
Abstract
Inflammation is a risk factor for the development of many types of neoplasia, including skin, colon, gastric, and mammary cancers, among others. Chronic pulmonary diseases, such as chronic bronchitis and asthma, predispose to lung neoplasia. We will review the mouse literature examining the role of inflammation in lung neoplasia, focusing specifically on genetic susceptibility, pharmacologic modulation of inflammatory pathways, and both transgenic and knockout mouse models used to assess pro- and anti-inflammatory pathways involved in lung neoplasia. Identification of molecular mechanisms that govern the association between inflammation and pulmonary neoplasia could provide novel preventive, diagnostic, and therapeutic strategies for a disease in which few biomarkers currently exist.
In the mid 1800s, Rudolf Virchow was the first to suggest a causal role for inflammation in neoplasia. 17 It is now clear that chronic inflammation predisposes humans to the development of many types of cancer, such as liver, breast, prostate, pancreas, ovary, skin, gastric, and colorectal. 40 Some of the causal agents or pathologic conditions include Helicobacter pylori with gastric adenocarcinoma, human papillomavirus with cervical cancer, and gastroesophageal reflux disease with adenocarcinoma of the esophagus. 169 Since we will focus on pulmonary neoplasia, please refer to Schottenfeld and Beebe-Dimmer, 169 and Coussens and Werb 40 for more global reviews on inflammation and cancer. This review will discuss human and mouse lung neoplasia, cell types involved in inflammation, epidemiologic studies supporting a role for inflammation in lung cancer, genetic similarities between nonneoplastic and neoplastic pulmonary diseases in mice, and finally specific inflammatory pathways involved in either exacerbating or inhibiting inflammation in the tumor microenvironment.
Pulmonary Neoplasia in Humans
Lung cancer has the highest mortality rate among all types of cancer in both males and females and was second in new cancer cases in 2006. 83 In 2007, approximately 160,000 patients succumbed to lung cancer, greater than that of colon/rectal, breast, and prostate cancer deaths combined. 83 While the majority of lung cancer deaths can be attributed to cigarette smoking, approximately 13% of those individuals afflicted with this disease do not smoke, accounting for ∼22,000 deaths annually. 183 Second-hand smoke, air pollution, or other effectors such as radon and asbestos are all possible causes in this population. 18 By 2010, 1.5 million deaths/year due to lung cancer are projected worldwide, thus the public health burden is evident. 145
Lung cancer is divided into 2 clinical classes, small cell lung carcinoma (SCLC) and non-small cell lung carcinoma (NSCLC). Seventy-five percent of human lung cancer comprises NSCLC, which is then subdivided into squamous cell carcinoma, adenocarcinoma, large cell carcinoma, adenosquamous, and undifferentiated. Adenocarcinoma (AC), a peripheral secretory tumor, is now the predominant subtype in most countries (http://www.cancer.gov). AC is also the most common NSCLC subtype among smokers, and the only lung cancer found in nonsmokers. 168 Although there are several recent advances in lung cancer detection, such as the use of computed tomography (CT) scans, 14 and the use of genetic analysis, 178 these methods are still largely investigational. Unfortunately, for most patients afflicted with NSCLC, specifically AC, this disease is essentially undetectable until advanced stages, with no available biomarkers, making it the most clinically intractable of lung cancers.
Pulmonary Neoplasia in Mice
An approach to understanding the pathogenesis of chronic lung disease and lung neoplasia is to use experimentally manipulatable animal models. Mice are the preferred species because of the enormous number of inbred mice and genetic models (spontaneous, knockout, and transgenic). In addition, mice primarily develop lung AC, whether spontaneously, chemically (e.g., urethane, 3-methlycholanthrene, NNK), or environmentally (e.g., diesel exhaust) induced, with rare exceptions, such as chemically induced squamous carcinoma or a conditional knockout (both Trp53 and Rb)-induced SCLC. 115, 116, 126, 192 Histopathologically, mouse and human AC are indistinguishable; molecularly, both are associated with mutations in the oncogene KRAS and tumor suppressor genes such as TRP53, and the progenitor cell types involved in both are alveolar Type II pneumocytes and bronchiolar Clara cells. 115, 116 Kim et al. recently identified bronchioalveolar stem cells that express both a type-II cell-specific marker (prosurfactant apoprotein C) and a Clara cell-specific marker (Clara cell 10 kD protein [CC10]). 95 While 2 main morphologies exist with respect to lung AC, the type-II cell is the proposed progenitor for solid morphology tumors, and the Clara cell, the progenitor for papillary morphology tumors; however, mixed subtypes can exist. 115 Papillary adenomas are considered more aggressive and more likely to progress to malignancy. 115 However, based on recent stem cell evidence, they both may originate from the same stem cell. 95
In primary mouse lung tumor models, lung cancer develops through a series of definable lesions, starting from the normal epithelium to the malignant tumor. Tumorigenesis begins with a permanent DNA alteration in an oncogene or tumor suppressor gene through either spontaneous, chemically, or environmentally induced initiation events. The initiated cell containing this mutation or gene inactivation event clonally expands to become an adenoma during the promotion stage via changes in gene expression. This is the only reversible stage during cancer development. Progression consists of further modifications to DNA structure that result in the benign to malignant transition. Therefore, the promotion stage, which is the only reversible stage of neoplasia, is most amenable to chemopreventive strategies.
Inflammatory Cell Types Involved in Lung Neoplasia
The most common inflammatory cell type in a tumor-bearing tissue is the macrophage or tumor-associated macrophage (TAM). These cells are more often associated with a worse prognosis in human cancers, such as breast, prostate, bladder, kidney, esophageal, squamous cell carcinoma, and endometrial cancers, 107 although for several types of cancer, elevated levels of TAMs are associated with a favorable prognosis, such as gastric, colorectal, and melanoma. 107 In either case, the macrophage phenotype and activation are likely a critical determinant of whether the macrophages are pro- or anti-tumorigenic (to be discussed in detail in a later section; see inducible nitric oxide [iNOS] and arginase pathways) Macrophages contribute to tumorigenesis in several key ways, including tumor progression, tumor growth, tumor angiogenesis, metastasis, and immunosuppression. 107 In mouse pulmonary tumorigenesis models, macrophages are typically elevated compared with control mice during the promotion stage and thereafter. 20, 21, 158 Depletion of macrophages from the 2-stage carcinogenesis model using 3-methylcholanthrene (MCA) (initiator) and butylated hydroxytoluene (BHT) (promoter) significantly reduced tumorigenesis, implying a critical role for these cells in tumorigenesis. 21 Of interest, in urethane-induced A/J mice, TAMs did not enter the tumor parenchyma but remained on the outside periphery of the tumors. 158 Similarly, CD45+ lymphocytes also remain on the outside parenchyma. 158 The diversity among lymphocyte populations (i.e., multiple regulatory T-cell subtypes, multiple T-cells, etc.) suggests the role of the lymphocyte in neoplasia is more diverse than once expected. 191, 212 Lymphocytic infiltrate analysis in NSCLC tumors determined high numbers of these cells are CD3+ T-lymphocytes, primarily TCRα/β+, CD8+, CD28−. 47, 86, 207 Increases in regulatory T-cells (Tregs) in lung cancer patients were observed as well as increased expression of FOXP3 (a key regulatory gene for Tregs) in NSCLC patients. 79, 203 Tregs suppress other lymphocyte populations and presumably may inhibit anti-tumorigenic properties of these other lymphocytes. 203 Several stromal cells were observed to enter lung tumors, namely, polymorphonuclear neutrophils (PMNs) (just inside the tumor parenchyma) and endothelial cells (within tumor parenchyma and microvessels). 158 PMNs, while considered an acute response cell type, can induce nucleotide excision repair 63 and are one of the primary cell types involved in the inflammation elicited by environmentally induced AC, such as diesel exhaust 102 or particle exposure (carbon black, 48 vanadium pentoxide, A. K. Bauer and E. A. Rondini, unpublished data). PMNs are therefore likely involved at every stage of tumor development. Other stromal cell types do not change during tumor development; both mast cells and fibroblasts. 158 However, several cell types have not been fully investigated, such as dendritic cells, and based on their importance in many other pulmonary diseases, 39, 198 these cells are likely important in pulmonary neoplasia as well.
