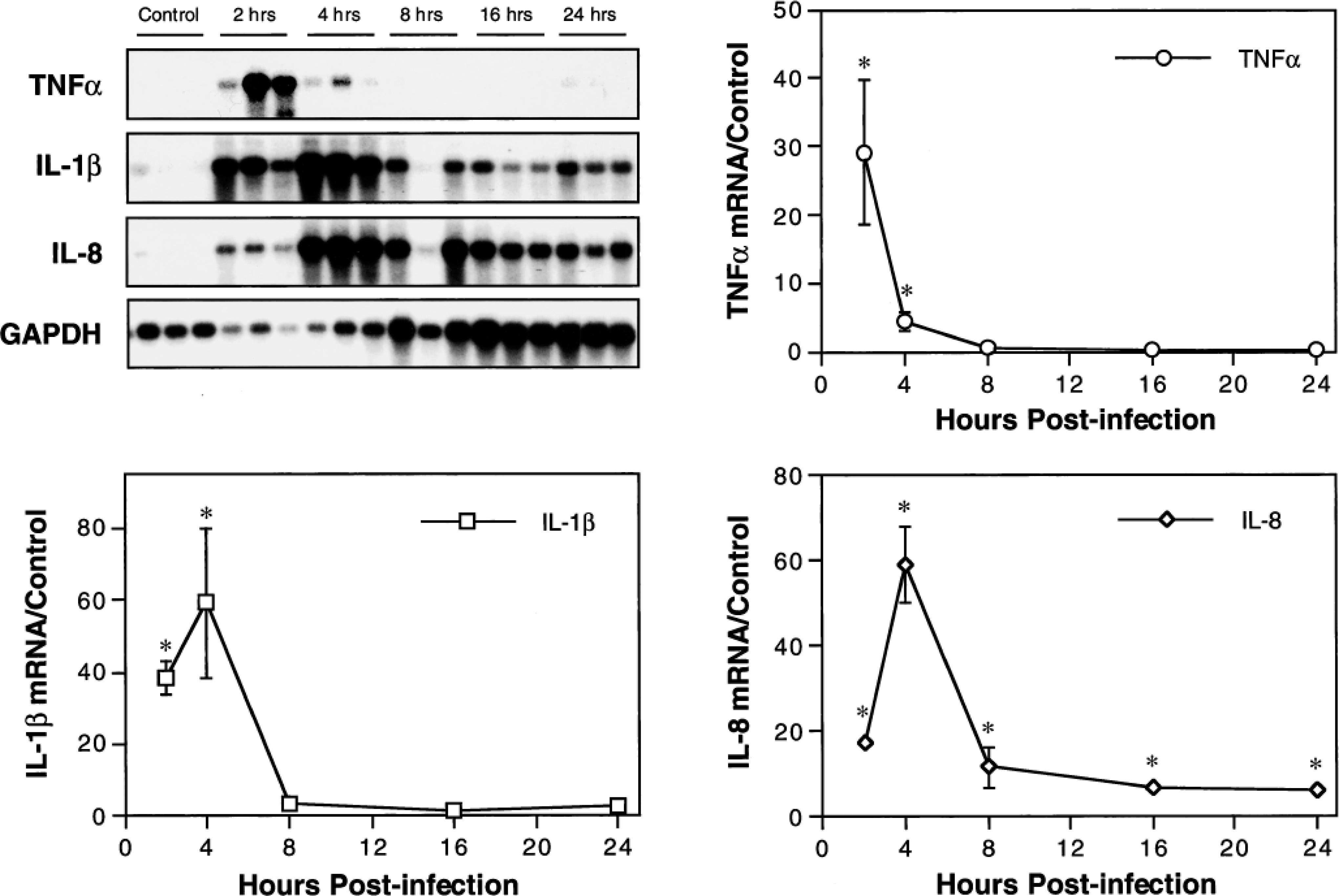
Kinetics of TNFα, IL-1β, and IL-8 mRNA expression in BAL cells from calves with acute pneumonic pasteurellosis. Inflammatory cytokine gene expression was quantitated by densitometric analysis of northern blots. For each cytokine, data were normalized to the expression of GAPDH mRNA and presented relative to mean normalized expression in BAL cells from mock-infected control calves. Values represent the mean ± SEM (n = 3). ∗P < 0.05 compared with mock-infected controls.
Kinetics of cytokine mRNA expression in pneumonic lung
Northern analysis was used to characterize inflammatory cytokine gene expression within pulmonary lesions of pneumonic pasteurellosis during the first 24 hours of disease. Steady-state levels of TNFα, IL-1β, and IL-8 mRNA in lesional lung tissues collected at 2, 4, 8, 16, and 24 hours PI were compared with those in grossly normal lung tissues from mock-infected control calves (Fig. 2). For all three cytokines, postinfection mRNA levels were significantly higher than those of controls (P < 0.05), and peak levels occurred within 8 hours of disease onset. At maximum expression, levels of IL-1β and IL-8 mRNA were roughly 5- and 60-fold greater, respectively, than those of TNFα mRNA. By 24 hours PI, mRNA for each of the cytokines had declined to control (TNFα and IL-1β) or near-control (IL-8) values. To address the possibility that experimental results were influenced by differences in gene expression among the groups due to factors unrelated to P. haemolytica infection, tissues collected from the unaffected right lung of all calves at necropsy were subjected to identical northern analysis. Results showed no significant differences in cytokine mRNA levels among groups (data not shown).
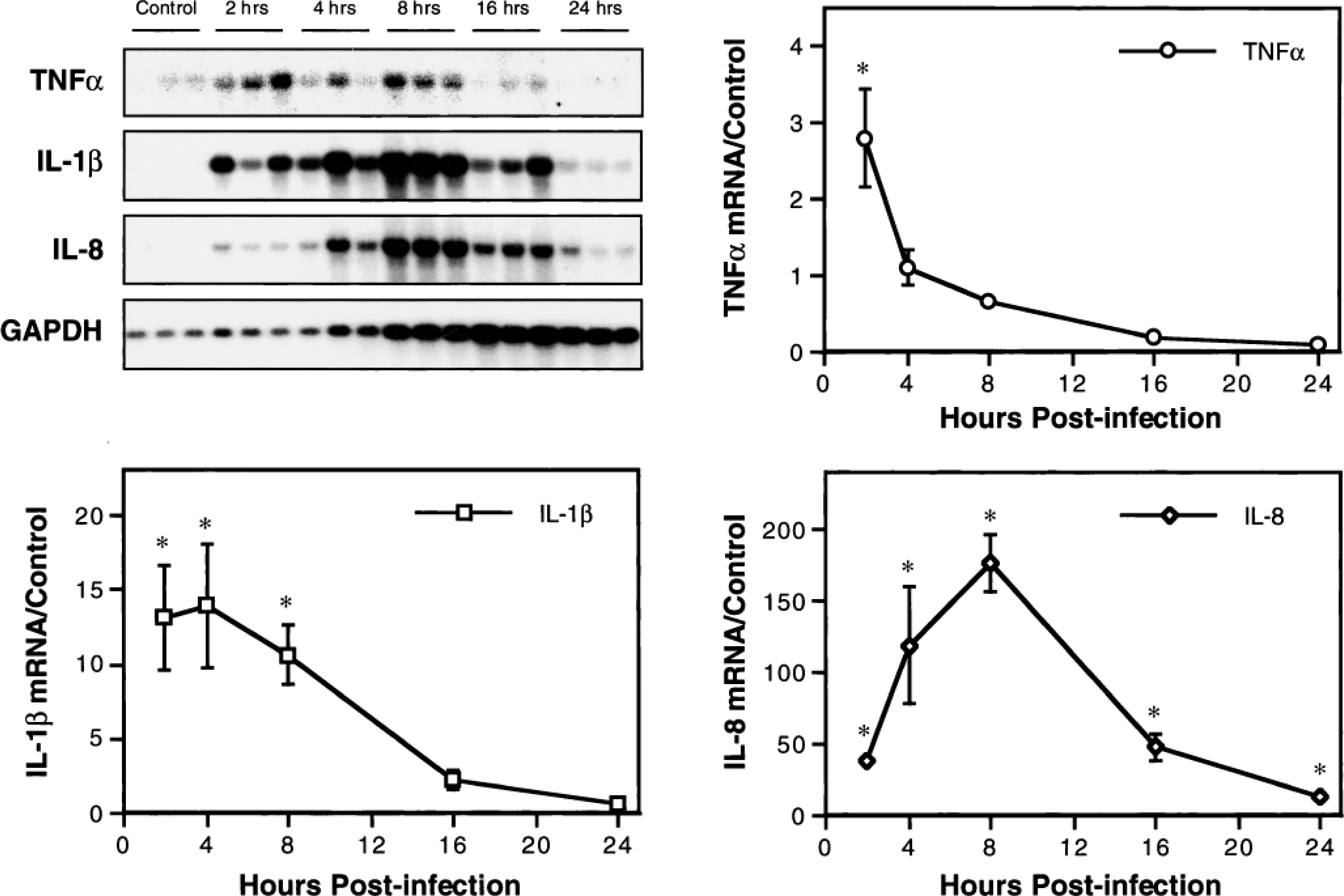
Kinetics of TNFα, IL-1β, and IL-8 mRNA expression in the lungs of calves with acute pneumonic pasteurellosis. Inflammatory cytokine gene expression was quantitated by densitometric analysis of northern blots. For each cytokine, data were normalized to the expression of GAPDH mRNA and presented relative to mean normalized expression in lung tissues from mock-infected control calves. Values represent the mean ± SEM (n = 3). ∗P < 0.05 compared with mock-infected controls.
