
Abstract
The pathogenesis of porcine reproductive and respiratory syndrome virus (PRRSV) infection in ovary was studied in sexually mature, cycling, nonsynchronized gilts infected with the PRRSV 16244B, a virulent field strain. Previous studies have shown that PRRSV can be isolated from ovaries and is transplacentally passed from gilts to the fetuses. The cause of infertility following PRRSV infection is not known. In this study, we identified the tropism of PRRSV in ovarian tissue from experimentally infected gilts in samples collected between 7 and 21 days postinfection (DPI). Tissues were collected and examined by virus isolation, in situ hybridization (ISH), immunohistochemistry (IHC), and double labeling to identify PRRSV-infected cell types. PRRSV was isolated in ovarian follicles at 7 days DPI. The IHC and ISH indicated that PRRSV-positive cells in ovaries were predominantly macrophages, which were numerous in atretic follicles. No evidence of infection and/or perpetuation of PRRSV in ova was observed, indicating that the female gonad is an unlikely site of persistence. No alteration of the normal ovarian architecture that would support a possible role of PRRSV infection in porcine female infertility was observed.
Keywords
The arterivirus porcine reproductive and respiratory syndrome virus (PRRSV) has a single-stranded polyadenylated RNA genome of positive polarity and is now classified in the new family Arteriviridae, order Nidovirales. 8 24 34 Other viruses of this group are lactate dehydrogenase–elevating virus (LDV) of mice, equine arteritis virus, and simian hemorrhagic fever virus. 23 28 The PRRSV affects pigs of all ages and causes respiratory disease, infertility, early embryonic death, late-term abortion, and a high incidence of stillborn, mummified, and weak newborn pigs. PRRSV is known to infect individual animals for long periods of time, and during that time, the virus may be transmitted by direct, close animal–animal contact or through semen. 10–12, 26 38 Recently, studies of the pathogenesis of male and female reproductive system have been reported. We have previously reported that PRRSV infects germinal epithelial cells in the testis implicating a cause for male infertility. In addition, PRRSV has been shown to induce apoptosis in germ cells from testes, alveolar macrophages, and mononuclear cells from lymphoid tissues. 36 37 Moreover, it has been reported that PRRSV can be isolated from the ovary and may be responsible for episodes of female reproductive failure. 7 13 20 29–32, 39 The etiologic role of PRRSV in the ovary has not been elucidated, and little is known about the mechanisms of pathogenesis underlying ovarian infection by PRRSV.
The purpose of this study was to demonstrate the sites of PRRSV replication in the ovary of experimentally infected gilts to obtain a more complete understanding of the pathogenesis of PRRSV infections in gonads. Macrophages of the ovary have been shown to be involved in most ovarian events such as follicle development and atresia, ovulation, and corpus luteum formation and regression. 2 4 11 15 17 Evidence presented in this report demonstrates that PRRSV predominantly infects resident macrophages in the ovary.
Materials and Methods
Experimental design
Nine 6-month-old gilts of conventional pigs were obtained from a PRRSV-free, specific-pathogen-free herd. Serum was collected from all gilts before challenge and was tested and confirmed negative (S/P < 0.4) for the presence of PRRSV antibodies using a commercial PRRSV enzyme-linked immunosorbent assay (Idexx Laboratories, Portland, ME). The strain used for challenge was PRRSV 16244B, which was isolated in February 1997 from a case of porcine reproductive failure in Johnson County, Nebraska. 1 The inoculum was prepared after three passages in MARC-145 cells. Six gilts were inoculated intranasally with 1 ml of PRRSV (median tissue culture infective dose = 107/ml), which was delivered in two aliquots of 0.5 ml per nostril. Three uninfected pigs were inoculated in a similar manner with PRRSV-free MARC-145 cell culture supernatant. Groups of infected and control gilts were euthanatized and necropsied according to the following schedule. Two infected and one control gilt were necropsied at 7, 14, and 21 days postinfection (DPI), respectively, and in all cases reproductive tissues were removed and examined by histopathology, virus isolation (VI), in situ hybridization (ISH), and regular and double-labeling immunohistochemistry (IHC). Ovary samples for VI were frozen at −70 C. The remaining ovarian samples and other reproductive tissues were fixed in 10% neutral buffered formalin.
VI
The MARC-145 cell line was used for VI assays on the ovarian samples. The growth medium consisted of minimal essential medium, 10% fetal calf serum, and gentamicin (50 µg/ml). Tissue samples, consisting of representative areas of the entire ovary, were promptly homogenized in sterile plastic bags using a mechanical tissue homogenizer. Culture medium was added to achieve a 20% (w/v) suspension, which was clarified by centrifugation (2,000 × g, 15 minutes), and supernatants were filtered (0.22 µm). Supernatants were adsorbed on confluent monolayers of MARC-145 cells grown on six-well plates (No. 3506, Costar Co., Cambridge, MA) at 37 C for 1 hour and then removed, and 4 ml of fresh culture medium was added. The culture plates were then incubated at 37 C in a humidified 5% CO2–air atmosphere. Monolayers were examined for cytopathic effect (CPE) at 5–7 days postinoculation. If CPE was not evident after 7 days of incubation, a second passage was performed. The plates were frozen and thawed twice, the cell debris was removed by centrifugation at 600 × g for 10 minutes, and the supernatant was inoculated onto fresh monolayers of MARC-145 cells. The monolayers were observed for 7 additional days. If no CPE was evident after the additional 7 days of incubation, the cells were fixed and stained for indirect immunofluorescence (using PRRSV nucleocapside antigen–specific monoclonal antibody SDOW-17 25 as first antibody and goat anti-murine fluorescein isothiocyanate conjugate as second antibody), examined with a Leica DM IRB inverted epifluorescence microscope, and discarded if negative.