Epidemiological Studies Associating Chronic Inflammation and Pulmonary Neoplasia
Epidemiology studies suggesting a positive association of chronic inflammatory lung diseases including asthma, chronic obstructive pulmonary disease (COPD) (emphysema and chronic bronchitis), tuberculosis, and pneumonia to lung cancer risk in humans have recently been reviewed. 49 Overall, there is indication of a causal relationship; however, complexities in assessing the clinical course and severity of pulmonary inflammation in humans as well as controlling for confounding variables limits definitive conclusions. More consistent associations on lung cancer risk were observed among asthmatics and individuals with serologic evidence of Chlamydia pneumoniae infection. 109, 167 Studies evaluating use of nonsteroidal anti-inflammatory drugs (NSAIDs) also suggest that chronic inflammation promotes lung carcinogenesis. In a recent meta-analysis, Khuder et al. (2005) 94 reported a 32% reduced risk of lung cancer among users of NSAIDs when limiting analysis to studies that adjusted for smoking. Collectively, these and other epidemiologic studies imply that pulmonary inflammation is a risk factor for lung neoplasia in humans.
Genetic Similarities between Nonneoplastic and Neoplastic Pulmonary Diseases
While an estimated 87% of lung cancers are the result of smoking, less than 20% of cigarette smokers develop lung cancer. 205 Many factors play a role in interindividual variation, such as extrinsic factors including previous disease history, nutritional factors, and exposure to pollutants, and intrinsic factors including gender, age, and genetic susceptibility. There is an increased risk of developing lung cancer among relatives of patients with the disease. 85 It is difficult to study genetics in human populations due to the large variance in responses; however, the inbred mouse is homogeneous in nature, making it an important model for genetic studies. 149 Humans and mice share ∼99% of their genes, demonstrating the similarities between the 2 species. 193
Mouse studies have also demonstrated a genetic component of lung cancer. 15, 114, 115 Table 1 contains the genes found in common between pulmonary inflammation and pulmonary neoplasia models in mouse gene mapping studies, which identified quantitative trait loci (QTL), or regions on the mouse chromosome responsible for certain traits (i.e., inflammation or tumor phenotypes).∗ For more details on these specific studies, please refer to Bauer et al. (2004). 23 Only one mouse model is able to directly test the role of inflammation in lung tumor promotion. In BALB/By mice, the MCA/BHT 2-stage carcinogenesis model induces tumors only in mice receiving both the initiator (MCA) and the promoter (BHT). Therefore, those tumors that arise are the result of promotion. Chronic BHT alone induces pulmonary inflammation characterized by significant increases in alveolar macrophages and lymphocytes, as well as increased hyperpermeability. QTL sites on chromosomes 17 and 18 (see Table 1) were identified for both BHT-induced inflammation and promotion implicating the involvement of these 2 sites in the mechanism underlying inflammation-induced promotion. For all of the 8 sites identified in Table 2, candidate genes have been suggested. In later sections of the review, we will discuss the additional studies done for some of these candidate genes (Tnf, Nos2, and Tlr4).
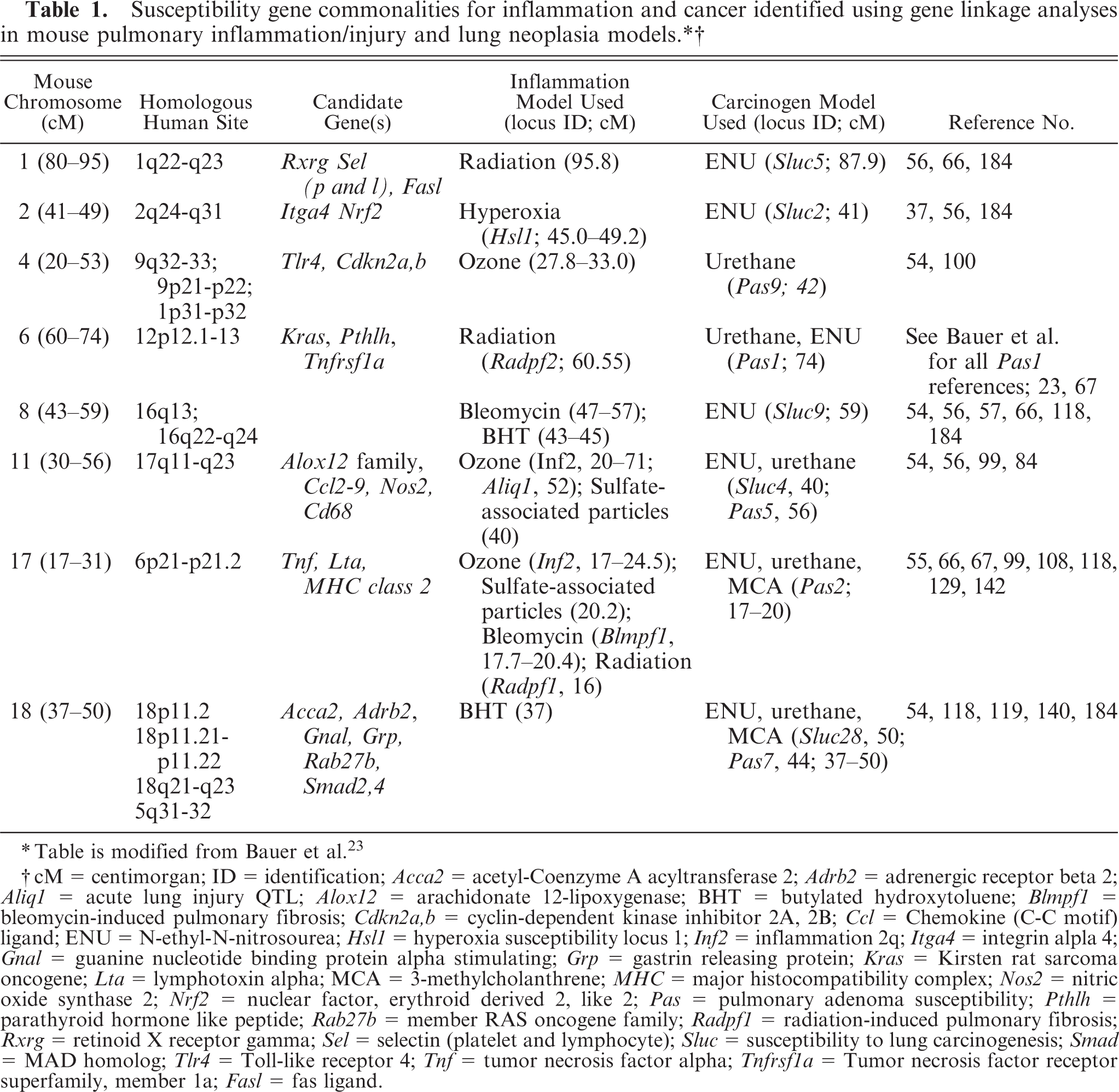
∗ Table is modified from Bauer et al. 23
† = centimorgan; ID = identification; Acca2 = acetyl-Coenzyme A acyltransferase 2; Adrb2 = adrenergic receptor beta 2; Aliql = acute lung injury QTL; Aloxl2 = arachidonate 12-lipoxygenase; BHT = butylated hydroxytoluene; Blmpfl = bleomycin-induced pulmonary fibrosis; Cdkn2a,b = cyclin-dependent kinase inhibitor 2A, 2B; Ccl = Chemokine (C-C motif) ligand; ENU = N-ethyl-N-nitrosourea; Hsll = hyperoxia susceptibility locus 1; Inf2 = inflammation 2q; Itga4 = integrin alpla4; Gnal = guanine nucleotide binding protein alpha stimulating; Grp = gastrin releasing protein; Kras = Kirsten rat sarcoma oncogene; Lta = lymphotoxin alpha; MCA = 3-methylcholanthrene; MHC = major histocompatibility complex; Nos2 = nitric oxide synthase 2; Nrf2 = nuclear factor, erythroid derived 2, like 2; Pas = pulmonary adenoma susceptibility; Pthlh = parathyroid hormone like peptide; Rab27b = member RAS oncogene family; Radpfl = radiation-induced pulmonary fibrosis; Rxrg = retinoid X receptor gamma; Sel = selectin (platelet and lymphocyte); Slue = susceptibility to lung carcinogenesis; Smad = MAD homolog; Tlr4 = Toll-like receptor 4; Tnf = tumor necrosis factor alpha; Tnfrsfla = Tumor necrosis factor receptor superfamily, member la; Fasl = fas ligand.