Although overall kinetic patterns in lung lesions were similar to those in BAL cells, some important differences were observed. Upregulation of TNFα mRNA was 10-fold greater in BAL cells than in lung tissue, suggesting that these cells constitute the major cellular source of TNFα in affected lungs. At 2 hours PI, the time at which TNFα mRNA levels were highest in both samples, BAL cells are comprised largely of alveolar macrophages. 43 This finding provides indirect evidence that alveolar macrophages are important sources of TNFα in P. haemolytica-infected bovine lung. Similarly, upregulation of IL-1β gene expression was fourfold greater in BAL cells than in lung lesions, suggesting that BAL cells are important sources of IL-1β within affected lung. Because BAL cells consist largely of neutrophils at 4–8 hours PI, 43 the period during which peak IL-1β mRNA levels were observed, this finding provides indirect evidence that neutrophils are important pulmonary sources of IL-1β in BPP. Upregulation of IL-8 mRNA was threefold greater in lung lesions than in BAL cells, suggesting that pulmonary cell types not present in BAL fluid produce significant amounts of IL-8.
Kinetics of cytokine secretion in ELF
ELISAs were used to characterize inflammatory cytokine secretion within airways. Concentrations of immunoreactive TNFα, IL-1β, and IL-8 in BAL fluid collected prior to infection and at 2, 4, 8, 16, and 24 hours PI were compared with those in BAL fluid from mock-infected controls. Urea was used as an endogenous marker of dilution to calculate the extent to which ELF was diluted during BAL in each calf. Measured values were then corrected for dilution and expressed as cytokine concentration per milliliter of ELF (Fig. 3). We consider this the best practical method by which to control for variable dilution of ELF constituents during BAL. Samples collected prior to inoculation with P. haemolytica and those from mock-infected controls contained no detectable TNFα, IL-1β, or IL-8. Concentrations of all three cytokines were significantly increased by 2 hours PI and remained elevated throughout the 24-hour study period (P < 0.05). Peak concentrations of TNFα (6.5 ± 0.4 ng/ml), IL-1β (48.0 ± 11.8 ng/ml), and IL-8 (5.4 ± 1.3 µg/ml) occurred at 4, 8, and 16 hours PI, respectively.
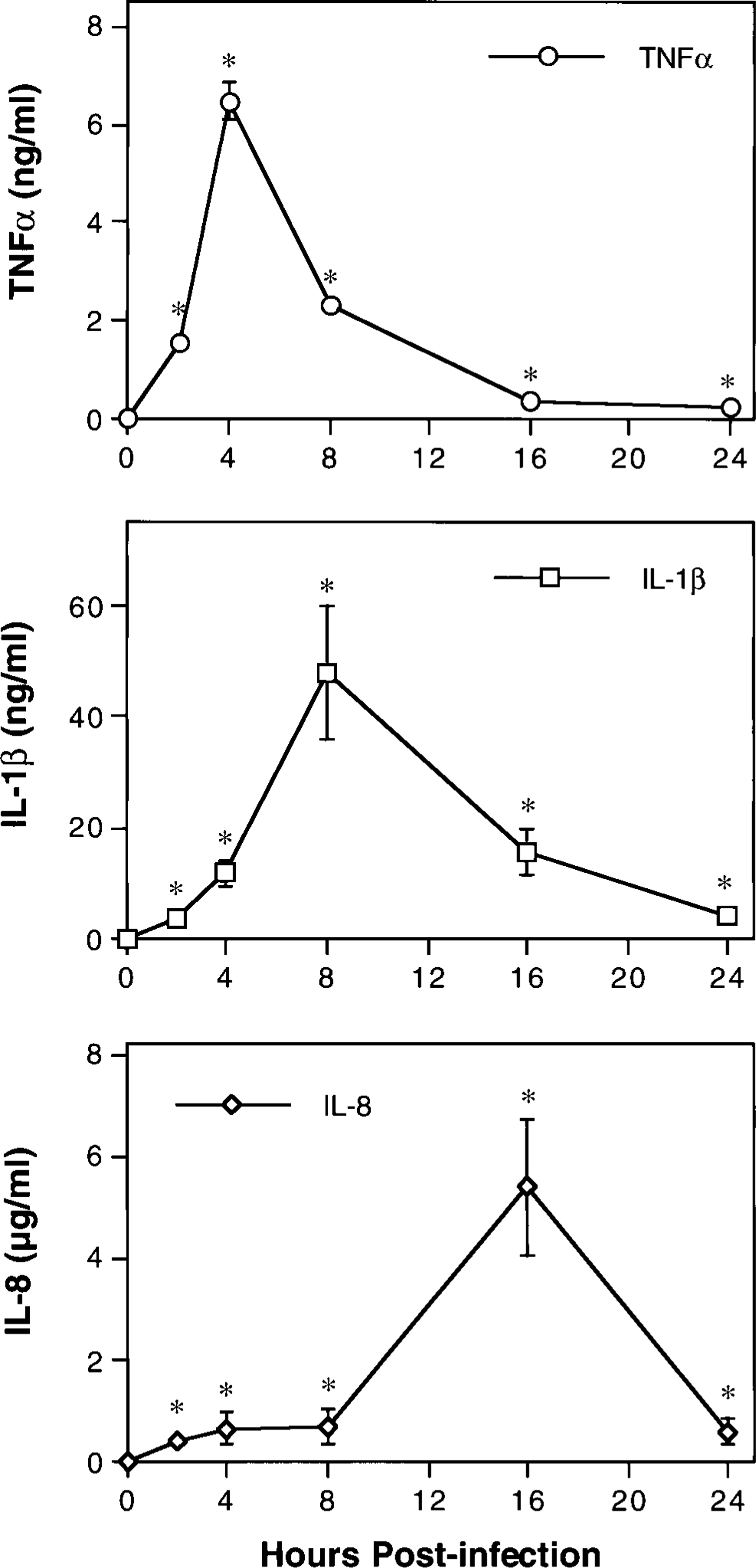
Kinetics of TNFα, IL-1β, and IL-8 secretion in the airways of calves with acute pneumonic pasteurellosis. Immunoreactive cytokines in BAL fluid were quantitated by antigen-capture ELISA. Urea was used as an endogenous marker of dilution to calculate the extent to which ELF was diluted during the BAL procedure in each calf. Measured values were corrected for dilution and expressed as cytokine concentration per milliliter of ELF. Values represent the mean ± SEM (n = 3). ∗P < 0.05 compared with mock-infected controls.
