
Abstract
Glannzmann's thrombasthenia (GT) is an autosomal recessive bleeding disorder caused by qualitative or quantitative deficiencies of the platelet membrane glycoprotein αIIbβ3. This is the first report of a molecular genetic basis for type I GT in dogs. As previously reported, a thrombasthenic Great Pyrenees dog (dog No. 1) experienced uncontrolled epistaxis despite results of coagulation screening tests, platelet quantitation, and von Willebrand factor quantitation that were within reference ranges. Platelet aggregation was minimal in response to agonists. Flow cytometry, autoradiography, and immunoblot experiments demonstrated either marked reduction or absence of glycoproteins αIIb and β3. In this study, we report the presence of a 14-base insertion in exon 13 and defective splicing of intron 13 in the αIIb gene of two thrombasthenic dogs (Nos. 1 and 8). The insertion disrupted the fourth αIIb calcium-binding domain, caused a shift in the reading frame and resulted in a premature termination codon. Possible consequences of this mutation include decreased αIIb mRNA stability and production of truncated αIIb protein that lacks the transmembrane and cytoplasmic domains and a large portion of the extracellular domain. We identified the dam, sire, and three littermates of dog No. 8 as carriers of the αIIb mutation. Canine αIIb and β3 genes share significant homology with the genes in human beings, making canine GT an excellent translational model for human GT. A defined molecular basis for canine GT will enhance ongoing gene therapy research and increase the understanding of structure-function relationships of this integrin.
The αIIbβ3 glycoprotein complex is the major platelet membrane–associated fibrinogen receptor and is one of the most extensively studied integrins. 29 The α and β subunits are products of different genes that have been mapped to the long arm of chromosome 17 in human beings. 6 36 38 Expression of αIIbβ3 on the platelet surface requires the presence of both subunits, 39 and maintenance of subunit association is calcium dependent. 5 When associated in a heterodimeric complex and expressed on the platelet surface, the amino termini of the αIIb and β3 subunits interact with each other to form a globular structure that contains the ligand-binding domain. 50 The cytoplasmic tails form intracellular complexes that are vital for bidirectional transmembrane signaling mechanisms. 9 12 22 40
The β3 subunit is synthesized and expressed as a single polypeptide. 32 In dogs and human beings, the β3 transcript is 2,286 base pairs (bp) in length 32 and is composed of 14 exons that range in size from 63 to 430 bp (GenBank accession no. AF116270). 33 52 The αIIb subunit is initially synthesized as a single polypeptide that forms a dimeric complex with the β3 subunit while associated with the rough endoplasmic reticulum and is cleaved into two disulfide-linked peptide chains after transport to the Golgi complex. 14 34 39 43 In human beings, the αIIb transcript is 3.3 kb in length and is composed of 30 exons that range in size from 45 to 249 bp. 43 The αIIb transcript consists of 1,008 codons in human beings 43 and 1,005 codons in dogs (GenBank accession no. AF153316).
Glanzmann's thrombasthenia (GT) is an autosomal recessive bleeding disorder caused by qualitative or quantitative deficiencies of the platelet membrane glycoprotein αIIb β3. 17 19 Bleeding is due to defective hemostatic plug formation and is characterized by in vitro failure of platelets to aggregate in response to physiologic agonists such as collagen, adenosine diphosphate (ADP), and thrombin. 17 19 GT has been classified into three categories, based on αIIbβ3 deficiency: type I, characterized by severe (<5% of normal) quantitative deficiency; type II, characterized by moderate (10–20% of normal) quantitative deficiency; and variant, characterized by a relatively normal quantity but reduced function. 19
The first diagnosis of type I GT in a dog (No. 1) was based on results from platelet function studies, flow cytometry, immunoblots, and autoradiography of two-dimensional gel electrophoresis. 4 Here, we describe the molecular genetic basis for type I GT in the same dog (No. 1) and include nucleotide sequence analysis of a separate family of Great Pyrenees dogs, including the dam, sire, and five littermates (dog Nos. 2–8). Of the seven Great Pyrenees dog family members, only one dog (No. 8) demonstrated clinical bleeding. The α IIb gene was amplified by polymerase chain reaction (PCR) from genomic DNA and platelet-derived cDNA for normal (control) dogs and dog No. 1 and from genomic DNA for dog Nos. 2–8. Nucleotide sequence analysis of α IIb from the two thrombasthenic dogs (dog Nos. 1, 8) showed a 14-base insertion in the region encoding the fourth calcium-binding domain of exon 13 and a splicing defect of intron 13 (dog No. 1). A shift in reading frame occurred, and after 42 aberrant codons, a premature termination codon was reached. The insertion was located −20 bases from the original 5′ splice site of intron 13, and although the 5′ and 3′ splice sequences and putative branch sequence of intron 13 were present, efficient splicing of αIIb mRNA did not occur.