KRAS is a signaling mediator involved in many pathways, including proliferation, cell death, and inflammation. 2, 189 Recent studies have demonstrated that mutant Kras in mice (a conditional mutant with both CC10-Cre recombinase and the Lox-stop-Lox KrasG12D alleles) induces significant inflammation characterized by infiltration of alveolar macrophages and neutrophils (PMNs), inflammatory chemokine release (Cxcl2 [MIP-2], Cxcl1 [Gro-α; KC], monocyte chemoattractant protein-1 [MCP-1], and Cxcl5 [LIX]), and adenomas by 3 weeks of age. 84
Some single nucleotide polymorphism (SNP) associations exist for human lung neoplasia and inflammation genes, such as IL1b, TNF, and KRAS (see Table 2).† However, many of the gene association studies in humans have not fully evaluated the functionality of the SNPs, therefore the relevance to human lung cancer is unclear (PTGS2, CSF2, ICAM1, IL1a, IL8R1, IL12a, PPARG, and PPARD) (Table 2). Four genes (GSTT1, IL1b, KRAS, and TNF) have SNPs that are functional and involve increased risk of lung neoplasia. TNF-308 has also recently been identified as a significant contributor to other pulmonary diseases, such as COPD, fibrosing alveolitis, and idiopathic pulmonary fibrosis. 159, 165, 199 Activating KRAS mutations, including G12D, are found in 20–40% of human patients. 163 Unfortunately, despite much effort, KRAS-targeted drugs have been unsuccessful. 43 Myeloperoxidase (MPO), which is a phase 1 enzyme that can convert carcinogens to more active and toxic compounds, such as benzo[a]pyrene, is associated with decreased lung cancer risk in individuals with an SNP that decreases MPO activity. 181 One study recently demonstrated that IL6-634 was not significantly associated with lung neoplasia unless the individual was also asthmatic or atopic. 171 Thus, these association studies as well as mouse linkage studies demonstrate that genetic similarities exist between nonneoplastic and neoplastic pulmonary diseases in both mouse and man.
Significant polymorphisms in inflammation genes associated with either increased or decreased lung cancer risk in humans. ∗
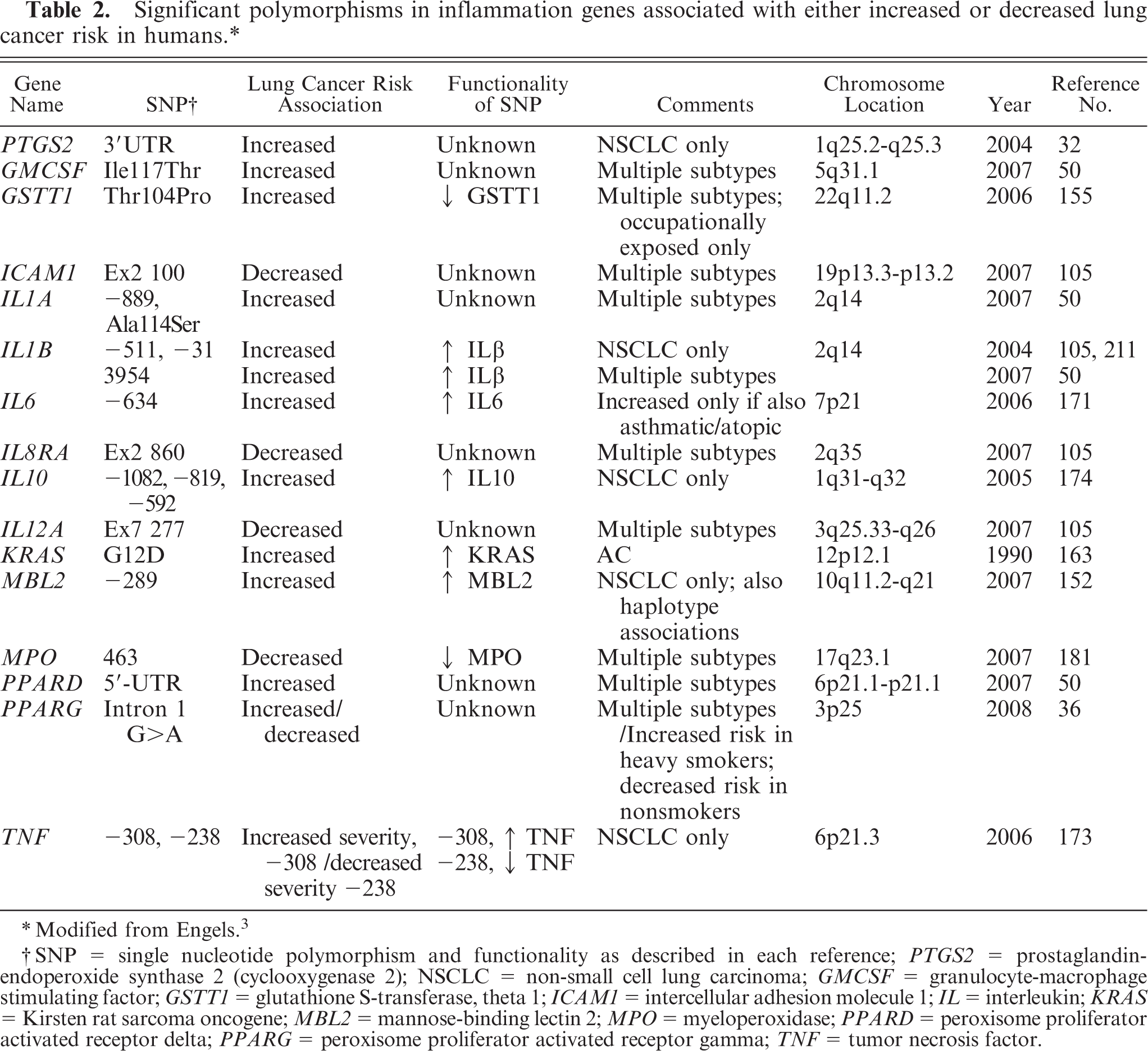
∗ Modified from Engels.3
†SNP = single nucleotide polymorphism and functionality as described in each reference; PTGS2 = prostaglandin-endoperoxide synthase 2 (cyclooxygenase 2); NSCLC = non-small cell lung carcinoma; GMCSF = granulocyte-macrophage stimulating factor; GSTT1 = glutathione S-transferase, theta 1; IC AM1 = intercellular adhesion molecule 1; IL = interleukin; KRAS = Kirsten rat sarcoma oncogene; MBL2 = mannose-binding lectin 2; MPO = myeloperoxidase; PPARD = peroxisome proliferator activated receptor delta; PPARG = peroxisome proliferator activated receptor gamma; TNF = tumor necrosis factor.
While there are now many studies that demonstrate the involvement of inflammatory mediators in in vitro models, such as Dwyer-Nield et al., 46 we will now focus on in vivo models using pharmacologic methods to inhibit inflammation, both general inhibition and specific mediator inhibition, or null and transgenic models of inflammatory mediators in primary lung tumorigenesis studies.
Pharmacologic Inhibitors of Inflammation
Studies examining pharmacologic inhibitors of inflammatory-mediated pathways provide further evidence for a causal role of inflammation in lung carcinogenesis. A summary of the results from these studies is presented in Table 3.