Kinetics of cytokine expression in pneumonic lungs
ELISAs were used to characterize the expression of inflammatory cytokines within pulmonary lesions of pneumonic pasteurellosis. Concentrations of immunoreactive TNFα, IL-1β, and IL-8 in extracts of pneumonic lung collected at 2, 4, 8, 16, and 24 hours PI were compared with those in extracts of grossly normal lung from mock-infected controls (Fig. 4). Control extracts contained 68.0 ± 6.0 ng/ml IL-8 but no detectable TNFα or IL-1β. As in ELF, concentrations of all three cytokines were significantly increased in tissue extracts by 2 hours PI and remained elevated throughout the 24-hour study period (P < 0.05). Peak concentrations of TNFα (131 ± 18 pg/ml) occurred at 8 hours PI, and peak concentrations of IL-1β (56.9 ± 6.5 ng/ml) and IL-8 (3.4 ± 0.6 µg/ml) occurred at 16 hours PI. Except that expression of TNFα and IL-1β was more prolonged in lung extracts than in ELF, overall kinetic patterns were similar. Relative concentrations in the two samples varied among cytokines, however. Whereas maximal concentrations of IL-1β and IL-8 were on roughly the same order of magnitude in either sample, TNFα concentrations were 50-fold higher in ELF than in lung extracts. This observation was consistent with the results of northern analysis and provides further evidence that TNFα expression in BPP occurs primarily within airways.
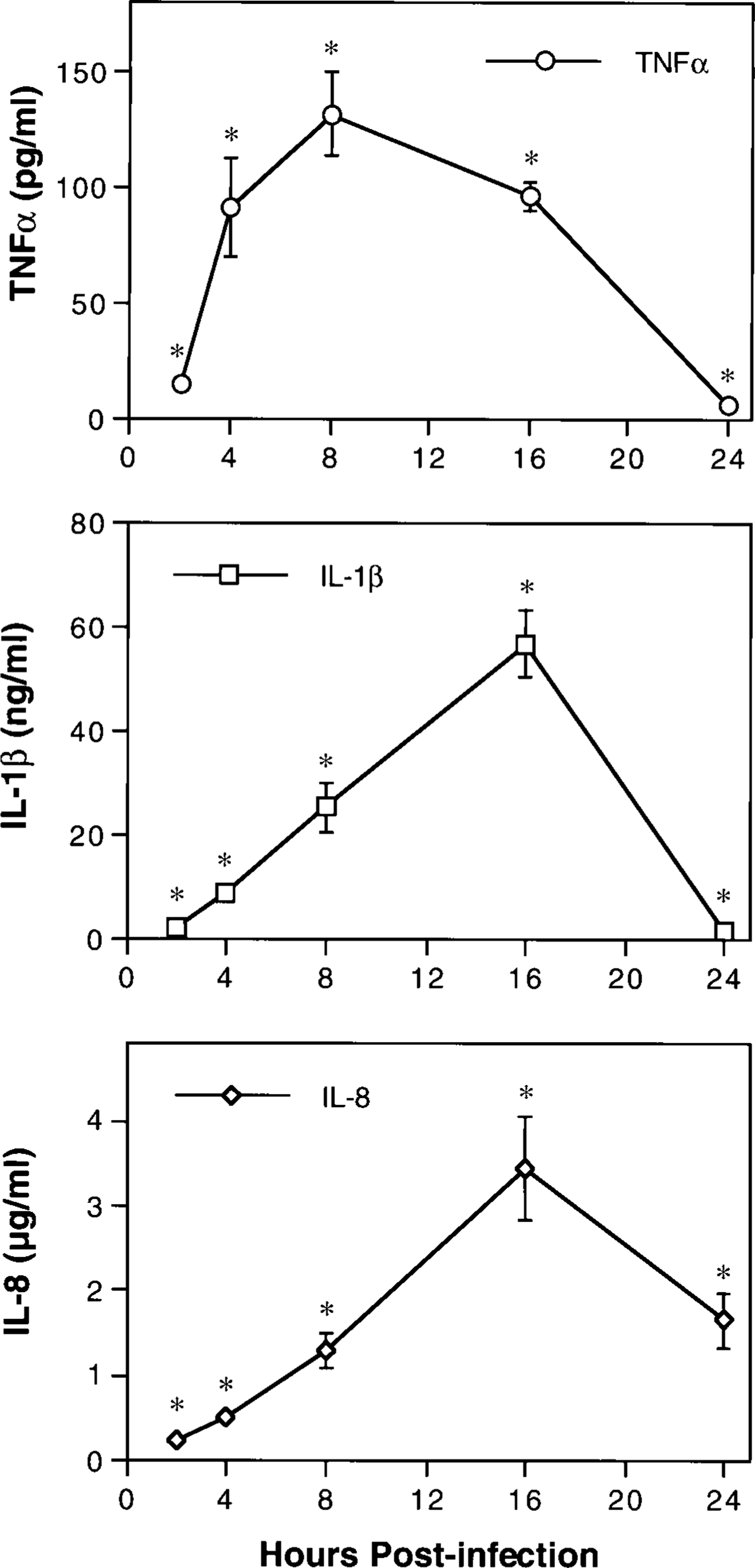
Kinetics of TNFα, IL-1β, and IL-8 expression in the lungs of calves with acute pneumonic pasteurellosis. Immunoreactive cytokines in extracts of lesional lung tissue were quantitated by antigen-capture ELISA. Values represent the mean ± SEM (n = 3). Extracts of mock-infected control lung tissue contained 68.0 ± 6.0 ng/ml IL-8 but no detectable TNFα or IL-1β. ∗P < 0.05 compared with controls.
In situ hybridization analysis of pulmonary cytokine mRNA expression
In situ hybridization with nonradioactive DIG-labeled riboprobes was used to localize inflammatory cytokine mRNA in infected and mock-infected lung tissues. Cells staining positive for TNFα, IL-1β, and IL-8 mRNA were enumerated by quantitative morphometric analysis using a combination of light microscopy and digital image analysis (Fig. 5). Tissue sections from mock-infected controls contained 2.6 ± 0.3 cells/mm2 staining positive for TNFα mRNA and no detectable staining for IL-1β or IL-8 gene expression. In postinfection lung, the number of cells expressing mRNA specific for each of the cytokines was significantly increased throughout the 24 hour period following inoculation with P. haemolytica (P < 0.05). Peak numbers of cells expressing TNFα mRNA (15.9 ± 1.4 cells/mm2) occurred at 2 hours PI, and peak numbers of cells expressing mRNA specific for IL-1β (640 ± 208 cells/mm2) and IL-8 (7,392 ± 2,519 cells/mm2) occurred at 8 hours PI. These data were consistent with the results of both northern analysis and ELISA and provide further evidence that IL-8 is expressed to the greatest extent within P. haemolytica-infected lung, whereas TNFα is least expressed.