Preparation of tissues for histopathology
For histopathology, ISH, IHC, and double-labeling IHC, tissue sections were adhered to Superfrost/plus slides (Fisher Scientific, Pittsburgh, PA). Sections were deparaffinized by heating for 20 minutes at 65 C and were rehydrated through graded alcohols and washed with phosphate-buffered saline (PBS) (pH 7.4). Ovaries were fixed in 10% neutral buffered formalin, embedded in paraffin, sectioned, stained with hematoxylin and eosin for routine microscopic examination. Tissue sections (5 µm) were used for ISH, IHC, and double-labeling IHC.
ISH
The hybridization probe (a 433-bp cDNA) was produced by reverse transcription polymerase chain reaction as previously described. 37 The cDNA probe represents a region of the PRRSV genome in open reading frame 7 that is also present in all subgenomic mRNAs produced by active replication of PRRSV within a cell. The ISH was carried out as previously described. 37 The probe was labeled via random priming reaction with digoxigenin (DIG)-dUTP (Boehringer Manheim Co., Indianapolis, IN). All the solutions used for pretreatment, prehybridization, and hybridization were prepared with 0.1% diethyl pyrocarbonate (DEPC)-treated water. Tissue sections were rehydrated and equilibrated in PBS, deproteinized in 0.2 N HCl for 20 minutes at room temperature, washed with DEPC-treated water for 1 minute, and digested with proteinase K (Gibco BRL, 20 µg/ml) for 20 minutes in PBS. After digestion, all tissues were fixed in 4% paraformaldehyde in PBS for 5 minutes. After rinsing with PBS, the slides were acetylated in 200 ml of 0.1 mM triethanolamine HCl buffer (pH 8.0) to which 0.5 ml of acetic anhydride (0.25%) was added. After 5 minutes, 0.5 ml of acetic anhydride was added for an additional 5 minutes, and the slides were then rinsed in 2× standard saline citrate (SSC; 1× SSC, 150 mM NaCl + 15 mM sodium citrate, pH 7.0). The prehybridization mixture contained 50% deionized 4× SSC, 10% dextran sulfate, 1× Denhardt's solution (0.02% Ficoll 400, 0.02% polyvinylpyrrolidone, 0.02% bovine serum albumin), 2 mM ethylenediaminetetraacetic acid, and 500 µg/ml salmon testis DNA. The slides were covered with 200 µl of prehybridization mixture and were incubated in a humidified chamber for 1 hour at 65 C. The labeled probe (0.1 ng/µl) was diluted in 300 µl of the prehybridization mixture and then heated for 5 minutes in a 95 C heating block and quenched on ice before being applied to the sections. The slides were rinsed briefly in 2× SSC, and 60 µl of probe mixture for tissues was applied to each slide. The hybridization was performed overnight at 55 C. After hybridization, the tissue sections were washed twice in 4× SSC for 5 minutes at room temperature, once in 2× SSC for 10 minutes at 56 C, once in 0.2× SSC containing 60% formamide for 10 minutes at 56 C, twice in 2× SSC for 5 minutes at room temperature, twice in 0.2× SSC for 5 minutes at room temperature, and once in buffer I (100 mM maleic acid, 150 mM NaCl [pH 7.5]) for 5 minutes at room temperature. For immunohistologic detection of hybridized PRRSV RNA, slides were preincubated with buffer II (1% blocking reagent in buffer I, Boehringer Manheim, Indianapolis, IN) for 1 hour at room temperature to reduce background staining. The anti-DIG–alkaline phosphatase conjugate was freshly diluted 1:500 in buffer II and was then added to the tissue sections for 2 hours at room temperature. All incubations were followed by washes in two changes of buffer I for 5 minutes at room temperature and one wash in buffer III (100 mM Tris HCl, 100 mM NaCl, 50 mM MgCl2 [pH 9.5]) for 5 minutes at room temperature. Sections were then incubated with color substrate solution consisting of 45 µl of nitroblue tetrazolium salt (75 mg/ml in 70% dimethyl formamide) and 35 µl of 5-bromo-4-chloro-3-indolylphosphate, toludinium salt (50 mg/ml in dimethylformamide) in 10 ml of buffer III. The reaction was incubated for 2–3 hours in the dark. The color reaction was stopped with distilled water. Counterstaining was done with 0.5% methyl green for 10 seconds, and the slides were then washed in distilled water for 1 minute and dried completely. Slides were dipped in xylene and coverslips were applied before microscopic examination.
IHC
IHC was performed essentially as described previously. 35 Tissue sections were first treated with 3% hydrogen peroxide in PBS for 20 minutes, followed by washes in PBS and digestion with 0.05% Protease XIV (Sigma Chemical Co., St. Louis, MO) for 7 minutes at 37 C. After several washes in PBS, sections were incubated in a blocking solution (5% normal goat serum in PBS) for 30 minutes at room temperature. Monoclonal antibodies (MAbs) were the primary antibodies in all cases. The hybridoma ATCC 142.1, which produces an MAb against the SWC3a antigen of swine macrophages was used as a marker for swine macrophages. 22 For PRRSV antigen, SDOW17 (National Veterinary Services Laboratories, USDA/APHIS, Ames, IA), a MAb specific against PRRSV nucleocapsid antigen, 25 was used. A MAb against proliferating cell nuclear antigen (PCNA; Novacastra Laboratories, New Castle Upon Tyne, UK) was also used. PCNA is a 36-kd nuclear protein that has been associated with the cell cycle 18 21 27 and characterizes proliferating cells in different tissues. When used to stain ovarian tissues, this MAb detects proliferating granulosa and thecal cells. 27 MAbs were coated on the slides at dilutions of 1:50 (SWC3a), 1:500 (SDOW17), and 1:200 (PCNA). Sections were incubated with the MAb, at the corresponding dilution in PBS overnight at 4 C. After two 10-minute washes with PBS, the slides were incubated with biotinylated goat anti-mouse linking antibody for 40 minutes at room temperature. After further washes with PBS, slides were incubated with either alkaline phosphatase–conjugated antibody or peroxidase-conjugated streptavidin antibody for 20 minutes at room temperature. After the last wash in PBS, slides were developed by incubation with either the alkaline phosphatase substrate or 3,3′-diaminobenzidine tetrahydrochloride (DAB; Zymed, South San Francisco, CA) until color development was seen. The color reaction was stopped by washing the slides in distilled water, and the slides were then counterstained with Mayer's hematoxylin.