Effect of Pharmacologic Inhibitors on Mouse Lung Neoplasia. ∗
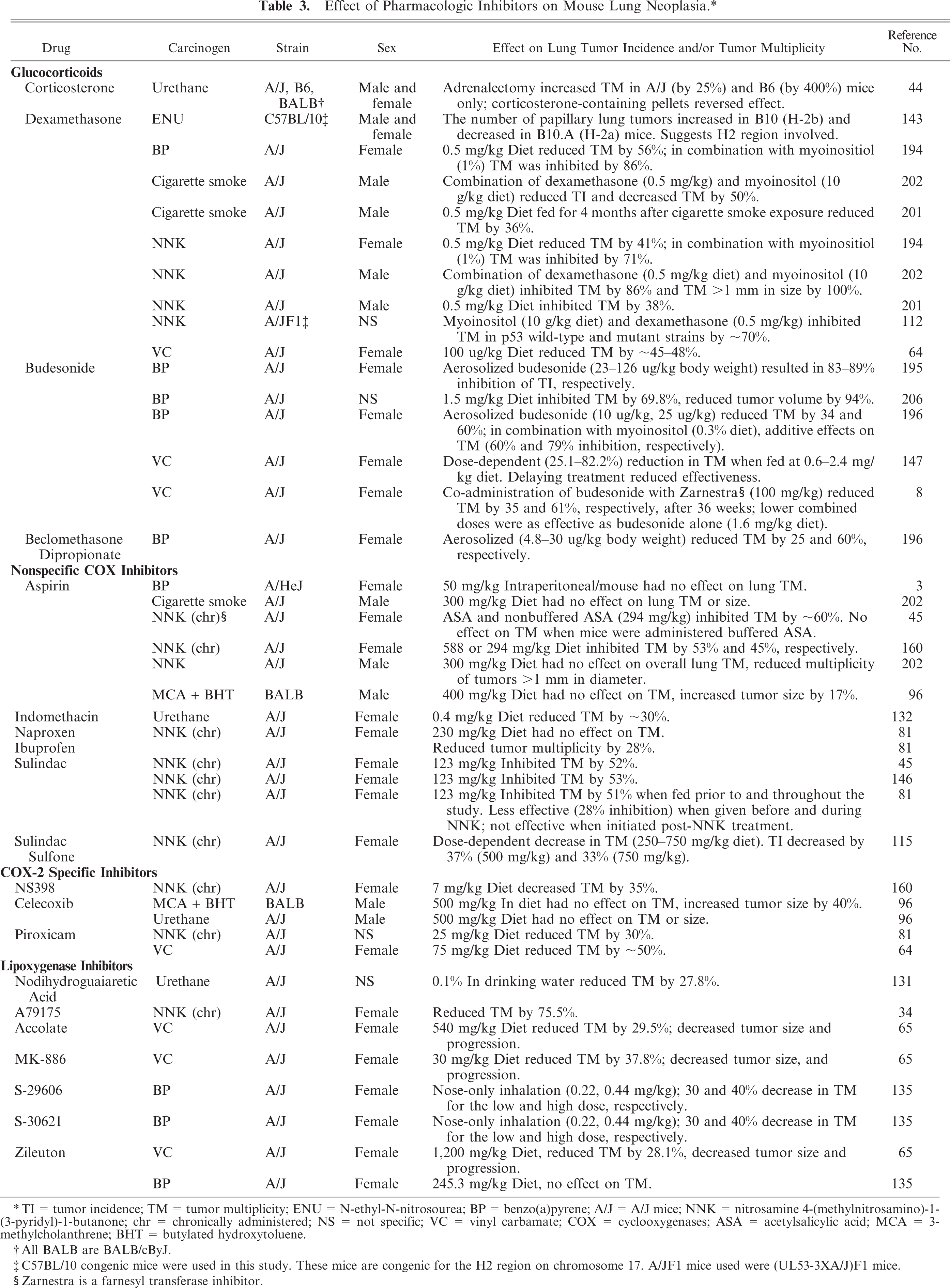
∗
† All BALB are BALB/cByJ.
‡ C57BL/10 congenic mice were used in this study. These mice are congenic for the
§ Zarnestra is a farnesyl transferaser inhibitor.
Glucocorticoids
Synthetic glucocorticoids (GCs) are widely used for treatment of chronic inflammatory conditions due to their ability to suppress a number of immune- and inflammatory-mediated responses. 137, 177 For example, they stimulate apoptosis in certain immune cells and inhibit the expression of pro-inflammatory chemokines, cytokines, adhesion molecules, and enzymes such as inducible nitric oxide synthase (NOS2; iNOS) and cyclooxygenases (PTGS; COX). 6, 137, 177 The biologic effects of GC are thought to be mediated primarily through nuclear receptor–dependent regulation of gene transcription, although posttranscriptional mechanisms have been reported. 138, 161 Binding of GC to its cognate receptor releases it from an inactive complex in the cytoplasm and facilitates nuclear translocation, dimerization, and subsequent modulation of gene expression. 25, 137, 177 The GC-receptor complex can activate or repress transcription of target genes by binding glucocorticoid responsive elements (GREs) on promoter regions of DNA. 25, 82, 134, 164 Alternatively, the GC receptor can interfere with signaling pathways through direct interaction with transcription factors including AP-1 and NFκB. 137, 157, 177, 200 Several pro-inflammatory mediators are regulated in this manner. 6, 177
Due to the ability to suppress a number of inflammatory indices, GCs have been evaluated as chemopreventative agents in various mouse models of lung carcinogenesis (Table 3). Initial studies demonstrated dietary administration of the synthetic GC dexamethasone (0.5 mg/kg diet) or budesonide (0.6–2.6 mg/kg diet) reduced tumor multiplicity, with effectiveness ranging from 41 to 82% of controls depending on the dose given.‡ Synergistic effects up to 30% were observed when co-administered with dietary-derived myoinositol (1% of diet) or Zarnestra, a farnesyl transferase inhibitor. 8, 112, 194, 202 Subsequent studies examined the potential of aerosolized GCs to inhibit lung tumorigenesis. 196, 197 This type of targeted delivery is beneficial as it requires lower doses than needed by oral route, thereby limiting the potential for chronic, adverse systemic effects. 196 At doses calculated to be similar to those used to treat moderately severe asthma in humans (5.7–8.6 µg/kg body weight), budesonide inhibited tumor formation by 34 to 60% 196 ; reductions of up to 79% were observed when administered with myoinositol, and up to 90% inhibition was achieved at higher doses. 195
Despite promising results from these studies, the mechanisms of chemoprevention have not been examined in detail. In addition to anti-inflammatory effects, GCs are known to affect cell cycle regulation; they enhance differentiation of alveolar type-II cells 38, 117 and reduce proliferation of bronchial and alveolar epithelial cells. 44, 147 Tumor tissue appears to be sensitive to growth inhibitory properties of budesonide, 147 potentially through induction of growth arrest and/or activation of apoptotic pathways. 206 Due to the association of inflammation on cell cycle indices, however, it is difficult to evaluate the independent contribution of each to tumorigenesis in vivo. Recently, Malkinson 117 proposed a model for budesonide inhibition of mouse lung tumor development whereby transcriptional repression of iNOS by budesonide results in decreased synthesis of nitric oxide (NO) from arginine. This subsequently restores the balance of pro- and anti-tumorigenic prostaglandins (PGs). 117 Of interest, a recent epidemiology study suggests that individuals with COPD using inhaled steroids are at reduced risk of developing pulmonary neoplasia. 144
Nonsteroidal anti-inflammatory drugs
Nonsteroidal anti-inflammatory drugs (NSAIDs) inhibit the COX enzymes and are among the most extensively studied drugs (Fig. 1). There are 2 main isoforms of COX, which share 60% homology at the protein level. 176 PTGS1 (COX-1) is constitutively expressed in many tissues, whereas PTGS2 (COX-2) can be induced by pro-inflammatory cytokines, growth factors, lipopolysaccharides (LPS), and mitogens. 70, 176 In the lung, both isoforms are abundant in the epithelial cells of the upper airways, 22, 51 but present with less intensity in the bronchiole epithelium, type-II alveolar cells, smooth muscle, and macrophages. 22 Increased expression of COX-2 has been detected in human lung cancers, 51, 71, 204 and both isoforms are elevated in urethane-induced lung tumors in mice, 22 making them attractive targets for cancer prevention.