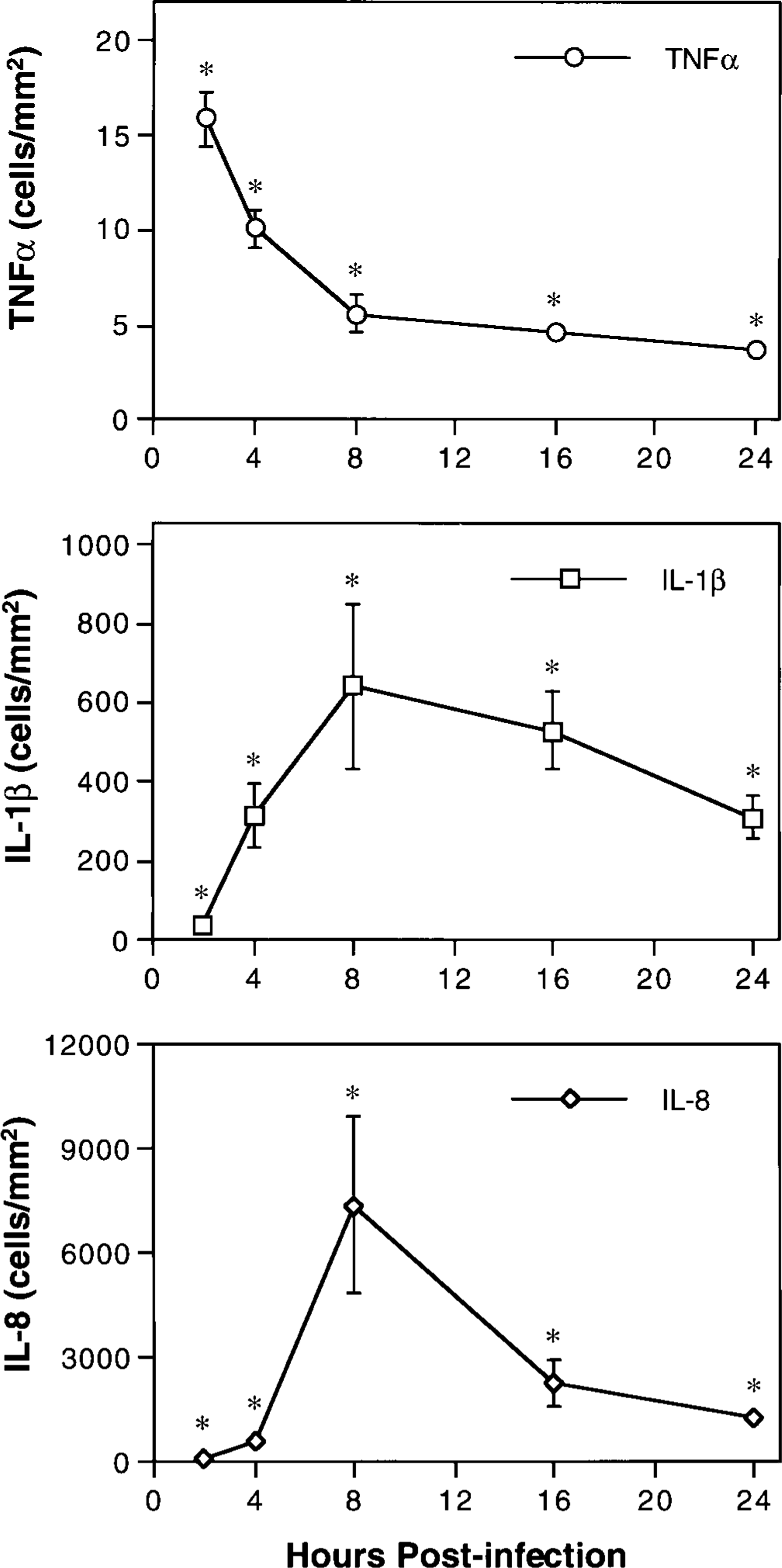
Changes over time in the number of cells expressing TNFα, IL-1β, and IL-8 mRNA in the lungs of calves with acute pneumonic pasteurellosis. Pulmonary expression of cytokine mRNA was localized by in situ hybridization. The number of positively staining cells per square millimeter of lung tissue was determined by quantitative morphometric analysis. Values represent the mean ± SEM (n = 3). Mock-infected control tissues contained 2.6 ± 0.3 cells/mm2 staining positive for TNFα mRNA and no detectable staining for IL-1β or IL-8 mRNA. ∗P < 0.05 compared with controls.
In situ hybridization with the TNFα antisense riboprobe revealed that pulmonary expression of TNFα mRNA was restricted to alveolar macrophages in both infected and mock-infected lungs (Fig. 6). TNFα was the only cytokine for which mRNA-expressing cells were detected in control tissues. No staining was observed in lung tissues hybridized with the TNFα sense riboprobe (Fig. 7). Hybridization of tissues with the IL-1β antisense probe showed that at 2 hours PI, expression of IL-1β mRNA was localized to alveolar macrophages and cells within the alveolar septum, likely intravascular and/or interstitial macrophages (Fig. 8). Definitive identification of cell types at later time points was hindered by suboptimal tissue morphology, but comparison of serial sections routinely stained with hematoxylin and eosin confirmed that neutrophils within exudate in alveolar spaces and the lumina of bronchi and bronchioles became the dominant cellular source of IL-1β mRNA within 4–8 hours of disease onset (Fig. 9). Hybridization of lung tissues with the IL-1β sense riboprobe yielded no detectable staining (Fig. 10). At 2 and 4 hours PI, IL-8 mRNA was detected in alveolar macrophages, macrophages within the alveolar septum, bronchial epithelial cells, bronchiolar epithelial cells, and neutrophils (Fig. 11). At 8 hours PI and later time points, the majority of staining for IL-8 mRNA occurred in neutrophils within alveolar exudate and exudate in the lumina of bronchioles and bronchi (Fig. 12). Staining was also observed in fibroblasts within interlobular septa. No staining was observed in tissues hybridized with the IL-8 sense riboprobe (Fig. 13). A quantitative analysis of the cell types represented among cells staining positive for TNFα, IL-1β, and IL-8 at each time point was performed. The results for IL-1β and IL-8 mRNA are summarized in Fig. 14. Expression of TNFα mRNA was restricted to alveolar macrophages.
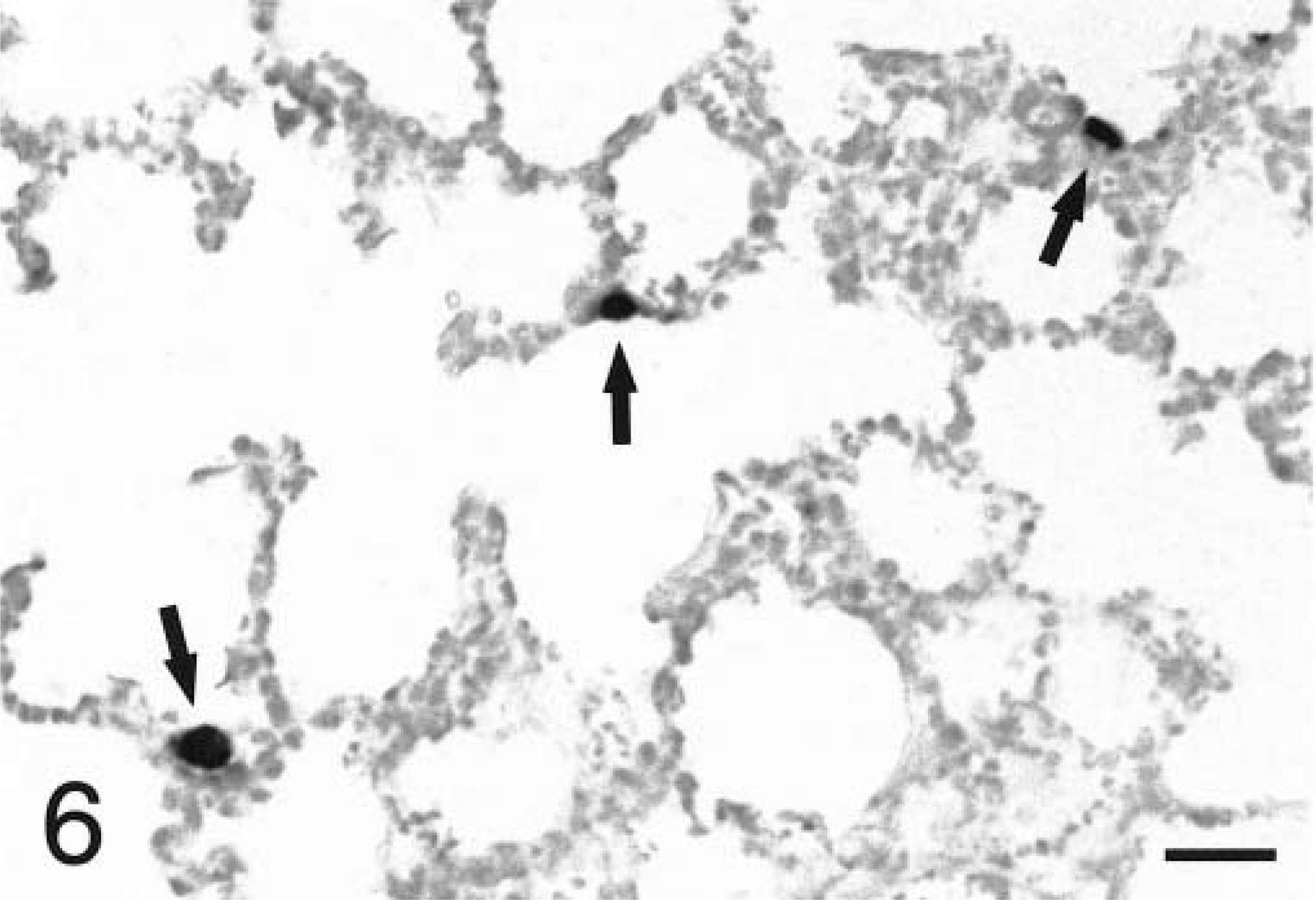
Lung; P. haemolytica-inoculated calf, 2 hours PI. Blue-black cytoplasmic staining indicates that alveolar macrophages (arrows) express TNFα mRNA in the peracute phase of pneumonic pasteurellosis. In situ hybridization with antisense probe, nuclear fast red counterstain. Bar = 25 µm.