Analysis of α IIb showed that the nucleotide sequence of genomic DNA isolated from dog Nos. 2–6 matched the sequence obtained from the two thrombasthenic dogs (Nos. 1, 8) and also matched that of normal (control) dogs. The presence of α IIb sequence data matching both the normal (control) and thrombasthenic genotypes indicates that dog Nos. 2–6, including the dam and sire, are carriers of the mutation. The nucleotide sequence of α IIb for dog No. 7 was identical to that of the normal (control) dogs.
Materials and Methods
Case report
Type I GT was reported previously in a Great Pyrenees dog. 4 This 8-month-old female (dog No. 1) was presented for evaluation of a bleeding diathesis first observed by the owners when the dog was shedding deciduous teeth between the ages of 3 and 6 months. Excessive gingival hemorrhage ceased after eruption of all permanent teeth; however, epistaxis began shortly thereafter. Results of laboratory tests, including routine coagulation screening tests (activated partial thromboplastin time, prothrombin time, thrombin time), whole blood platelet quantitation, and von Willebrand factor antigen quantitation, were within reference ranges.
Affected platelets underwent shape change in response to ADP, collagen, thrombin, and platelet-activating factor (PAF); however, platelet aggregation in response to these agonists was minimal. Clot retraction was markedly reduced. Flow cytometry and immunoblot experiments were performed using antibodies to human β3 (MoAb Y2/51), canine platelet-bound fibrinogen (CAP1), and canine αIIbβ3 (MoAb 5G11). Results indicated absence of activated platelet-bound fibrinogen and minimal quantities of β3 and αIIbβ3. Autoradiography of two-dimensional electrophoresis of 125I-surface-labeled platelet membrane glycoproteins demonstrated that glycoproteins αIIb and β3 were markedly reduced or absent.
Canine subjects
For cDNA and genomic DNA studies, canine subjects included two normal adult mixed-breed dogs (control dogs) and the thrombasthenic Great Pyrenees dog (dog No. 1). Genomic DNA studies were performed on a family of seven other Great Pyrenees dogs that consisted of the dam (dog No. 2), sire (dog No. 3), and five littermates, all 1 year of age: one female (dog No. 5) and four males (dog Nos. 4, 6–8). Of dog Nos. 2–8, only dog No. 8 had a history of clinical bleeding that consisted of excessive gingival hemorrhage during tooth eruption. Results of laboratory tests, including platelet function studies, flow cytometry, and two-dimensional gel electrophoresis/autoradiography, were not available for dog Nos. 2–8.
Preparation of genomic DNA
Whole blood (1–2 ml) was collected from dogs via jugular venipuncture into 15% (w/v) ethylenediaminetetraacetic acid (EDTA) or 3.8% (w/v) sodium citrate. Genomic DNA was prepared from whole blood according to the Qiaamp Blood Kit (Qiagen, Valencia, CA) and was stored at 4 C or −80 C.
Preparation of platelet RNA
Whole blood (100–200 ml) was collected from dog No. 1 and normal (control) dogs via jugular venipuncture into 15% (w/v) EDTA. Platelet-derived total RNA was prepared as previously described. 33 The pellet of total RNA was resuspended in a small volume (<100 µl) of diethylpyrocarbonate-treated water and stored at −80 C.