Double IHC for simultaneous detection of granulosa cells and PRRSV antigens
To determine the phenotypic characterization of the PRRSV-infected cells, double labeling IHC techniques were used. Endogenous peroxidase activity was removed by 20 minutes of incubation in 3% hydrogen peroxide in PBS (pH 7.4) and then washing three times in PBS. Nonspecific antibody binding was blocked by incubation with 5% normal goat serum (Sigma Chemical Co.) for 20 minutes at room temperature. The slides were then incubated at room temperature with PCNA (diluted 1:200) MAb for 2 hours. The sections were washed three times in PBS and incubated with biotinylated secondary antibody (Dako Corp., Carpinteria, CA) for 30 minutes at room temperature. The sections were then treated with peroxidase-conjugated streptavidin (Dako Corp.) for 20 minutes. After three subsequent washes in PBS, the slides were incubated in DAB (Zymed) until color development was seen. The color reaction was stopped by washing the slides in distilled water, and the sections were then transferred to PBS. For detection of PRRSV antigen, endogenous biotin was quenched by pretreatment of the slides with an avidin–biotin blocking reagent (Vector Laboratories, Burlingame, CA) at room temperature, followed by washes in PBS and digestion with 0.05% Protease XIV (Sigma Chemical Co.) in 50 mM Tris buffer (pH 7.6) for 6 minutes at 37 C. The sections were then washed three times in PBS and incubated with 5% normal goat serum for 20 minutes at room temperature. Subsequently, PRRSV nucleocapsid-specific MAb SDOW17 (diluted 1:200 in PBS) was applied and incubated for 16 hours at 4 C. The slides were then washed in PBS and incubated with biotinylated goat anti-mouse Ig linking antibody for 40 minutes at room temperature. After washing three times in PBS, the slides were incubated with alkaline phosphatase–conjugated antibody for 30 minutes at room temperature. After washing three times in PBS, color reaction was developed with the alkaline phosphatase substrate (Vector Laboratories) until color development was seen. The color reaction was stopped by washing the slides in distilled water, and the slides were then counterstained with methyl green and mounted for light microscopy.
Results
Morphologic findings in atretic follicles
Ovaries of control and infected gilts appeared normal. Ovaries in both groups had morphologic changes consistent with those developed during the course of the normal estrous cycle. In individual ovaries, follicular atresia was characterized by cessation of mitotic activity in the granulosa cell layer, separation of granulosa cells from the basilar layer, and death of oocytes. Many granulosa cells were apoptotic as indicated by shrunken nuclei and clumping of chromatin. As follicular atrophy proceeded, there was cytoplasmic shrinkage of the oocyte and retraction of the zona pellucida. The antrum became filled with cellular debris and phagocytic cells, including macrophages. With time, the granulosa cell layer of individual ovaries underwent further degeneration and became indistinguishable from the underlying thecal cell layer.
Distribution of macrophages in the ovary
Staining with MAb SWC3a was evident in the cytoplasm of macrophages of several different areas of ovary. Tissue distribution of macrophages in the ovaries of control (data not shown) and infected (Figs. 1–3) gilts was similar. Macrophages were not detected in primordial, primary, or secondary follicles. Concurrently, PRRSV antigen was not observed in any of these areas. Macrophages were commonly identified in the ovarian medulla and regressing corpora lutea (Fig. 1). Few to moderate numbers of macrophages stained by MAb SCW3a were detected in the thecal and granulosa cell layers of luteal follicles (Fig. 2). However, the greatest number of macrophages were seen in atretic follicles (Fig. 3a). SWC3a staining was also intense in the macrophages in atretic follicles, which often appeared to contain phagocytosed cells (Fig. 3b).
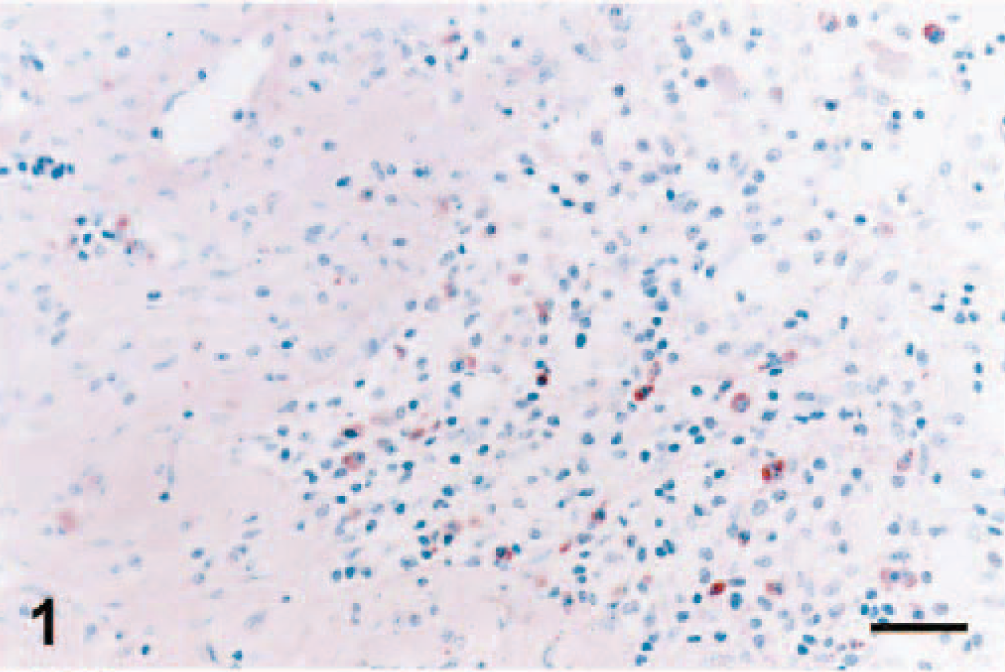
Ovary; pig infected with PRRSV 16244B. Macrophages of the corpus luteum at 7 DPI. IHC; alkaline phosphatase method, methyl green counterstain. Bar = 45 µm.