Lung; P. haemolytica-inoculated calf, 2 hours PI. No staining is evident in tissues hybridized with TNFα sense probe. In situ hybridization, nuclear fast red counterstain. Bar = 25 µm.
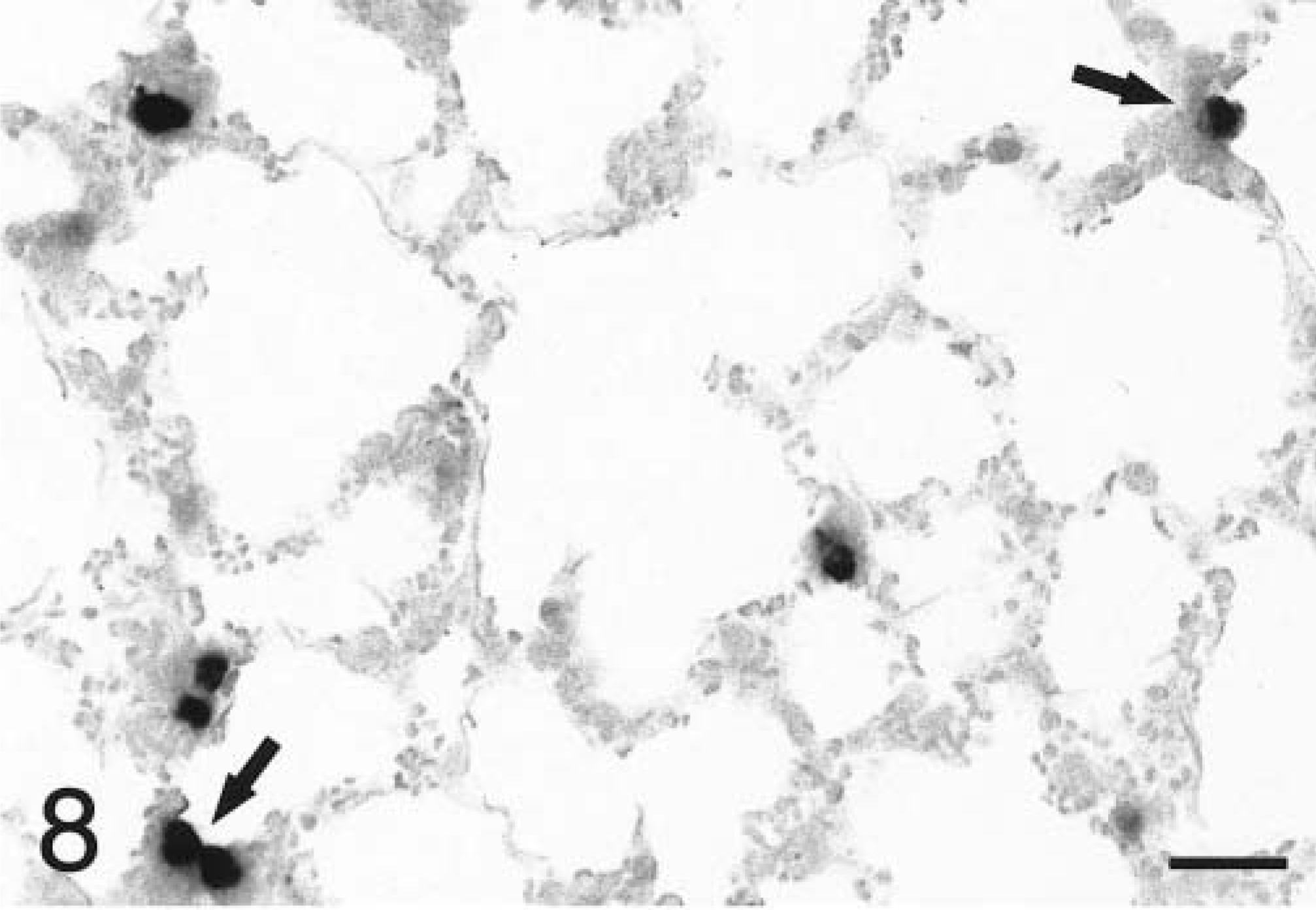
Lung; P. haemolytica-inoculated calf, 2 hours PI. Alveolar macrophages (arrows) and cells within the alveolar septum, likely intravascular macrophages, express IL-1β mRNA in the peracute phase of pneumonic pasteurellosis. In situ hybridization with antisense probe, nuclear fast red counterstain. Bar = 25 µm.
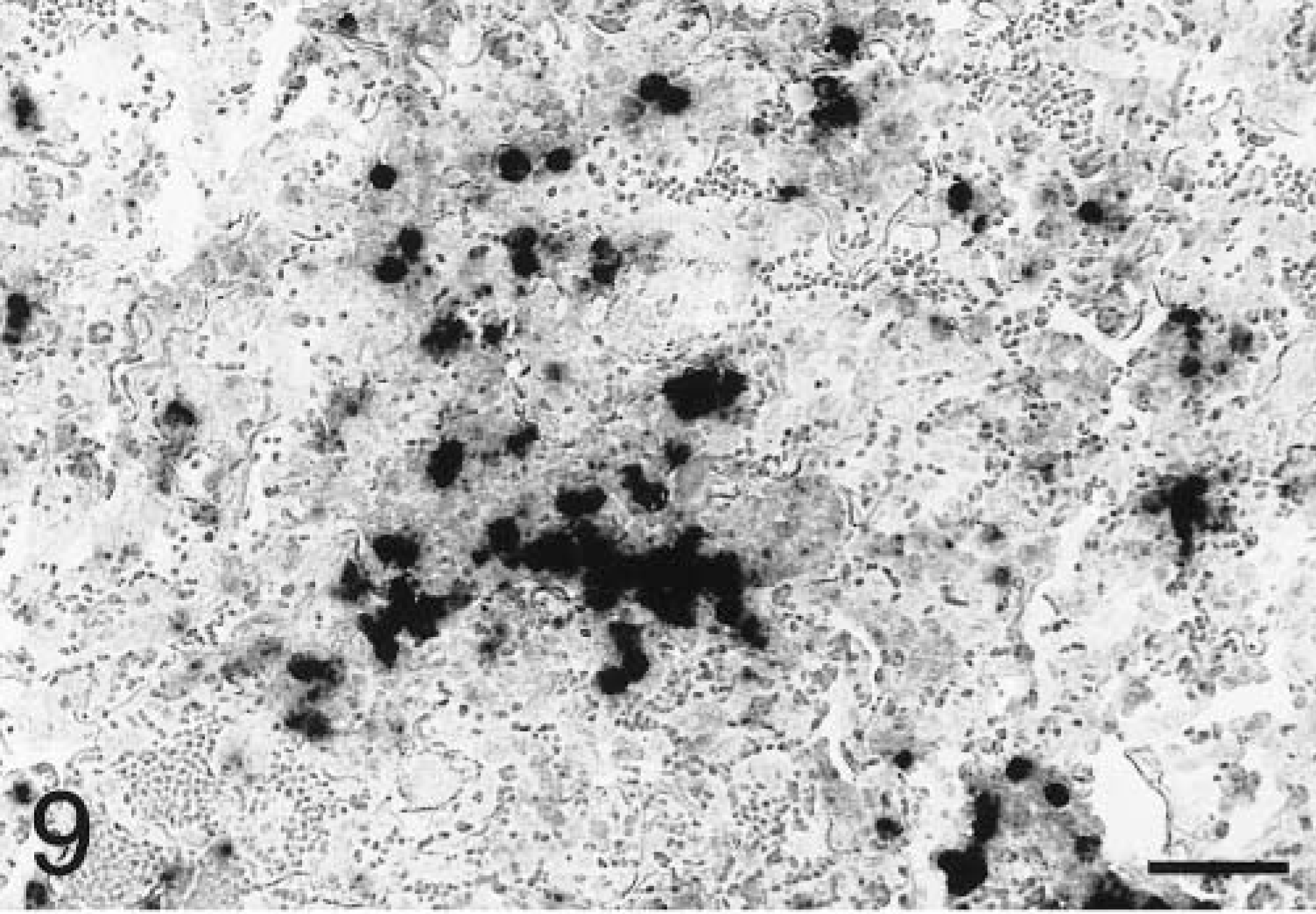
Lung; P. haemolytica-inoculated calf, 8 hours PI. Neutrophils within alveolar exudate express IL-1β mRNA in the lesions of acute pneumonic pasteurellosis. In situ hybridization with antisense probe, nuclear fast red counterstain. Bar = 50 µm.