First-strand cDNA synthesis
First-strand cDNA was synthesized from total RNA as previously described 33 and stored at 4 C or −20 C.
Amplification by PCR
For dog No. 1 and normal (control) dogs, all exons of α IIb were amplified using PCR. Primers used for PCR were designed so that the products of amplification overlapped in successive reactions; thus, exons were amplified and sequenced multiple times for each dog. Exons associated with the α IIb gene were amplified using PCR according to Taq Master Mix (Qiagen) as previously described 33 using 2 µl of prepared cDNA as template. For amplification of genomic DNA, 4 µl of DNA was used as template. Touchdown-type PCR amplification was performed using a programmable thermocycler (GeneAmp PCR System 2400, Perkin-Elmer, Norwalk, CT) set for initial denaturation of template (94 C, 4 minutes) and then five rounds of denaturation (94 C, 1 minute), hybridization of primers to complementary sequences of cDNA (1 minute; Table 1), and DNA synthesis (72 C, 2 minutes) for 2 cycles each. The thermocycler was programmed to decrease the hybridization temperature every 2 cycles until the final hybridization temperature, which proceeded for 20 additional cycles (Table 1). After the 30 cycles were completed, the PCR mixture was subjected to final extension at 72 C for 7 minutes. The reaction was terminated (4 C), and the sample was stored at 4 C. Amplified PCR products were purified as previously described 33 and stored at −80 C. For dog Nos. 2–8, only genomic DNA was used in PCR reactions (Table 1).
Amplification (PCR) criteria of αIIb exon 13 using platelet-derived cDNA and genomic DNA from dog No. 1 and genomic DNA from dog Nos 2-8 as template.
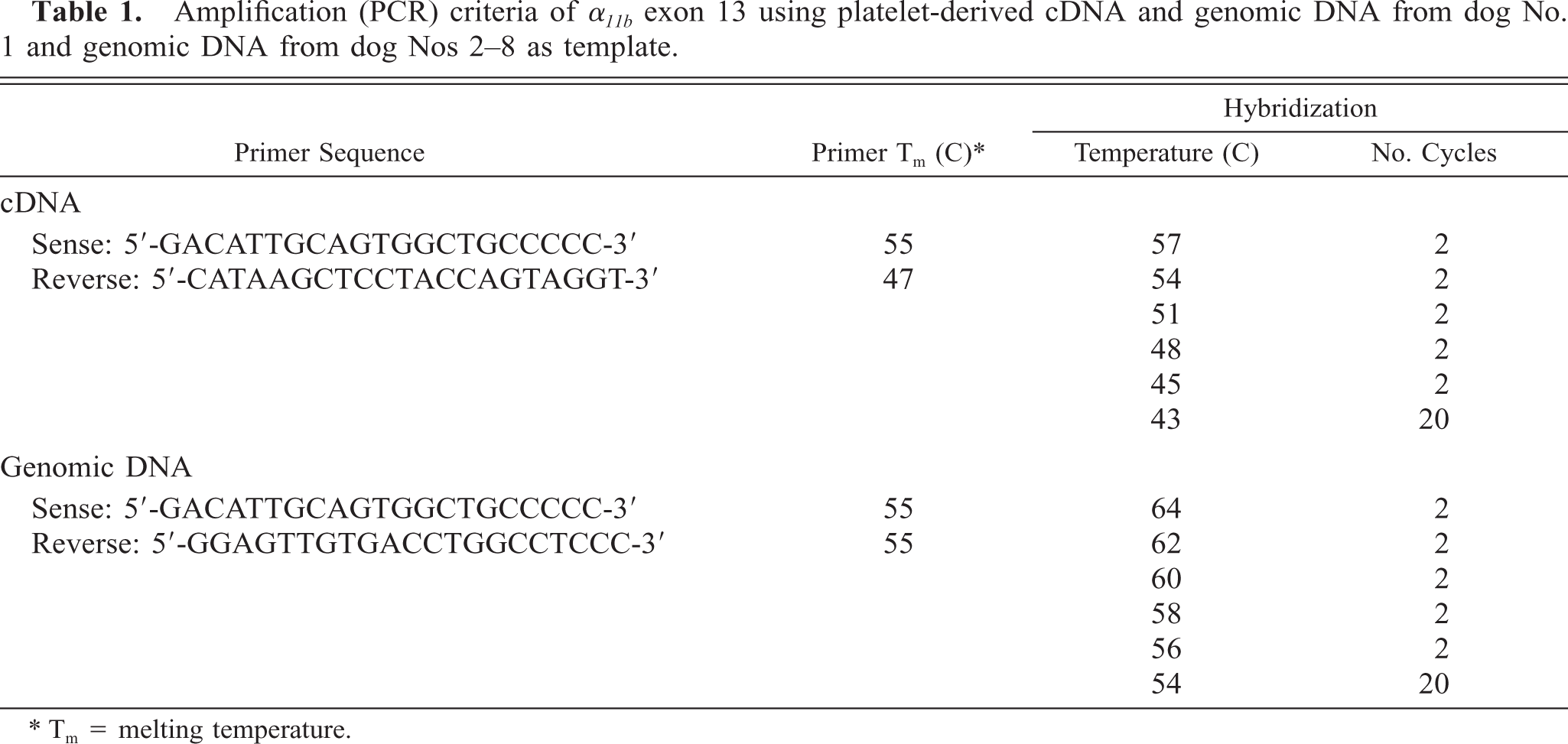
Tm = melting temperature.
DNA sequencing