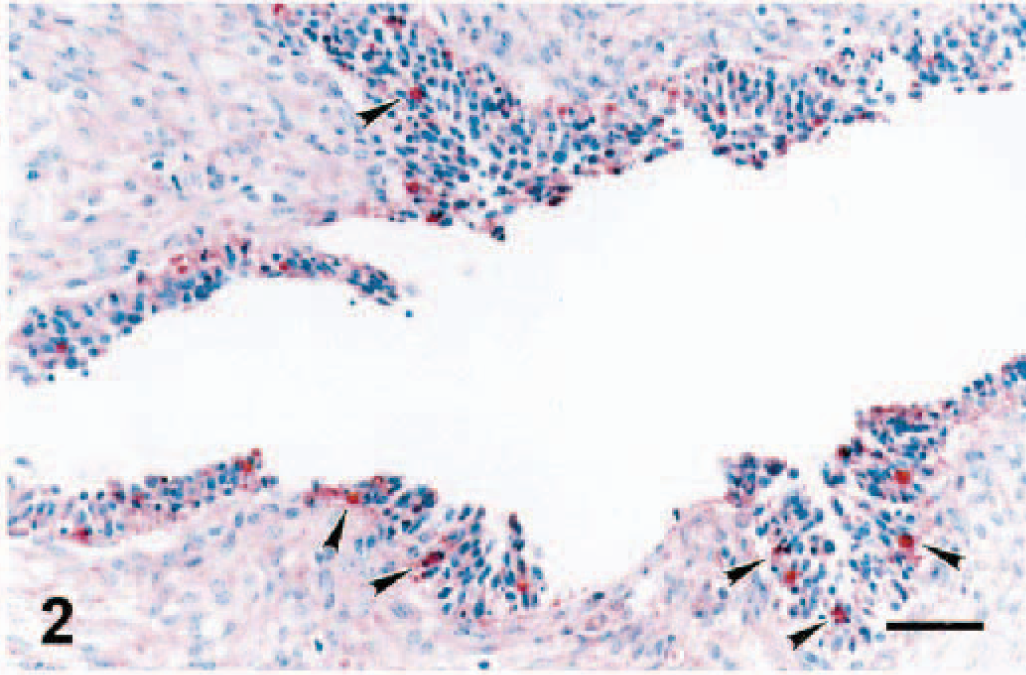
Ovary; pig infected with PRRSV 16244B. Macrophages of a ruptured follicle at 14 DPI. Macrophage SCW3a antigen is detected in the granulosa cell layer (arrowheads). A similar pattern of distribution of macrophages was observed in ruptured follicles of the uninfected controls (not shown). IHC; alkaline phosphatase method, methyl green counterstain. Bar = 60 µm.
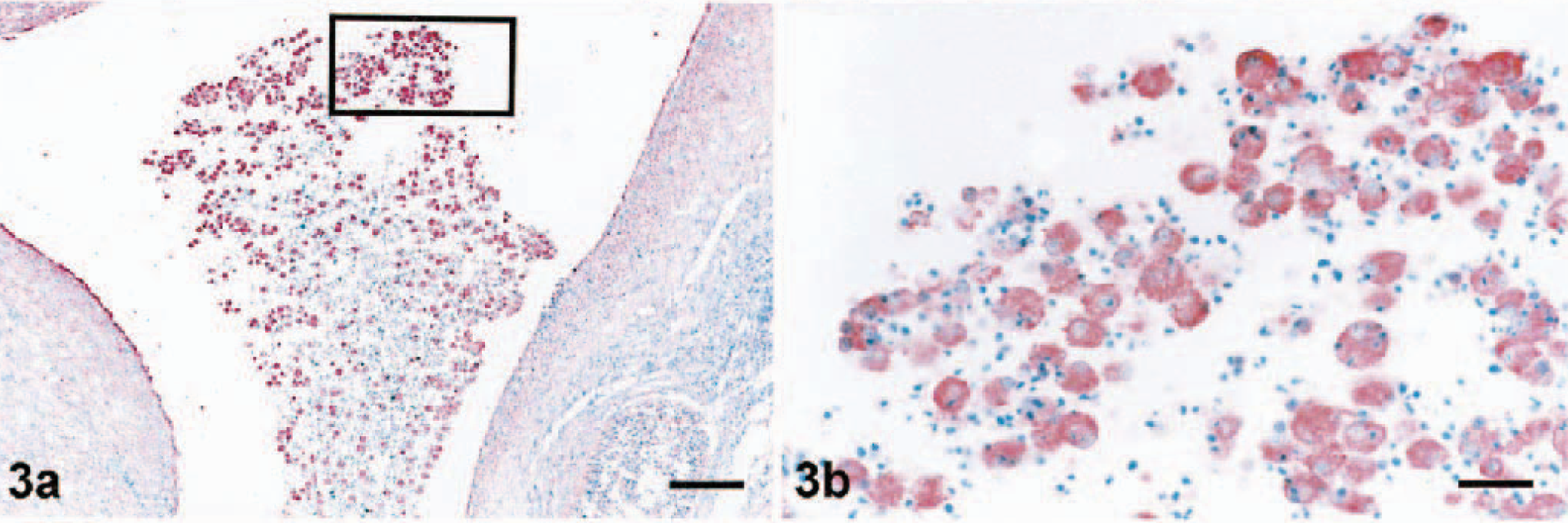
Ovary; pig infected with PRRSV 16244B. Macrophages of the atretic follicle at 21 DPI.