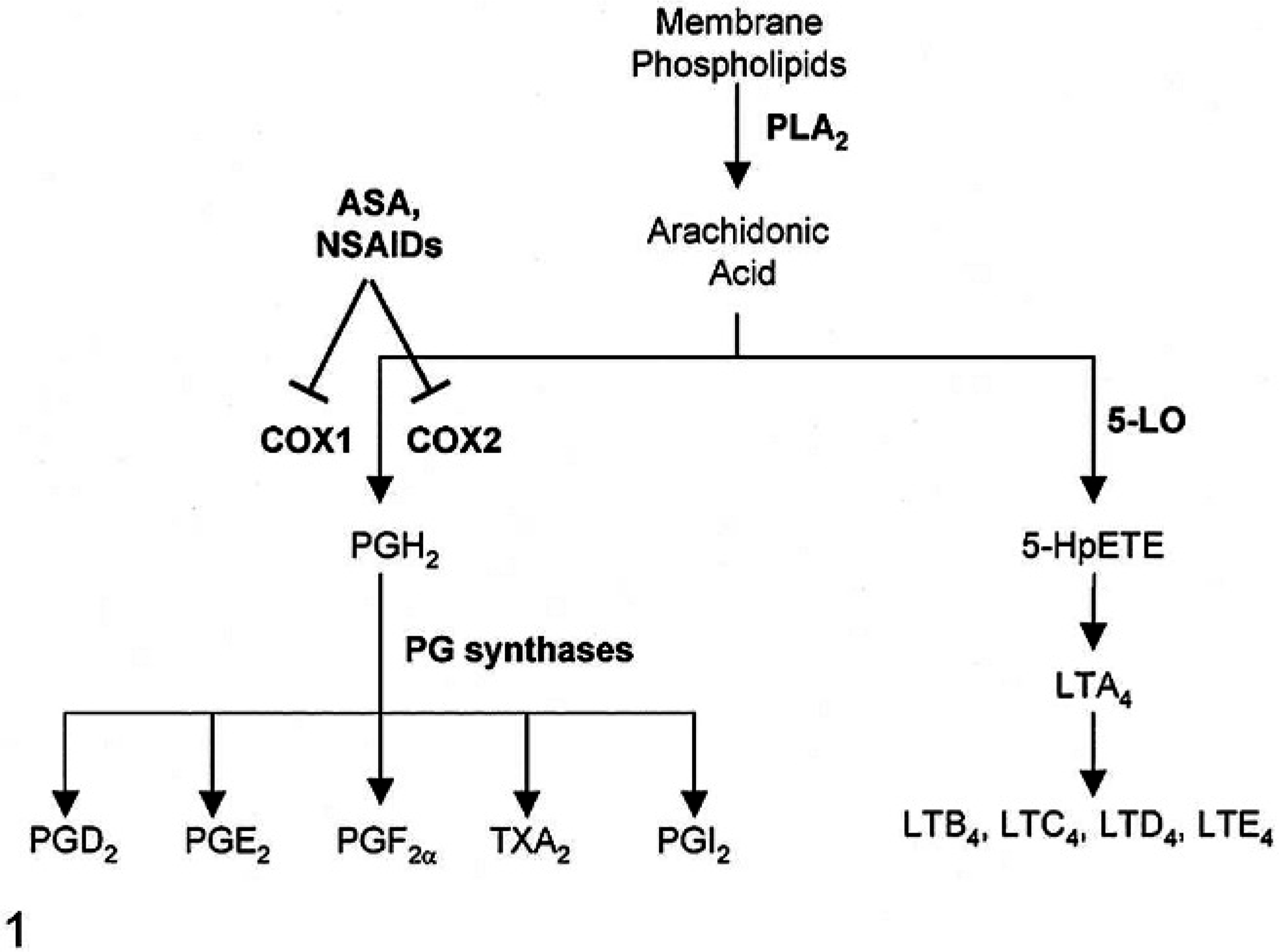
Arachidonic acid (AA) pathways evaluated in mouse lung neoplasia models. Phospholipase A2 (PLA2), cyclooxygenase (COX), prostaglandin E2 (PGE2), prostacyclin (PGI2), 5-Lipoxygenase (5-LO), leukotrienes (LT).
Inhibition of COX activity leads to a reduction in prostaglandins, prostacyclins, and thromboxane synthesis, although COX-independent actions have also been described. 69, 150, 187 The enzyme phospholipase A2 releases arachidonic acid (AA) from membrane phospholipids, which can then be metabolized to prostaglandin H2 (PGH2) by COX isoforms or to 5-hydroperoxyeicosatetraenoic (5-HpETE) by 5-lipoxygenase (5-LO, Fig. 1). 33 PGH2 is further metabolized by specific synthesis to produce PGE2, PGF2α, PGI2, or thromboxanes. whereas 5-HpETE metabolism produces leukotrienes. 33 Lipid mediators downstream of COX have differing effects on cell growth. PGE2 has been the most extensively studied prostaglandin and is generally considered pro-tumorigenic by promoting cell proliferation and angiogenesis and blocking apoptotic cell death.§ In contrast, PGI2 exerts anti-tumorigenic properties by suppressing inflammation, platelet aggregation, and metastasis. 76, 122
Studies examining the effect of NSAIDs on tumor incidence and/or multiplicity in mice are presented in Table 3.∥ Results are inconsistent depending on the protocol used to initiate tumorigenesis. Most studies used tobacco-derived NNK as the carcinogen in a chronic design in which it is fed in drinking water for 7 weeks. In these studies, treatment with NSAIDs was initiated 2 weeks prior to NNK administration and continued throughout the course of the study. With the exception of one study, 81 reductions in tumor multiplicity ranging from 30 to 88% to that of controls were reported. The relative sensitivity of this model may be limited, in part, to initiation events, 81 as some NSAIDs were found to inhibit NNK bioactivation by p450 45 or COX isozymes. 160 Less conclusive results were observed when different protocols were used to initiate carcinogenesis; some NSAIDs reduced, 132, 202 while others had no effect 3, 96, 202 on tumor multiplicity.
Although much attention has focused on a promotional role of COX-derived PGE2, this does not consistently correlate to tumorigenesis in murine models of lung cancer. Rioux 160 examined varying doses of acetylsalicylic acid (ASA) (73–588 mg/kg diet) along with the selective COX-2 inhibitor NS-398 (7 mg/kg diet) using the chronic NNK tumorigenesis model. They found ASA reduced tumor multiplicity in a dose-dependent manner. Plasma levels of PGE2 also dose-dependently decreased, and reductions correlated positively to tumor multiplicity (r 2 = 0.98). Two other studies found no predictive value of PGE2 levels on tumor growth. 35, 98 Castonguay et al. 35 reported reduced plasma PGE2 levels in ASA-, sulindac-, and Naproxen-treated mice compared with controls; however, only sulindac and ASA were effective in reducing tumor multiplicity. Similar results were found by Kisley et al. 98 who reported a 60% reduction in PGE2 by Celexicob in urethane-induced tumors, but no effect on tumor multiplicity or size. From these studies, it appears that pharmacologic inhibition of COX enzymes does not strongly predict reduced tumorigenesis. This may be related to concomitant reductions in anti-tumorigenic PGI2 91, 92 and/or enhanced production of leukotrienes 130 when COX activity is blocked.
5-Lipoxygenase inhibitors
5-LO, an alternative pathway in AA metabolism produces the leukotrienes LTA4, LTB4, LTC4, LTD4, and LTE4 (Fig. 1). 33 Inhibitors of this pathway reduce lung cancer cell growth 13 and enhance apoptotic cell death. 13, 131 In murine models of lung cancer, 5-LO inhibitors reduced tumor multiplicity by up to 40%, regardless of the carcinogen used (Table 3). 34, 65, 131, 135 In most of these studies, inhibitors were administered following carcinogen treatment, indicating an antipromotional role for these compounds. The effectiveness of some 5-LO inhibitors as therapeutic drugs in humans, however, may be limited by hepatotoxicity. 148 Recently, Myrdall et al. 135 tested 2 compounds by nose-only inhalation in mice and found both to be effective in reducing tumor development at much lower doses than orally administered Zilueton.