All DNA sequencing of purified PCR products was performed as previously described. 33 Primers (Table 1) used for PCR were also used in sequencing reactions. The nucleotide base sequences were obtained by manual and automated interpretation of electropherograms.
Results
DNA sequencing
To identify the mutation(s) associated with the canine thrombasthenic phenotype (dog No. 1), all exons of the α IIb gene were amplified using first-strand platelet-derived cDNA and genomic DNA as templates. Except for exon 13, the results of all sequencing reactions of α IIb derived from genomic and cDNA of dog No. 1 and genomic DNA of dog No. 8 were identical to that of normal (control) dogs. Amplification and sequence analysis of α IIb from platelet-derived cDNA of the normal (control) dogs showed that exons 1–13 contained 1,392 nucleotides encoding 433 amino acids in the mature protein. Sequence analysis of amplified segments of exon 13 and intron 13 showed that the linear nucleotide sequence of genomic DNA and platelet-derived cDNA from the dog No. 1 and of genomic DNA from dog No. 8 were identical to each other but shared little similarity with the linear sequence data of normal (control) dogs (Figs. 1, 2). Beginning at nucleotide 1,374, exon 13 amplified from platelet-derived cDNA of dog No. 1 contained additional intron-derived nucleotide bases encoding 35 additional amino acids before the appearance of a premature termination codon (Figs. 2, 3). Amplified segments of exon 13 from genomic DNA and platelet-derived cDNA of dog No. 1 and from genomic DNA of dog No. 8 showed a 14-base insertion after nucleotide 1,373 that consisted of a direct repeat of nucleotides 1,360–1,373 (Figs. 1, 3). The nucleotide sequences of genomic DNA and platelet-derived cDNA obtained from dog No. 1 were identical to the sequence of genomic DNA from the normal (control) dog for the 3′ end of intron 13 adjacent to exon 14 and for the putative branch site located −23 bases from the 3′ end of intron 13.