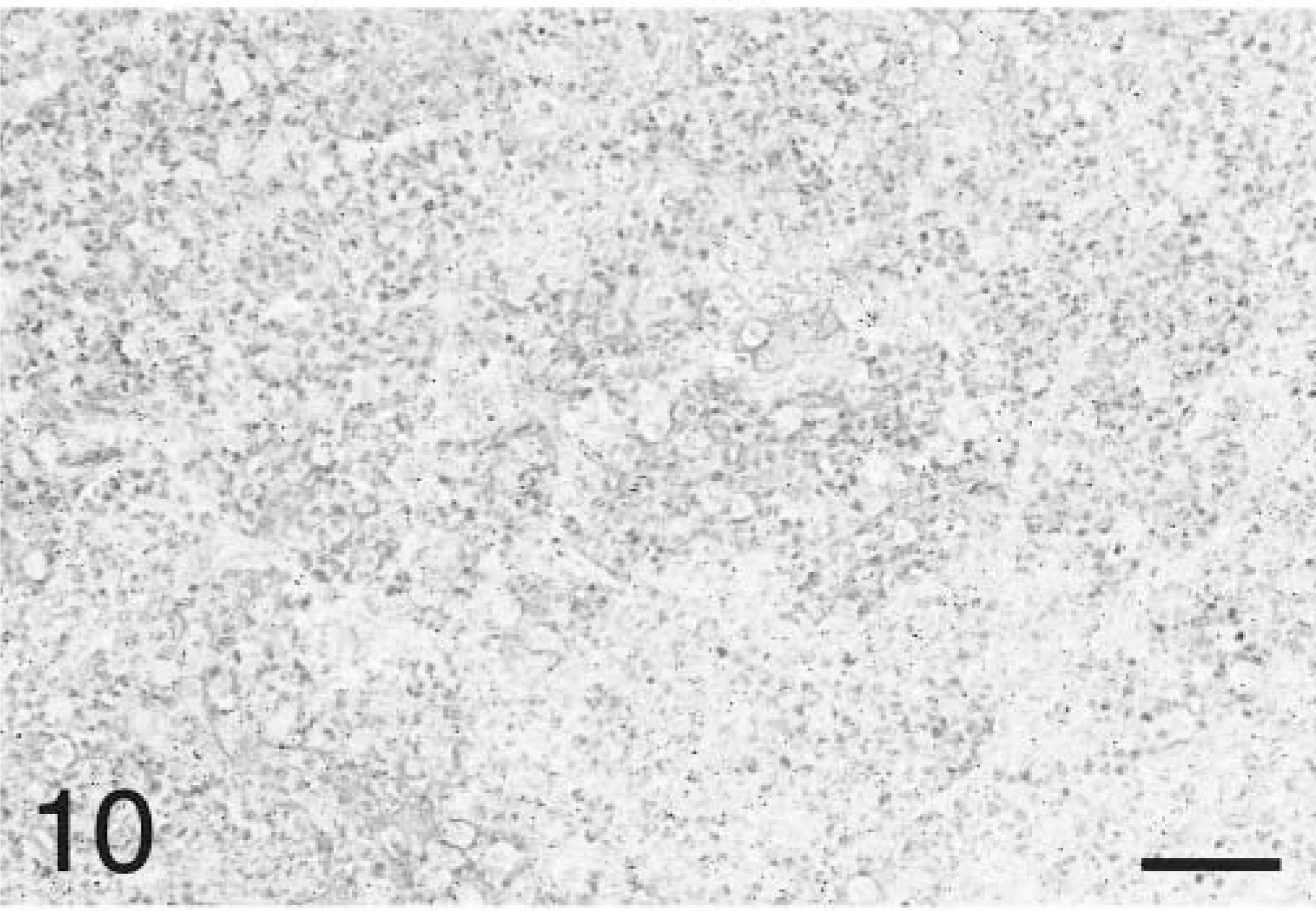
Lung; P. haemolytica-inoculated calf, 8 hours PI. No staining is evident in pneumonic lung tissues hybridized with IL-1β sense probe. In situ hybridization, nuclear fast red counterstain. Bar = 50 µm.