Genetic Modulation of Specific Inflammation Pathways That Augment or Protect from Pulmonary Tumorigenesis
AA pathway
As described above, the AA pathway is complex and involves both pro-inflammatory and anti-inflammatory mediators. cPLA2 –deficient mice developed significantly less tumors following urethane treatment than was observed in the wild type (wt) controls (Table 4). 127 Further down the pathway, metabolites of the cyclooxygenase pathway, namely, PGE2 and PGI2, have differing roles in inflammation. PGI2 synthase over-expressing mice developed significantly decreased tumor multiplicity compared with wt controls in response to urethane, 2-stage carcinogenesis (MCA/BHT), and a cigarette-smoking model (Table 4). 91, 92 In contrast, mice deficient in prostaglandin E2 receptor EP2 subtype (EP2), one of the receptors for PGE2, developed significantly less tumors than respective wt controls (Table 4). 90 However, mice over-expressing lung specific microsomal PGE synthase (mPGES)-1, producing elevated levels of PGE2, were not more susceptible to pulmonary carcinogenesis. 28 Although the two studies specific to the PGE2 pathway are perplexing, multiple isoforms of PGES exist as well as PGE2 receptors, which may explain some of these discrepancies. These findings may also explain why mice deficient in COX-1 did not develop less tumors than wt controls after urethane treatment (A. K. Bauer and R. Langenbach, unpublished results). In most tissues, the COX-1 null findings may be expected, but in pulmonary tissues, activation of both COX-1 and COX-2 elicits chronic inflammation and both enzymes are expressed in pulmonary tumorigenesis. 20, 22 The balance between PGI2 : PGE2 levels likely determines the inflammatory nature of this pathway. Thus, if COX-1 or COX-2 is inhibited (either pharmacologically or genetically), both PGI2 and PGE2 are also inhibited, likely canceling out or decreasing the effectiveness of the inhibition.
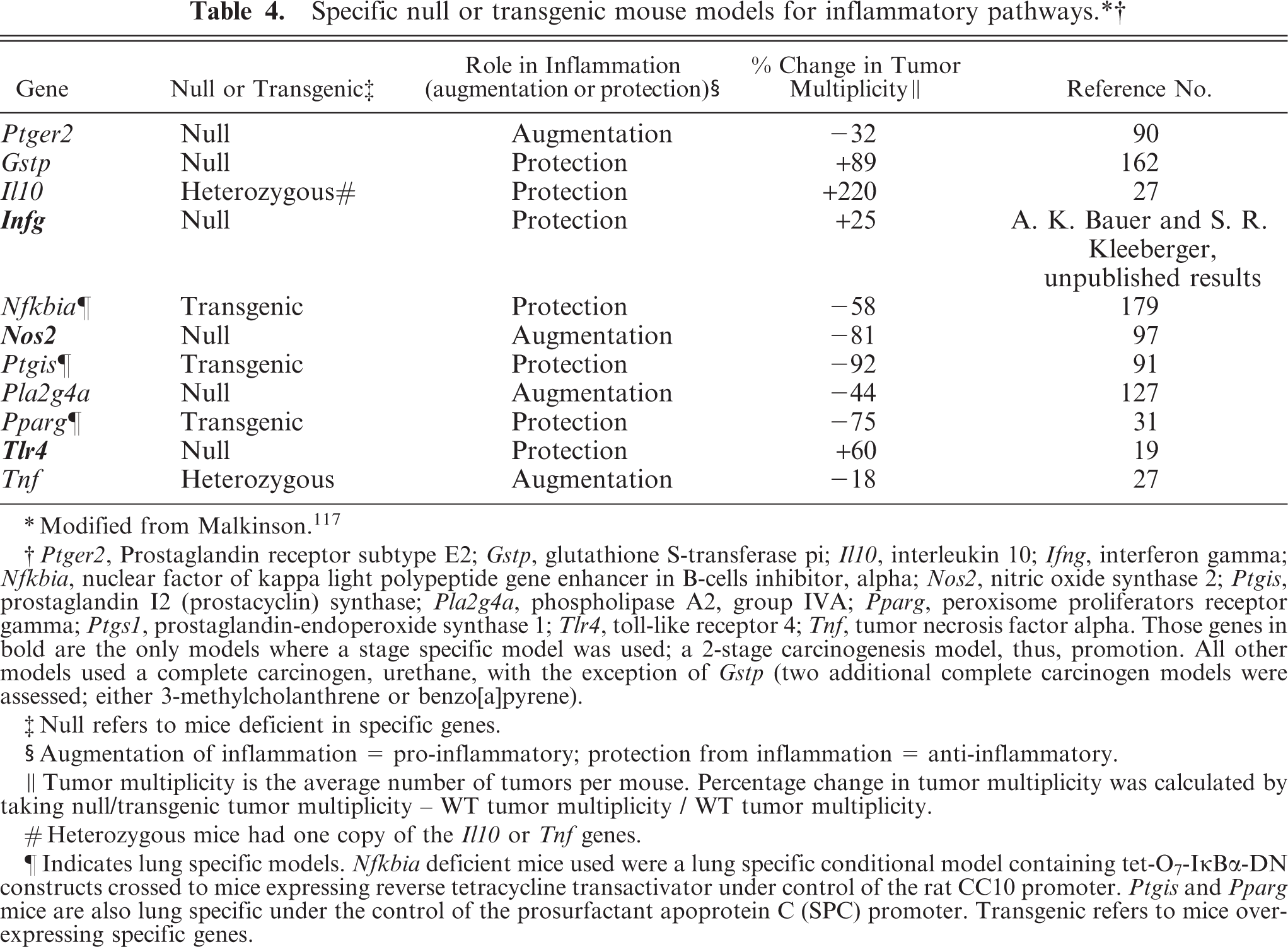
∗ Modified from Malkinson. 117
† Ptger2, Prostaglandin receptor subtype E2; Gstp, glutathione S-transferase pi; Il10, interleukin 10; Ifng, interferon gamma; Nfkbia, nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha; Nos2, nitric oxide synthase 2; Ptgis, prostaglandin 12 (prostacyclin) synthase; Pla2g4a, phospholipase A2, group IVA; Pparg, peroxisome proliferators receptor gamma; Ptgs1, prostaglandin-endoperoxide synthase 1; Tlr4, toll-like receptor 4; Tnf, tumor necrosis factor alpha. Those genes in bold are the only models where a stage specific model was used; a 2-stage carcinogenesis model, thus, promotion. All other models used a complete carcinogen, urethane, with the exception of Gstp (two additional complete carcinogen models were assessed; either 3-methylcholanthrene or benzo[a]pyrene).
‡ Null refers to mice deficient in specific genes.
§ Augmentation of inflammation = pro-inflammatory; protection from inflammation = anti-inflammatory.
∥ Tumor multiplicity is the average number of tumors per mouse. Percentage change in tumor multiplicity was calculated by taking null/transgenic tumor multiplicity - WT tumor multiplicity /WT tumor multiplicity.
# Heterozygous mice had one copy of the Il10 or Tnf genes.
¶ Indicates lung specific models. Nfkbia deficient mice used were a lung specific conditional model containing tet-
Nuclear factor kappa B
Nuclear factor kappa B (NFκB) is a transcription factor involved in many signaling pathways, including those for proliferation, apoptosis, and differentiation. 89 In vivo evidence demonstrated a promoting role for NFκB in some cancers, such as hepatocarcinogenesis and colitis-associated cancer. 61, 113, 151 However, in other cancer models, such as squamous cell carcinomas, NFκB appears to protect against neoplastic development. 41, 170, 188 Ikkβ is required for NFκB activation. In one chemically induced hepatocarcinogenesis model using a hepatocyte-specific Ikkβ deficient mouse, the Ikkb −/− mice developed a marked increase in tumor development compared with wt controls. 113, 166 However, if the Ikkβ deficiency was in both hepatocytes and hematopoietic-derived Kupffer cells, chemically induced hepatocarcinogenesis was significantly reduced. 113 Thus, tissue and cell specificity of the models as well as the carcinogen used determines the outcome of the deficiencies in or the inhibition of the NFκB pathway.
A recent study in lung using mice over-expressing Ikbα, an inhibitor of NFκB, demonstrated that the inhibition of NFκB greatly decreased tumor multiplicity in urethane-induced carcinogenesis compared with those observed in wt mice (Table 4). 179 In this study, the authors also demonstrated that inflammation was inhibited in the lungs of mice over-expressing Ikbα. It is therefore expected that some of the pro-inflammatory mediators downstream of NFκB are involved in this mechanism, such as TNF (see Cytokines below and Table 4).