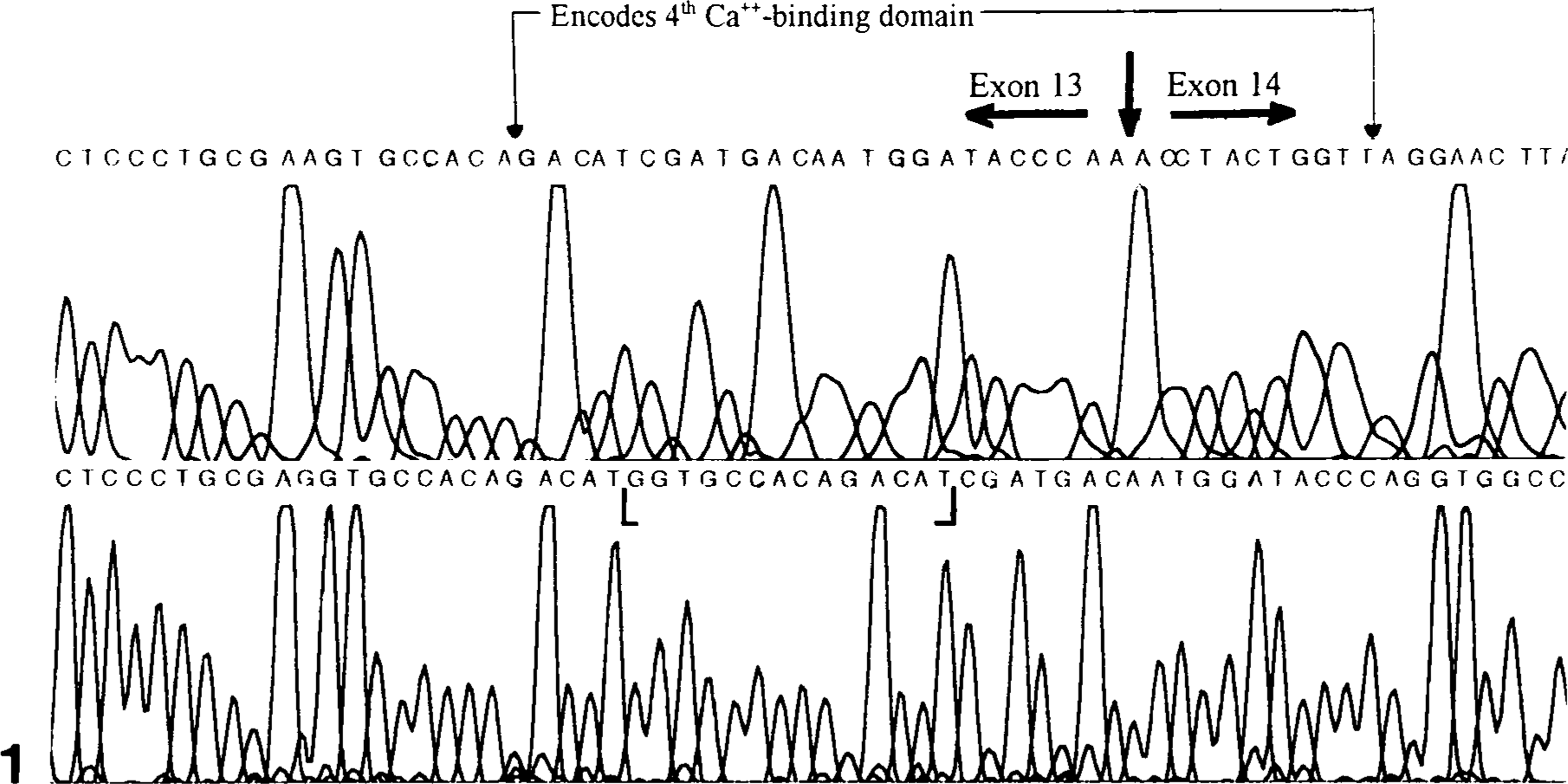
Nucleotide sequence electropherogram of α IIb exon 13 and exon 14 segments obtained from platelet-derived cDNA of normal (control) dogs (top) and from platelet-derived cDNA of dog No. 1 and genomic DNA of dog Nos. 1 and 8 (bottom). The 14-base insertion in exon 13 of dog Nos. 1 and 8 is designated by brackets. The α IIb segment that encodes the fourth calcium-binding domain is also designated.