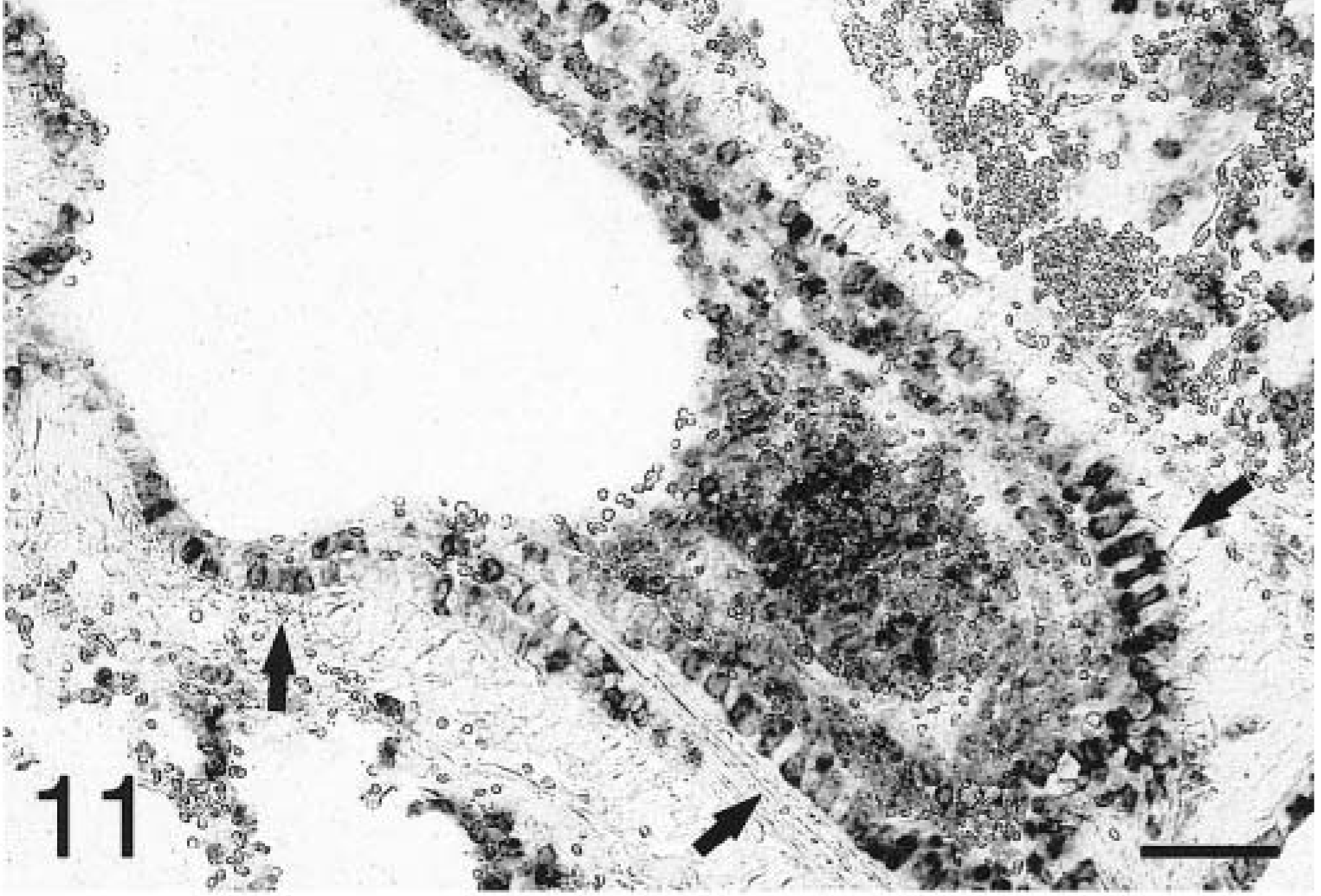
Lung; P. haemolytica-inoculated calf, 4 hours PI. Bronchiolar epithelial cells (arrows) and neutrophils within alveolar exudate and the bronchiolar lumen express IL-8 mRNA in the peracute lesions of pneumonic pasteurellosis. In situ hybridization with antisense probe, nuclear fast red counterstain. Bar = 50 µm.
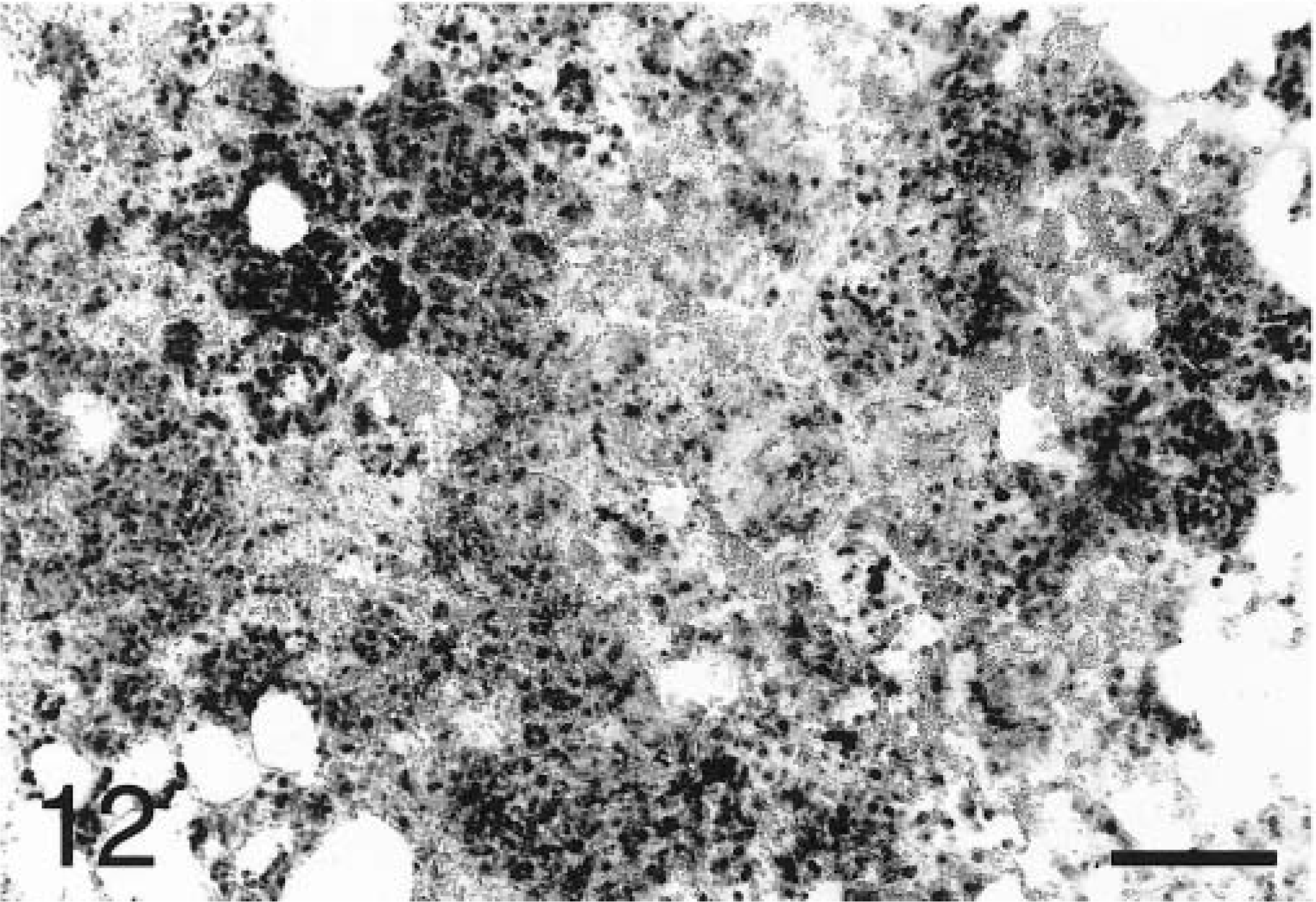
Lung; P. haemolytica-inoculated calf, 8 hours PI. Extensive staining of neutrophils within alveolar exudate reflects widespread expression of IL-8 mRNA in the acute lesions of pneumonic pasteurellosis. In situ hybridization with antisense probe, nuclear fast red counterstain. Bar = 100 µm.
Lung; P. haemolytica-inoculated calf, 8 hours PI. No staining is evident in pneumonic tissues hybridized with IL-8 sense probe. In situ hybridization, no counterstain. Bar = 50 µm.