PCNA in the ovarian follicles
As described for other species, 18 PCNA immunoreactivity was recognized in both healthy and apoptotic granulosa cells of ovaries of gilts. PCNA was almost entirely confined to the nucleus and had a diffuse or granular pattern or a mixture of both. In primary follicles, most granulosa cells were strongly positive for PCNA (data not shown). Even in atretic follicles, nuclear PCNA was present in granulosa cell layer and thecal cell layer of ovary (Fig. 4). Likewise, in advanced atretic follicles there was PCNA staining in nuclei of the phagocytosed granulosa cells (Fig. 5).
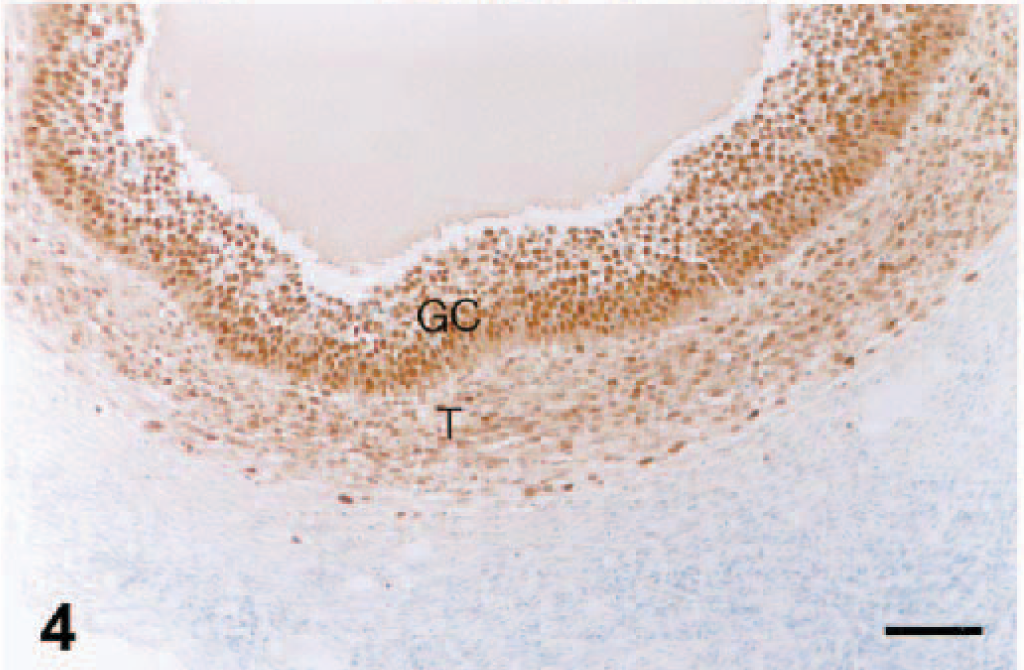
Ovary; pig infected with PRRSV 16244B. Atretic follicle at 7 DPI. Granulosa cells (GC) and thecal cells (T) are PCNA positive. An identical pattern of distribution of PCNA reactivity was observed in ovaries of uninfected pigs (not shown). IHC for PCNA; immunoperoxidase method, methyl green counterstain. Bar = 60 µm.
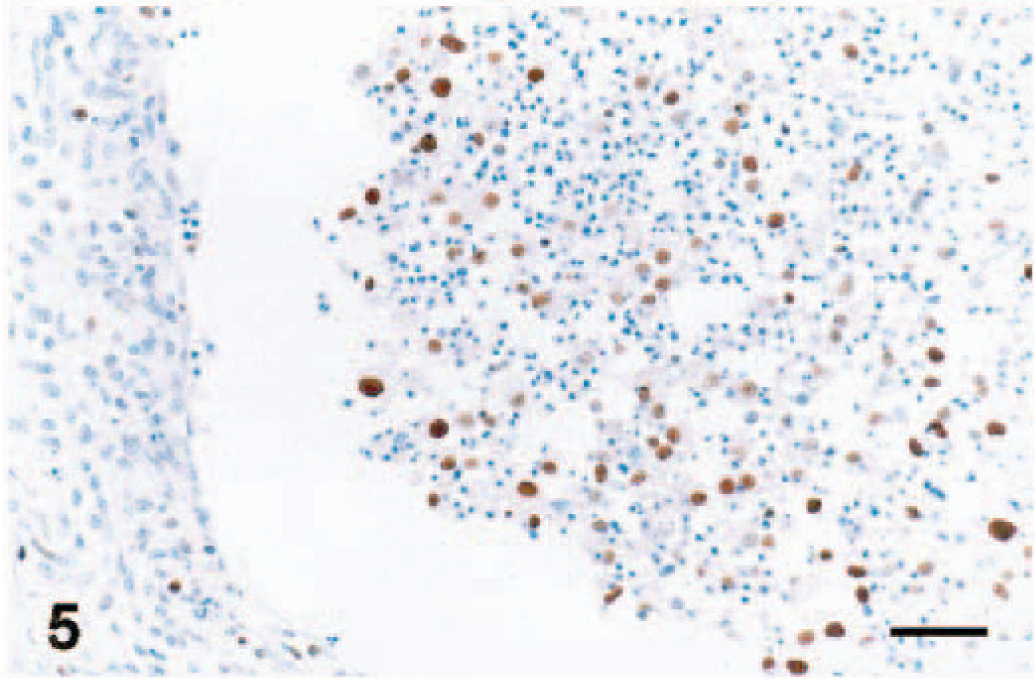
Ovary; pig infected with PRRSV 16244B. Atretic follicle at 14 DPI. Floating granulosa cells are PCNA positive. IHC for PCNA; immunoperoxidase method, methyl green counterstain. Bar = 45 µm.