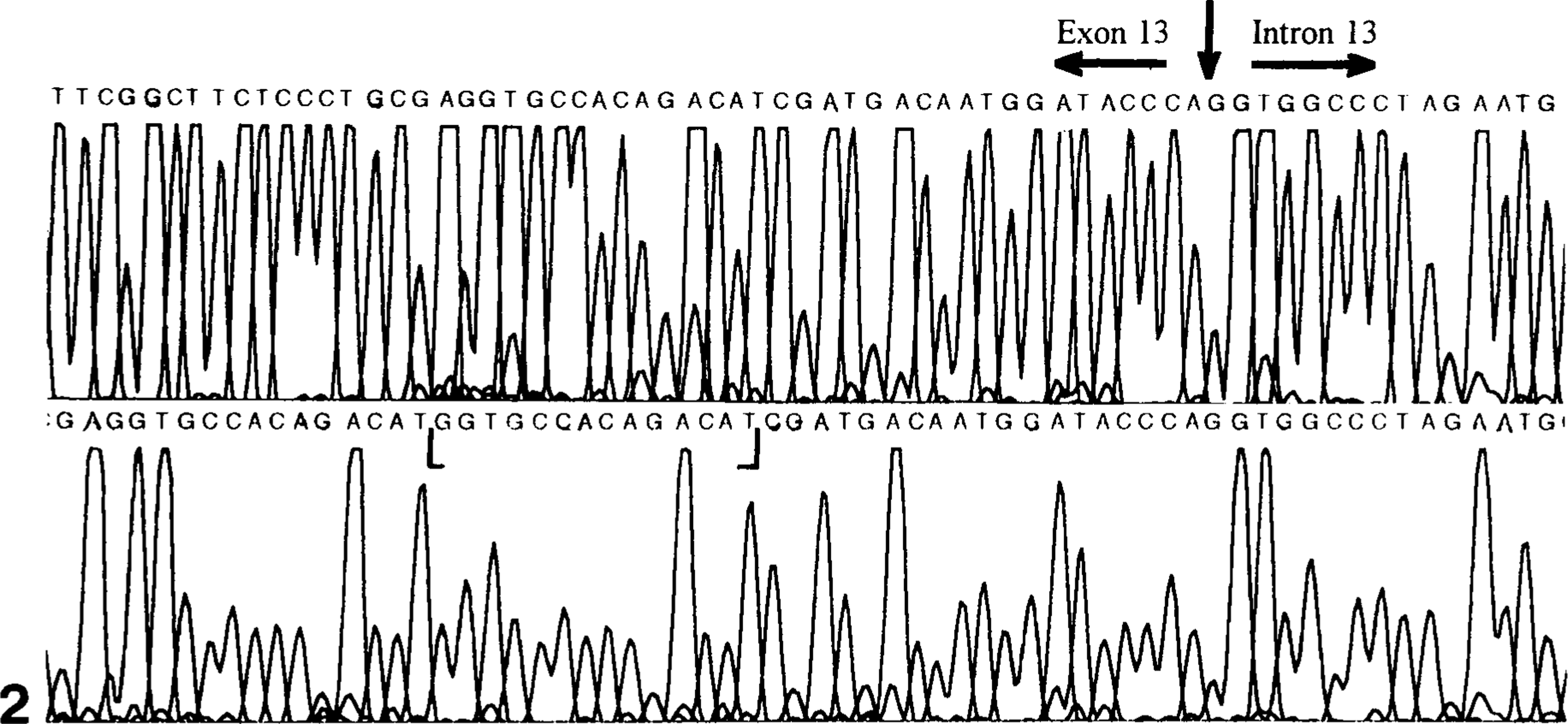
Nucleotide sequence electropherogram of α IIb exon 13 and intron 13 segments obtained from genomic DNA of normal (control) dogs (top) and from platelet-derived cDNA of dog No. 1 (bottom). The 14-base insertion in exon 13 of dog No. 1 is designated by brackets. Alignment of the top and bottom panels shows that the segment of α IIb from platelet-derived cDNA of dog No. 1 contains nucleotides that match the sequence of intron 13.