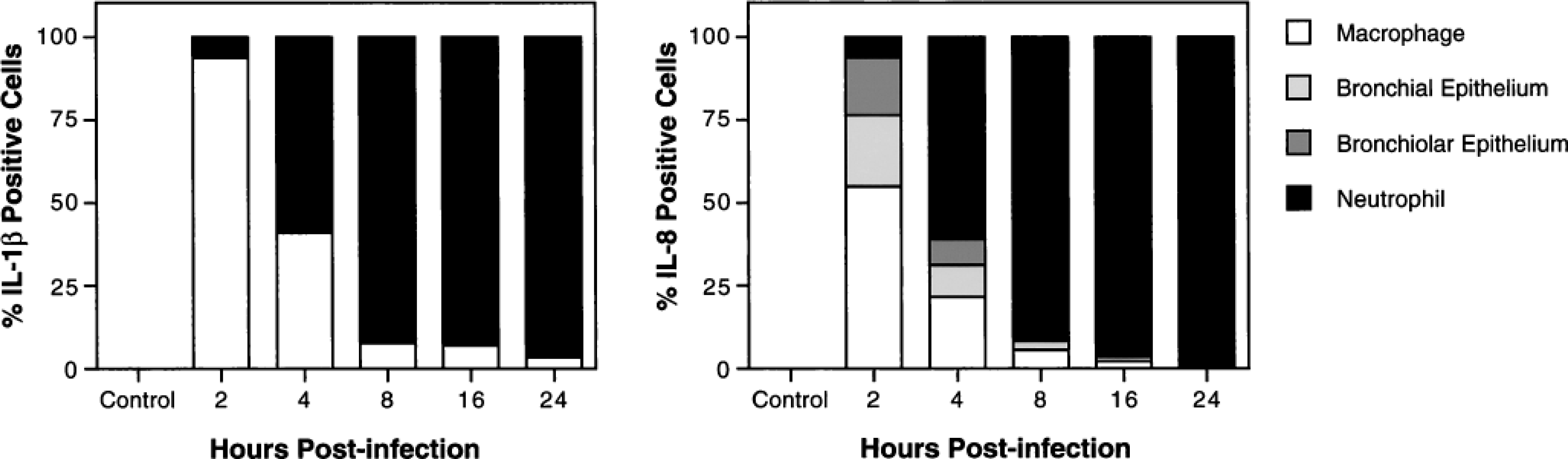
Cell types expressing IL-1β and IL-8 mRNA in the lungs of calves with pneumonic pasteurellosis. Pulmonary expression of cytokine mRNA was localized by in situ hybridization. Positively staining cells were identified by light microscopy on the basis of cell morphology and histologic location.
Discussion
Recent investigations implicate inflammatory cytokines in the pathogenesis of BPP. In this study, early and sustained expression of TNFα, IL-1β, and IL-8 mRNA and proteins was demonstrated within the airways and lung lesions of cattle experimentally infected with P. haemolytica. By contrast, samples collected from mock-infected control animals and the unaffected lung of infected cattle exhibited little or no such expression. These findings are consistent with earlier reports identifying an association between lung pathology and increased pulmonary expression of inflammatory cytokines in BPP. 8,45 However, because the present study addresses the peracute and acute phases of disease development, it provides stronger evidence in support of a causative role for TNFα, IL-1β, and IL-8 in disease pathogenesis. This study also extends previous work by characterizing the kinetics of pulmonary cytokine expression during the initial 24 hours of disease, comparing patterns of expression in airways with those in lung parenchyma, and identifying the major cellular sources of inflammatory cytokines within pneumonic lung.
The results obtained by northern analysis, ELISA, and in situ hybridization were closely correlated. Expression of TNFα, IL-1β, and IL-8 in the airways and lung parenchyma of infected calves was significantly upregulated at both the gene and protein levels. In all samples, IL-8 mRNA and proteins were upregulated to the greatest extent and those for TNFα were upregulated the least. Northern blots and ELISAs suggested that TNFα gene and protein expression occurred predominantly within airways, and in situ hybridization studies confirmed that mRNA expression was localized to alveolar macrophages. Expression of IL-1β and IL-8 genes and proteins, by contrast, was more generalized. Alveolar and interstitial macrophages were important early sources of both IL-1β and IL-8, and bronchial and bronchiolar epithelial cells were significant sources of IL-8 in the first 4 hours PI. Neutrophils, however, became the dominant source of both IL-1β and IL-8 within 4–8 hours of disease onset. These findings establish the existence of a spatial and temporal association between pulmonary expression of inflammatory cytokines and acute lung pathology, indirectly supporting the hypothesis that cytokines contribute to inflammatory lung injury in BPP.
In previous studies, pulmonary cytokine expression was measured between 2 and 4 days following endobronchial inoculation of P. haemolytica and changes over time were not addressed. 8,45 In this study, we demonstrated that cytokine upregulation occurs much earlier in the course of disease development than was previously recognized. TNFα, IL-1β, and IL-8 mRNA and proteins were significantly increased in all samples by 2 hours PI. Although kinetic patterns varied, peak levels of mRNA for all cytokines were achieved within 8 hours PI, and peak cytokine concentrations occurred within 16 hours PI. By 24 hours PI, mRNA specific for all three cytokines declined to near control values. Although cytokine concentrations in airways and lung lesions remained elevated throughout the study period, they were significantly decreased at 24 hours PI as compared with peak values achieved earlier. These observations suggest that TNFα, IL-1β, and IL-8 may exert their greatest pathogenetic effects within 16 hours of disease onset.
The specific mechanisms, if any, by which inflammatory cytokines mediate lung injury in BPP await clarification. It is reasonable to assume, however, that their biologic effects in bovine lung parallel those recognized in other systems. In most mammalian models, TNFα, IL-1β, and IL-8 are central components of a complex cytokine network that initiates, amplifies, and sustains the inflammatory response in tissue. Available evidence also supports the importance of this network in coordinating acute inflammatory responses within the lung. 27,35 TNFα and IL-1β are pleiotropic early response mediators that establish cytokine cascades through autocrine and paracrine activation of a broad array of cell types. 20,31 They initiate neutrophil transmigration and activation by upregulating the expression of adhesion molecules on neutrophils and microvascular endothelium. 7,38 Though not directly chemotactic for neutrophils, both TNFα and IL-1β induce the secretion of IL-8, the most potent neutrophil chemotactic and activating factor, and other chemokines by a variety of cell types. 4,27,35,37–39 In addition to their roles in neutrophil recruitment, TNFα, IL-1β, and especially IL-8 promote neutrophil-mediated tissue injury by stimulating neutrophil degranulation and the extracellular release of arachidonic acid metabolites, toxic oxygen radicals, and proteolytic enzymes. 6,10,13,28–30
Our findings indicate that IL-8 is the dominant inflammatory cytokine expressed within the lungs during the acute phase of BPP. Throughout the 24 hour period following inoculation of P. haemolytica, IL-8 was expressed in much greater quantities than either TNFα or IL-1β. At 2 hours PI, the earliest time point studied, concentrations of IL-8 in ELF were already roughly 250- and 100-fold greater than those of TNFα and IL-1β, respectively. In extracts of lesional lung parenchyma at the same time point, concentrations of IL-8 were roughly 17,000- and 100-fold greater than those of TNFα and IL-1β, respectively. Previously, abundant pulmonary expression of IL-8 has been considered a downstream event that is dependent, at least in part, on the prior secretion of early response cytokines such as TNFα and IL-1β. 18,27,35 The findings reported here do not exclude the possibility that a similar cascade of interactions is a necessary prerequisite for IL-8 production in BPP, but do indicate that the critical events in that cascade must occur well before 2 hours PI. These observations, together with the results of the kinetic analyses, have at least two important implications for therapeutic strategies based upon modulation of inflammatory cytokines. First, pharmacologic agents that inhibit the synthesis of IL-8 or antagonize its biologic effects are likely to be more effective in the management of BPP than those targeting only TNFα or IL-1β. Second, anti-cytokine agents may have to be administered very early in the course of disease, possibly even prior to colonization of the lung by P. haemolytica, to prevent or interrupt inflammatory lung injury.
This study demonstrated a correlation between early pulmonary expression of TNFα, IL-1β, and IL-8 and the development of acute BPP, further substantiating a role for these mediators in disease pathogenesis. Inflammatory cytokines may therefore represent drug targets that could be pharmacologically modulated in the management of this important disease of cattle. The results reported here, however, suggest that such strategies may have to be implemented very early in the course of disease if they are to be effective. Because pulmonary expression of IL-8 was much greater than that of TNFα and IL-1β at all time points studied, anti-cytokine agents targeting this mediator may be most useful in the prevention and treatment of BPP.
Footnotes
Acknowledgements
We thank Dr. T. Elsasser and the USDA for providing anti-TNFα antibodies, Dr. K. Heaney and Fort Dodge Animal Health for providing recombinant IL-1β, Dr. D. Godson and VIDO for providing recombinant TNFα, and Dr. M. Murtaugh for providing a human GAPDH cDNA. In addition, we are grateful to P. Ward, V. Lappi, and Dr. R. LaFleur for technical assistance. This research was supported by USDA-NRI competitive grant 95-37204-1963 and AVMA Foundation research grant 95-06.