Cytokines
Tumor necrosis factor α (TNF), a pro-inflammatory cytokine, is one of the most well studied cytokine pathways that induce other inflammatory pathways, including proteases. 16 TNF is involved in many features of tumorigenesis, from initiation through malignancy. 16 Deficiencies in TNF and TNF receptors decreased carcinogenesis of the skin and liver. 11, 101, 133 In the lung, TNF deficient heterozygous (−/+) mice developed significantly less tumors compared with the wt controls following urethane-induced carcinogenesis (Table 4). It is likely that the response in a homozygous TNF-deficient mouse would be significantly higher than observed using the heterozygous model. Tnf is located on chromosome 17, one of the major QTL sites identified for pulmonary carcinogenesis models as well as most pulmonary inflammation models. 23 Of interest, this gene is clustered with the two lymphotoxin (LT) genes (Lta and Ltb), which can also bind to TNF receptors, in addition to the LT receptor. 4, 125, 186 Lta is partly responsible for O3-induced pulmonary inflammation and injury. 190 Thus, the involvement of the TNF cluster in other pulmonary inflammation models as well as carcinogenesis models is probable.
Interleukin 10 (IL10) is considered an anti-inflammatory Th2 cytokine, because it suppresses macrophage and dendritic cell (DC) function, including production of pro-inflammatory cytokines and antigen presentation. 141 However, this suppression can result in feedback mechanisms of both the Th1 and Th2 responses. In extrapulmonary carcinogenesis, IL10 deficient mice were resistant to ultraviolet (UV)-induced skin carcinogensis. 111 IL10 is immunosuppressive to CD8+ T-cell effector functions and DC maturation, which can lead to impaired antitumor responses. 42, 141 In clinical use, recombinant IL10 used to suppress endotoxin effects resulted in immune stimulatory capacities as well as anti-inflammatory roles. 141 In an IL10 heterozygous (−/+) mouse model, enhanced lung tumor multiplicity was observed in female mice only in response to urethane-induced pulmonary carcinogenesis, compared with wt controls (Table 4). 27 Therefore, IL10 appears to both suppress pro-inflammatory pathways, such as TNF, and impair antitumor responses. Additional transgenic and transfectant IL10 tumor models have suggested that lower IL10 levels may be immunosuppressive, while higher IL10 levels may induce immune activation events, such as tumor rejection. 141 In lung, gender also appears to be involved in the IL10 response; the mechanism behind the involvement of gender remains unknown. NSCLC patients expressing elevated IL10 levels in tumor-associated macrophages (TAM) were associated with a worse prognosis. 209 Thus, the tumor microenvironment in addition to the amount of IL10 may determine the outcome of the response. More details on macrophage subtypes and activation will be discussed below (see Inducible NOS and arginase pathways section).
Pulmonary protection through the innate immune system
The innate immune response in pulmonary neoplasia is a novel area of research. The most commonly studied receptors involved in innate immunity are the toll-like receptors. There are now 13 of these receptors, although the function of 10, 12, and 13 is unknown. Two subfamilies exist; the TLR2, 4, 5, 6, and 11 subfamily is expressed on the surface of cells, has numerous agonists, and can be phagocytosed. 24 Another subfamily is TLR3, 7, 8, and 9, which is localized inside the cell in the endoplasmic reticulum, endosomes, or lysosomes and recognizes nucleic acids. 24 TLR4 can both exacerbate and inhibit pulmonary inflammation and injury. For example, TLR4 exacerbates ozone- and lipopolysaccaride (LPS)-induced lung injury and inflammation. 100, 153 TLR4 protects against pulmonary infection (Pseudomonas aeruginosa and Bordetella pertussis), and ovalbulmin-induced and oxidant-induced (hyperoxia) pulmonary inflammation and injury. 53, 72, 75, 210 Additionally, in extrapulmonary organs, TLR4 confers protection against gastric and cutaneous carcinomas, in humans and mice, respectively. 74, 208 Mechanistically, Tlr4 dominant negative mice (C3H/HeJ) have no ability to present antigen and instead degrade the antigenic material from phagocytosed dying tumor cells. 9 In addition, monocyte-derived DCs from humans with a nonfunctional TLR4 mutation exhibited a marked reduction in antigen presentation. 9 In breast cancer patients, the metastasis-free survival rate was significantly longer in breast cancer patients with functional TLR4 versus mutated TLR4 (either 299 or 399 mutations). 9 Thus, collectively, TLR4 is protective in carcinogenesis.
Several epidemiologic studies observed significant decreases in lung cancer risk in those individuals exposed to endotoxin, such as farm and textile workers. 12, 103, 104, 123, 124 TLR4 is the primary receptor that binds endotoxin (lipopolysaccharide; LPS) 153 ; thus it is likely involved in the protection observed with endotoxin exposure. We recently demonstrated that in the BHT-induced chronic inflammation model, Tlr4-deficient mice were more susceptible to pulmonary inflammation and injury than the wt controls. These significant increases in cellular infiltrates were macrophages and lymphocytes as well as increases in total BAL protein content. 19 In separate studies, we also found a 60% increase in tumor multiplicity in Tlr4-deficient mice compared with wt mice 20 weeks following the MCA/BHT protocol. Thus, it appears that in pulmonary neoplasia, TLR4 acts to protect the pulmonary epithelium possibly by decreasing inflammation and pulmonary injury elicited by the oxidative metabolites of BHT.
Interferon (IFN)γ, a Th1 cytokine, is another key immunoregulatory mediator involved in promoting innate and adaptive mechanisms of host defense. 29, 52 It can stimulate natural killer cells, upregulate MHC class II molecules, and most importantly, activate macrophages. 30 More specifically, IFNγ increases NOS, indoleamine (2,3)-dioxygenase, an enzyme that degrades tryptophan, and reactive oxygen species (ROS). 30 In Ifng-deficient mice treated with MCA, fibrosarcoma development was significantly enhanced compared with wt controls. 88 Of interest, certain human tumors become unresponsive to IFNγ, 88 supporting a more general role of this cytokine in protection against tumorigenesis. In the 2-stage pulmonary carcinogenesis model using MCA and BHT, Ifng-deficient mice developed significantly more tumors than wt mice (A. K. Bauer and S. R. Kleeberger, unpublished results). Significant increases in BHT-induced chronic inflammation were also observed in the Ifng-deficient mice compared with wt controls. Anti-angiogenic chemokines regulated by IFNγ, such as CXCL9 (MIG) and CXCL10 (IP10), are also up-regulated in human cancers, 1, 10, 172, 182 including NSCLC. These studies on specific innate immune pathways imply that individuals with defective innate immune systems may be more susceptible to pulmonary neoplasia.
Inducible NOS and arginase pathways
Arginine can be converted to either nitric oxide (NO) and citrulline by NOS, or ornithine and urea by arginases. 128 There are 3 isoforms of NOS; however, iNOS is the isoform most well studied in inflammatory diseases. 5 NO is important in normal physiology; however, in inflammatory diseases, depending on the model used, NO can be both a detriment or a critical component in host protection, inducing cytotoxicity, and causing DNA damage and lipid peroxidation, among other roles, such as metastasis. 73, 128, 136 High concentrations of NO are typically considered detrimental due to the combination with superoxide to form peroxynitrite, a free radical. 78 In human NSCLC patients, both iNOS and exhaled NO levels are elevated, supporting a role of this mediator in pulmonary neoplasia. 110 iNOS (Nos2)-deficient mice had an 81% decrease in lung tumor multiplicity compared with wt mice following urethane-induced carcinogenesis (Table 4). 97 Vascular endothelial growth factor content was reduced in tumors from iNOS-deficient mice, thus, reduced angiogenesis may account for some of the reduction in tumor number. 97
One of the major iNOS-expressing cell types in the lung is the alveolar macrophage. Macrophage activation and specific phenotypes of macrophages vary during tumorigenesis. 16, 120, 128 In the lung, the type of macrophage activation appears to depend on the stage of neoplasia. 158 To simplify, 2 main activation pathways exist, M1, or classical activation, and M2, alternative activation 128 (Fig. 2). IFNγ plus LPS, or TNF can stimulate M1 macrophages to induce NO production, among other cytokines, leading to Th1 responses, as well as tumor resistance. 120 M2 activation is more complicated and several M2 macrophage phenotypes exist. To simplify, IL4 and IL13 can stimulate the M2 macrophages to convert arginine to ornithine by induction of arginase 1, leading to polyamine synthesis. 128 Polyamines are a necessary component of DNA synthesis and deplete the arginine substrate required for NO production. 93 IL10 is also induced in the M2 macrophages. 120 M2 activation by IL4 and IL13 results in increased Th2 responses and increased proliferation, or tumor promotion. 120, 121 Recently, Redente et al. 158 described how macrophages expressed arginase 1 (M2 subtype), but not iNOS (M1 subtype) in lungs with premalignant lesions. In contrast, those lungs bearing carcinomas expressed only iNOS. Bone marrow monocytes adopted these expression patterns before entering the circulation. These different phenotypes of macrophages are likely involved in regulating the Th1 versus Th2 cytokine response in the tumor microenvironment and appear to influence tumor development. Because of the bone marrow involvement, these 2 markers may be early biomarkers for pulmonary AC.