VI, IHC for PRRSV, and PRRSV ISH data
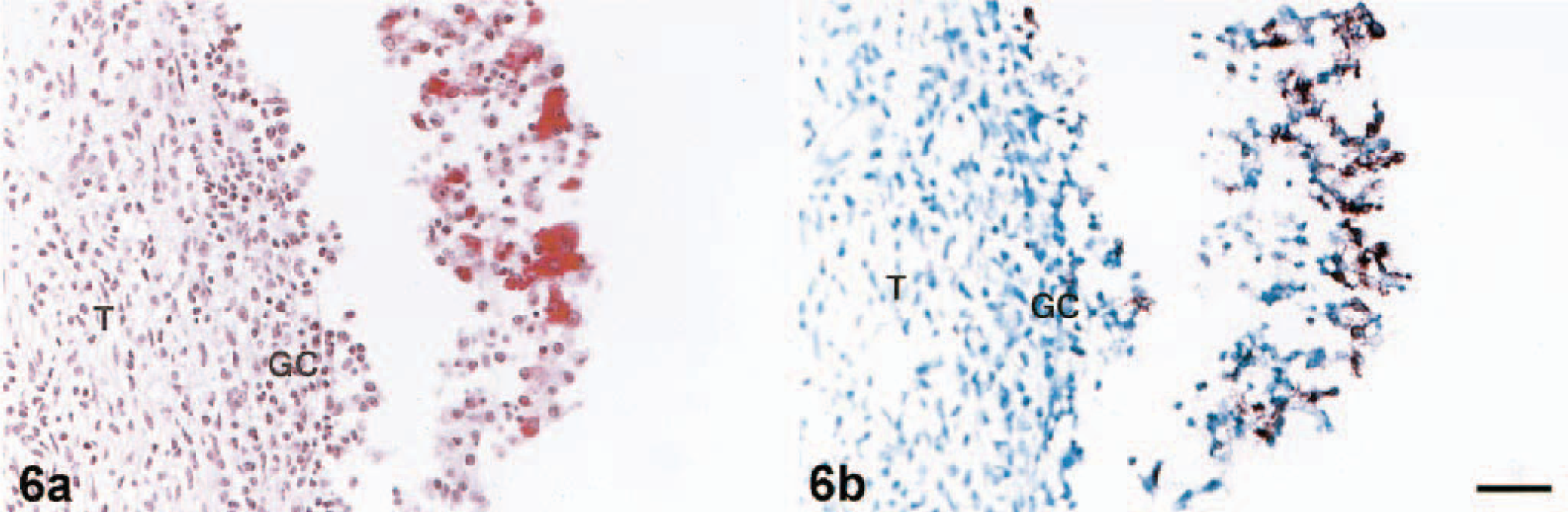
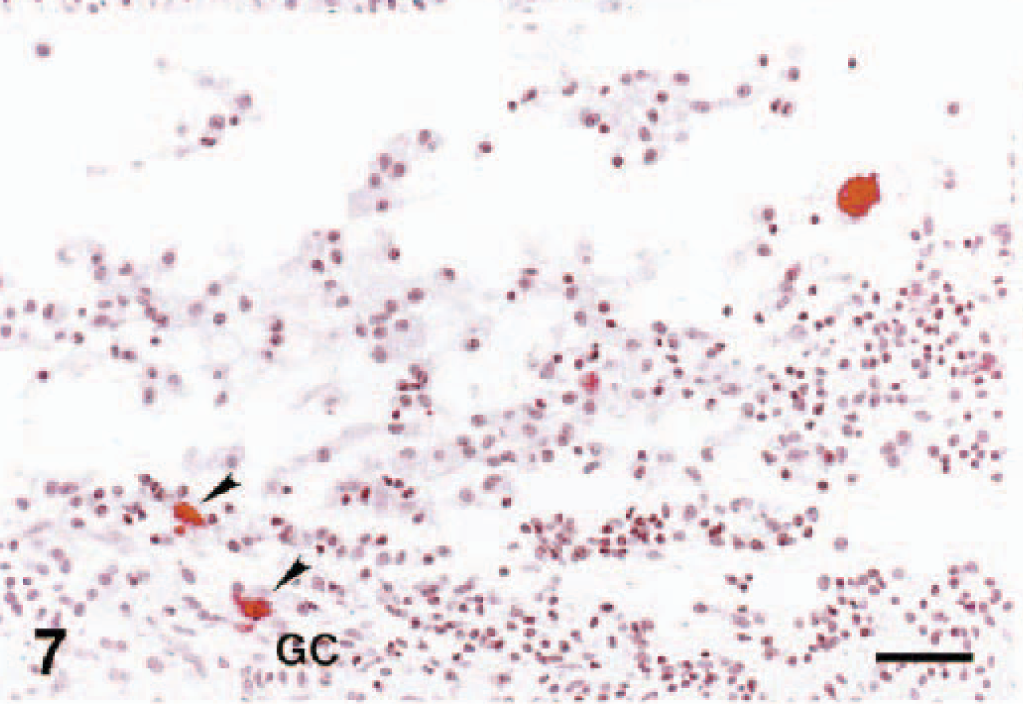
Table 1 contains a summary of the results of PRRSV detection in porcine ovary by VI, ISH, and IHC methods. Throughout the observation period, IHC and ISH were more consistent than the infectivity assay in MARC 145 cells for detecting PRRSV in porcine ovary. After two passages in MARC 145 cells, infectious PRRSV was isolated in 83% (5/6) of the PRRSV-infected gilts (Table 1). At 7 DPI, all attempts to detect PRRSV antigen and RNA by both IHC and ISH in ovary tissues were successful. The location, morphology, IHC results, and ISH signal of most PRRSV-positive cells in the atretic follicles suggested that these PRRSV-infected cells were macrophages. In an attempt to confirm the identities of PRRSV-infected ovarian cells, we performed ISH with serial sections of infected ovarian tissue. In all cases, the specificity of the signal reaction was confirmed by rigorous observance of two specificity controls on ISH: 1) predigestion of consecutive tissue sections with a solution of RNase A prior to the performance of ISH, which significantly decreased the development of the ISH signal, and 2) ISH performed with PRRSV probe on ovary tissues of uninfected pigs, which were consistently PRRSV negative (data not shown). The performance of IHC and ISH with serial sections of ovarian tissues indicated a close cell–cell correlation for the IHC and ISH signals (Fig. 6a, b). The PRRSV signal centered predominantly in macrophages. At 14 DPI, a few positive signals were observed in the granulosa cell layer and among clumps of floating granulosa cells by IHC (Fig. 7). In addition, double labeling experiments revealed that a few cells in the atretic follicles contained both PCNA and PRRSV antigen in the follicle at 7 DPI (not shown) and 14 DPI (Fig. 8).