Comparison of α IIb exon 13 (mutation area) nucleotide and deduced amino acid sequences from normal (control) dogs and platelet-derived cDNA and genomic DNA of dog No. 1. The exonic deduced amino acid sequence is shown as a one-letter abbreviation above the second base of each codon. For reference, several deduced amino acids have the corresponding numerical designation above the amino acid abbreviation. Variation of deduced amino acid sequences between normal (control) dogs and dog No. 1 is denoted by amino acid abbreviation: Normal Canine/Case 1. The deduced amino acid sequence of the segment affected by the splicing defect is shown below the second base of each codon for dog No. 1; for reference, numerical designations (in parentheses) are written below the corresponding deduced amino acid. Similarities in exonic nucleotide sequences of the normal (control) dogs and dog No. 1 are denoted by a dash and differences are denoted by nucleotide abbreviations in the sequence of dog No. 1. Exon 13 for normal (control) dogs ends with the first base of the codon for amino acid residue 434, and the intronic nucleotides are shown as lowercase letters. A 14-base insertion extends from nucleotide 1,374 through nucleotide 1,387 (denoted by brackets) in the sequence of dog No. 1 and consists of a direct repeat of nucleotides 1,360–1,373. The 5′ splice site consensus sequence is underlined in the normal dog sequence and in the corresponding sequence of dog No. 1. The nucleotide sequence for normal dogs is from GenBank (accession no. AF153316). Ter = termination codon.
Nucleotide sequence electropherograms of genomic DNA from dog Nos. 2–6 showed α IIb sequence data identical to that of normal (control) dogs superimposed over nucleotide designations that were identical to the sequences of the thrombasthenic dogs, Nos. 1 and 8. Sequence analysis of amplified genomic DNA showed that the α IIb nucleotide sequence of dog No. 7 was identical to that of the normal (control) dogs.
Discussion
This is the first report of a molecular genetic basis for canine type I GT. Previous studies provided valuable information regarding the diagnosis of type I GT in a Great Pyrenees dog (dog No. 1). 4 As a puppy, this thrombasthenic dog experienced excessive gingival hemorrhage followed by uncontrolled epistaxis. Results of coagulation screening tests, platelet quantitation, and von Willebrand factor antigen quantitation were within reference ranges; however, platelet aggregation was minimal in response to ADP, collagen, PAF, and thrombin. 4 Flow cytometry, autoradiography of two-dimensional gel electrophoresis, and immunoblot experiments demonstrated that glycoproteins αIIb and β3 were present in only minimal amounts and membrane-associated αIIbβ3 and fibrinogen binding to activated platelets was undetectable. 4
The goal of the present study was to characterize the molecular genetic basis of the thrombasthenic phenotype associated with the Great Pyrenees dog (dog No. 1). The nucleotide sequence of α IIb of dog No. 1 was compared with the sequence of normal (control) dogs. In addition, α IIb sequences from the seven members of a family of Great Pyrenees dogs (dog Nos. 2–8), including the dam and the sire, were analyzed. Only one of the seven dogs (dog No. 8) demonstrated clinical bleeding consisting of excessive gingival hemorrhage during tooth eruption. Our findings indicated an insertion and a splicing defect in platelet αIIb mRNA of dog No. 1. Genomic DNA studies of dog No. 8 showed the same 14-base insertion; however, cDNA was not evaluated. Using sequence analysis of amplified genomic DNA and platelet-derived cDNA from dog No. 1 and of genomic DNA from dog No. 8, we demonstrated the presence of a 14-base insertion in exon 13 and defective splicing of intron 13 (Figs. 1, 2). The 14-base insertion disrupts the consensus sequence that encodes the fourth calcium-binding domain, causes a shift in the reading frame and, after encoding 42 aberrant amino acids, results in a premature termination codon (Fig. 3). The 14-base insertion is located −20 bases from the original 5′ splice site of intron 13 (Fig. 3), and although the 5′ and 3′ splice sequences and putative branch sequence of intron 13 are present, efficient splicing does not occur. Conformational changes in the αIIb transcript, created by the 14-base insertion, probably cause the 5′ splice site to be inaccessible to the splicing mechanism and prevent efficient splicing of intron 13. Some pre-mRNA splicing of intron 13 may occur, but the 14-base insertion would cause a shift in reading frame and, after encoding 3 novel amino acids in exon 14, would result in the appearance of a premature termination codon. Possible consequences of this mutation include decreased αIIb mRNA stability and production of truncated, unstable αIIb protein. 2 13 23 28 41 48