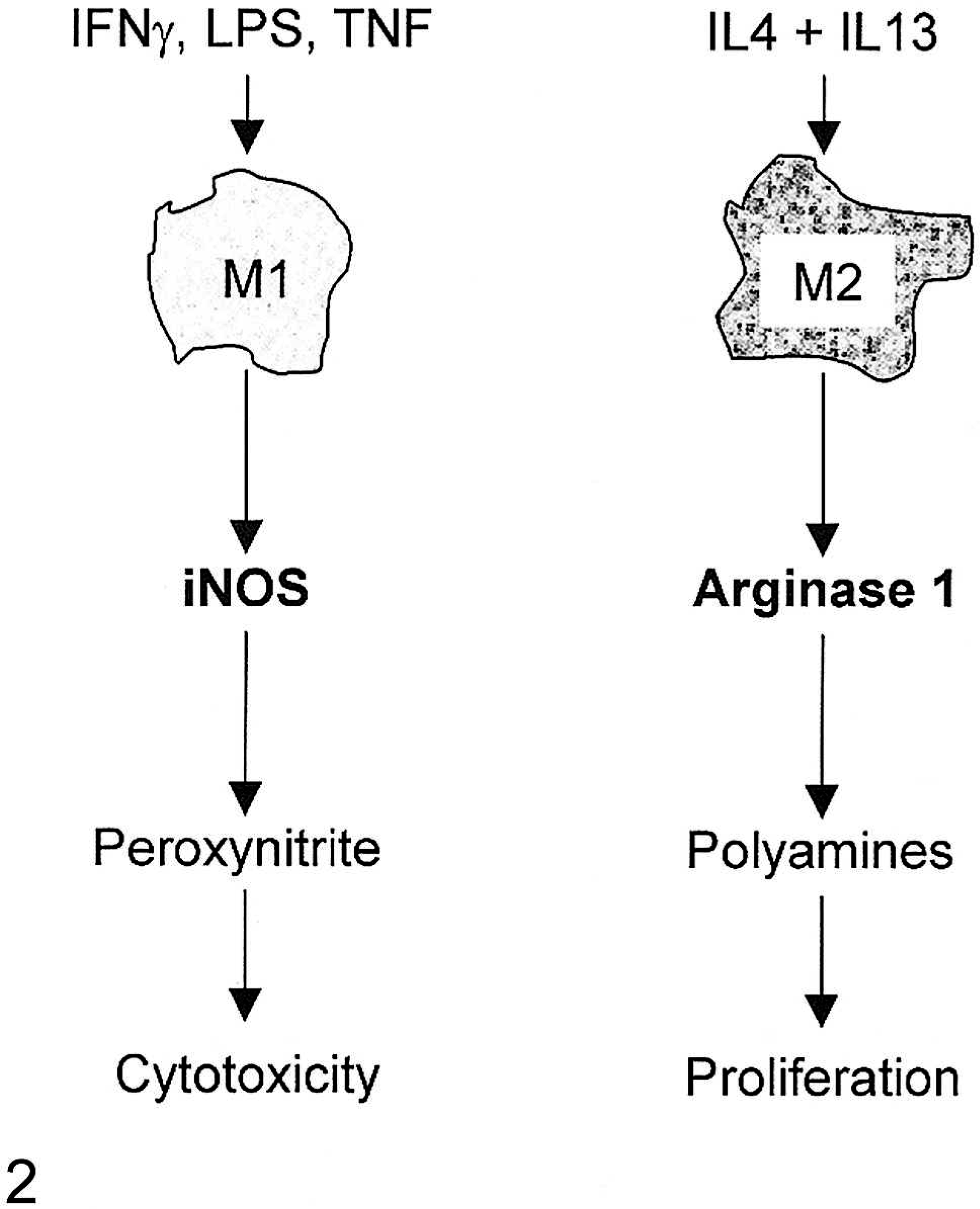
The 2 main macrophage activation phenotypes. Fig. 2A. Classical activation (left). Fig. 2B. Alternative activation (right). Interferon γ (IFNγ), interleukin 4 (IL4), interleukin 13 (IL13), inducible nitric oxide synthase (iNOS), lipopolysaccaride (LPS), tumor necrosis factor α (TNFα). (For a more detailed description of the many M2 phenotypes, please refer to Mantovani et al.) 120
Other anti-inflammatory pathways
Several other anti-inflammatory pathways have been evaluated in mouse pulmonary carcinogenesis. Peroxisome proliferator-activated receptor-(PPAR)γ inhibits TNF, NO, IL6, as well as proliferation of tumor cells. 26 Some PPARγ SNPs in lung are associated with decreased lung cancer risk, while others are associated with increased risk. 36 In lung-specific PPARγ–over-expressing mice, pulmonary tumorigenesis was significantly inhibited (75% reduction in tumor multiplicity), as well as suppression of COX-2 via NFκB. 31 Glutathione S-transferases (GSTs) are a family of enzymes (GSTα, µ, π, θ, σ, ζ, κ, ω) important in the detoxification of reactive electrophiles. 68 Some GST SNPs with decreased enzyme activity are also associated with increased pulmonary neoplasia in humans. 155 In mice, Gstp-deficient mice are more susceptible to pulmonary carcinogenesis (89% increased tumor multiplicity). 162 Thus, these studies also support the role of inflammation in pulmonary neoplasia and provide alternative pathways for prevention and therapeutics.
Other mouse models
Because metastasis is a topic deserving a review of its own, we will only briefly discuss mouse models assessing this component of pulmonary tumorigenesis. Matrix metalloproteinases (MMPs) are a family of enzymes involved in degrading the extracellular matrix proteins; many MMPs have been implicated in multiple tumor mechanisms, such as invasion and angiogenesis. 62 MMP9 (gelatinase B) promotes tumor metastasis in the Lewis Lung carcinoma model, a model for metastasis, while MMP12 (macrophage elastase) suppresses tumor metastasis. 77, 80 These diverse findings may explain why MMP inhibitors in lung cancer clinical trials have not been successful. 62 We refer the readers to Gueders et al. (2006) 62 for more details.
Summary
The evidence presented herein, both genetic and pharmacologic, supports a role for inflammation in pulmonary neoplasia. The mechanism behind the involvement of inflammation in this process is unclear; however, modulation of both pro- and anti-inflammatory pathways, as well as the involvement of specific cell types in inflammation, may explain how inflammation influences neoplastic growth. These novel pathways, including innate immune mediators, may lead to additional targets to explore in the prevention of lung cancer.
Footnotes
Acknowledgements
The authors would like to thank the intramural program NIEHS, Dr. Steven Kleeberger, and Michigan State University Department of PDI and the CIT.
∗ References 37, 54–57, 66, 67, 99, 100, 108, 118, 119, 129, 140, 142, 154, 184.
† References 32, 36, 49, 50, 105, 152, 155, 163, 171, 173, 174, 181, 211.
‡ References 7, 64, 143, 147, 194, 201, 206.
§ References 58–60, 87, 106, 139, 156, 175, 180, 185.
∥ References 3, 45, 64, 81, 96, 115, 132, 146, 160, 202.