Comparison of PRRSV detection in porcine ovary by VI, ISH, and IHC methods.
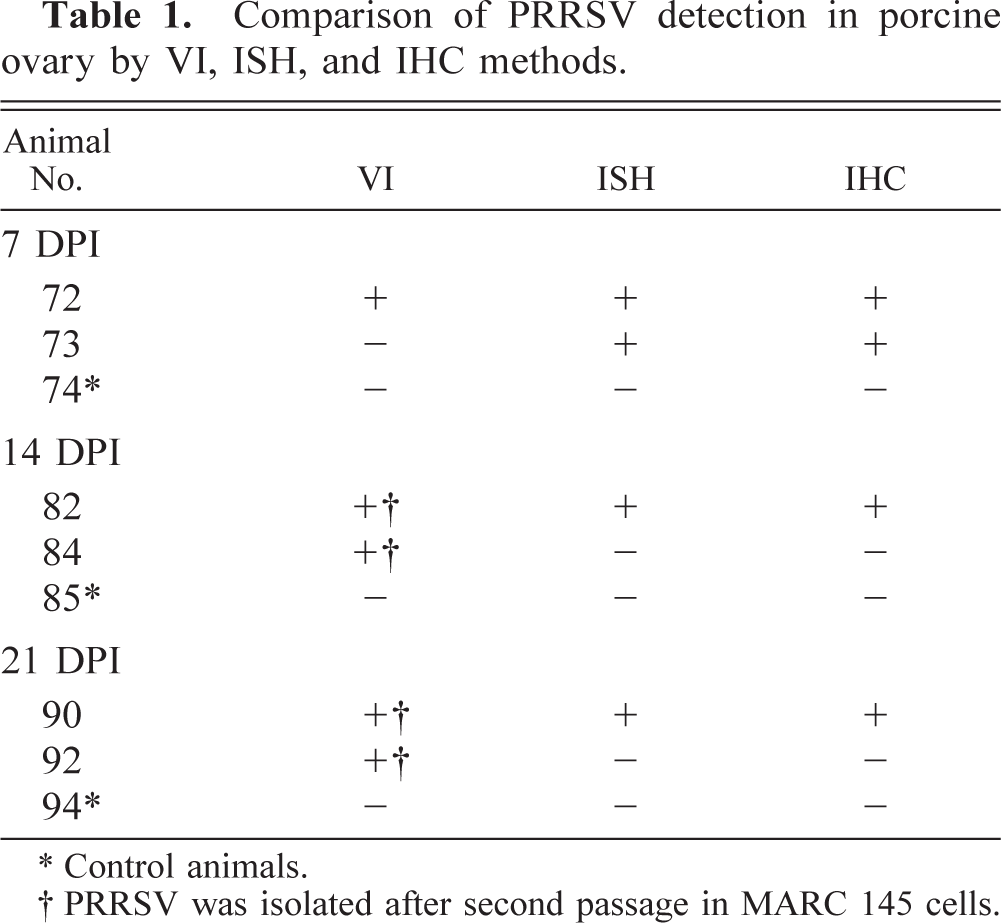
Control animals.
PRRSV was isolated after second passage in MARC 145 cells.
Ovary; pig infected with PRRSV 16244B. Atretic follicle at 7 DPI. PRRSV-infected cells in atretic follicle. GC = granulosa cell layer; T = thecal cell layer.
Ovary; pig infected with PRRSV 16244B. Atretic follicle 14 DPI. GC = granulosa cell layer. PRRSV-infected cell in atretic follicle (arrowheads). IHC; alkaline phosphatase method, hematoxylin counterstain. Bar = 45 µm.

Ovary; pig infected with PRRSV 16244B. Atretic follicle, 14 DPI. Arrowheads show cells colabeling for PRRSV antigen (red) and PCNA (brown). Double-labeling IHC; methyl green counterstain. Bar = 45 µm.
Discussion
Tropism of PRRSV and its persistence in susceptible gilts was examined using appropriate procedures for detection of PRRSV antigen and RNA. The distribution of macrophages in the ovary and their correlation with viral antigen were evaluated by IHC, and the presence of viral RNA was determined using ISH. The aim of this study was to address questions regarding detection of PRRSV in ovaries. Results of this study demonstrated the presence of PRRSV within the macrophages of the ovary, thecal cell layer, and granulosa cell layer and among detached granulosa cells in atretic follicles of infected gilts. Macrophages also were present in the ovarian medulla, corpora luta, and follicles. Previous studies have shown changes in macrophage populations within follicles of some animals following hormonal stimulation of the proliferation of granulosa cells. 2 15 17 41 It has been proposed that these normal resident macrophages constitute the major source of local factors that drive the process of follicular development. 17 41 In addition to participating in follicular development, invading macrophages, as well as granulosa cells, phagocytize dead cells in various stages of atretic follicle development. 5 9 17 41 Importantly, there was no evidence of PRRSV infection and/or perpetuation in ova, indicating that the female gonad (contrary to the situation in infected males 36 ) is an unlikely site of PRRSV persistence.