Our amplification studies were directed at individual and serial groups of exons and at the 5′ and 3′ ends of cDNA prepared from platelet-derived total RNA of dog No. 1. The 14-base insertion does not appear to significantly decrease the stability of αIIb transcript because the intensity of staining for each PCR product on agarose was similar for dog No. 1 and the normal (control) dogs. Truncation of the αIIb protein in the presence and absence of pre-mRNA splicing of intron 13 would completely eliminate the transmembrane and cytoplasmic domains and a large portion of the extracellular domain, including the fourth calcium-binding domain, which is important for intracellular processing of αIIbβ3 and transport to the platelet surface. 1 Maintenance of αIIbβ3 stability and function is dependent on the presence of calcium, 18 21 30 51 and binding of calcium to the fourth calcium-binding domain of αIIb may facilitate conformational changes that are required for transport of the αIIbβ3 complex to the platelet surface. 51 Results of several previous studies of αIIb and β3 showed that truncated subunits, lacking cytoplasmic and transmembrane domains, were able to form αIIbβ3 heterodimers; 21 31 however, the αIIbβ3 complexes were not expressed on the platelet surface. 16 Chaperone proteins, such as protein BiP, associate with truncated αIIb subunits and prevent assembly and transport of αIIbβ3 heterodimers. 3
The 14-base insertion in α IIb creates a shift in reading frame and leads to translation of novel amino acids and the appearance of a premature termination codon, whether splicing occurs or not. Mutations that impair the preassembly stage of αIIb (prior to assembly of the heterodimer complex) have been previously associated with type I GT of human beings; in all of these cases, little or no platelet membrane–associated αIIbβ3 was detected. 20 27 45 47 49 The degree of αIIb subunit and αIIbβ3 heterodimer stability has not been determined for this mutation. However, considering that the αIIbβ3 complex and fibrinogen binding to activated platelets were undetectable when previously assayed by flow cytometry, 4 we presume that either degradation of truncated αIIb subunit occurs prior to heterodimer assembly or unstable αIIbβ3 complex is formed and then degraded prior to surface expression.
Sequence analysis of α IIb showed that genomic DNA of the dam (dog No. 2), sire (dog No. 3), and three offspring (dog Nos. 4–6) matched that of the two thrombasthenic dogs (dog Nos. 1, 8) and the same DNA segment also matched the α IIb sequence of the normal (control) dogs, indicating that these five dogs are likely carriers of the mutation. Dog No. 8, having the thrombasthenic phenotype and being homozygous for the recessive α IIb mutation, would be predicted to have carrier parents, as demonstrated here. In addition the dam and sire, both being carriers of this recessive mutation, could be expected to produce some normal offspring, as observed with dog No. 7.
Numerous genetic defects have been previously described for type I GT in human beings, 1 7 8 10 11 15 20 24–27, 35 37 42 44–47, 49 51 but this is the first molecular genetic defect described for canine type I GT. Because the nucleotide sequences of canine α IIb (GenBank accession no. AF153316) and β 3 14 genes are similar to the sequence data reported for human beings, continued diagnosis of canine platelet disorders and identification of associated genetic defects will provide valuable information regarding regulation of α IIb β 3 gene expression. In addition, canine models of GT should facilitate gene therapy studies and further our knowledge and understanding of the biochemistry and function of this important integrin.
Footnotes
Acknowledgements
We thank Dr. J. Spano, Mrs. K. Worley, A. Hall, C. McDaniel, A. Mitchell, E. Whatley, and S. Waller for their assistance in obtaining blood samples and performing clinical laboratory tests and Dr. A. Samoylov (Scott-Ritchey Research Center, College of Veterinary Medicine, Auburn University) for performing all DNA sequencing. This work was supported by the Department of Pathobiology, Auburn University. This report represents a portion of the PhD dissertation by D. L. Lipscomb in the Biomedical Sciences graduate program, College of Veterinary Medicine, and is Auburn University publication no. 2575.