Variable numbers of macrophages were scattered among the detached granulosa cells in atretic follicles. The presence of PRRSV antigen-positive cells and PRRSV RNA-positive cells in degenerating follicles suggests that phagocytic macrophages in the ovary may deliver virus for subsequent infection of neighboring nonmacrophages, such as granulosa cells, by a “Trojan horse” mechanism, as has reported for the macrophage-tropic arteriviruses such as LDV 6 and PRRSV. 3
The localization of PRRSV antigen in ovarian tissues was most abundant in the cytoplasm of macrophages. More importantly, other unequivocal evidence that confirms the specificity of this observation was provided by simultaneous use of ISH and IHC with serial sections of the same tissue (Fig. 6a, b). Significant numbers of PRRSV-infected cells were consistently observed in the atretic follicles. Furthermore, the number of PRRSV-infected cells appeared to be much greater in atretic follicles than in other regions, such as regressing corpora lutea and medulla.
Most ovarian follicles mature and then degenerate through a well-described sequence of events. Natural atretic processes depend upon many factors. Atresia can vary among species depending on the physiologic condition of the animal. 9 14 19 40 Although the mechanisms underlying atretic processes are not well understood, the role of macrophages as the major source for proinflammatory mediators, particularly interleukin 1, interleukin 2, and tumor necrosis factor, is well established and therefore of critical importance during atresia. 4 Based on similarities we observed between infected and control gilts in our experiments, it appears that massive influx of PRRSV-infected macrophages into the atresic follicles does not cause infection of the ovum.
Of particular interest is how macrophages infiltrate follicles. Follicles undergo enormous changes throughout the estrous cycle. Angiogenesis, which is restricted to the thecal cell layer, is of special interest because both the theca interna and theca externa are extensively vascularized in atretic follicles. Follicles of several different stages of growth begin to mature each estrous cycle, but ordinarily only 2% of the total number of oocytes are discharged during the fertile period. 5 9 All remaining unsuccessful follicles, which include primordial follicles and follicles in various stages of growth, undergo degeneration (atresia). Follicles increase in size and become vascularized. Concurrently, macrophages invade the atretic follicle and remove degenerating material, including apoptotic cells, along with neighboring granulosa cells.
Recent studies have demonstrated that macrophages are involved in the atretic process during both pubertal and prepubertal development in the rat. 17 The current observation of macrophage distribution patterns in the gilt ovary agrees with existing reports on the localization of macrophages in the ovary, 4 in that the atretic follicle seems to be a major site of attraction for macrophages. However, the results described here for macrophages in the growing follicles of pigs differ significantly from those reported for rats. 17 In contrast to the rodent model, macrophages were absent from healthy developing swine follicles, in spite of the high number of macrophages that appeared in the atretic follicles. PRRSV-infected macrophages may be attracted to invade the granulosa cell layer and thus contribute to apoptosis of germinal epithelial cells. Alternatively, macrophages distributed within atretic follicles may be hormonally stimulated to produce cytokines, 2 4 16 41 which in turn cause germ cells to undergo apoptosis.
In addition to atretic follicles, PRRSV was observed in several different areas of the ovary, including medulla, thecal cell layer, and corpus luteum. However, the number of infected cells was significantly less than that in the atretic follicle. The relative number of PRRSV-infected cells in any given area of the ovary appeared to be directly dependent on the density of macrophages, which are the principal cells involved in the ovarian infection. The double labeling IHC in the present study indicated that both the PCNA-positive and PRRSV-positive signals could be colocalized within a few free-floating cells in the atretic follicular lumen but not in secondary or tertiary follicles. This finding could be interpreted as an actual PRRSV infection of degenerating granulosa cells of the atretic follicles due to the infectious input contributed by the significant influx of (infected) macrophages that occurs at this particular follicular stage. However, an alternative explanation for this colocalization of PCNA and PRRSV antigen signal would be the phagocytosis of PCNA material by PRRSV-infected macrophages. This ambiguity is a limitation of the double labeling technique per se. The technique cannot distinguish between PCNA material that has been phagocytosed by PRRSV-infected macrophages and actual PCNA-positive, PRRSV-infected cells. The low number of putative PRRSV-infected granulosa cells detected only at such advanced stage of follicular degeneration has questionable significance in relation to the functional activity and morphologic appearance of the ovary.
PRRSV infection within the porcine ovary appears to depend on the initial PRRSV replication within macrophages of regional lymph nodes, as is generally described for the pathogenetic spread of PRRSV. 3 These PRRSV-positive macrophages then distribute virus as they circulate through the animal. In particular, virus-infected macrophages infiltrate atretic follicles within the ovary and release virus, which can subsequently infect macrophages in the ovary. The failure to detect differences between the ovaries of PRRSV-infected and those of uninfected gilts does not support a possible role of PRRSV infection in female porcine infertility, as would be indicated by alteration of the normal ovarian architecture.
Footnotes
Acknowledgements
This research was supported by a grant from the US Department of Agriculture National Research Initiative Grant Program (project No. 99-35204-8041) and by a grant from the Center for Biotechnology of the University of Nebraska (Area of Concentration: Comparative Pathobiology). We thank G. Almond and M. Mc Caw for critical review of this manuscript. This article is published as paper No. 12,617 of the Agricultural Research Division, Institute of Agriculture and Natural Resources, University of Nebraska–Lincoln.