
Abstract
Ectonucleoside triphosphate diphosphohydrolase type 5 (ENTPD5, also CD39L4) is a soluble enzyme that hydrolyzes purine nucleoside diphosphates. Genetic inactivation of ENTPD5 in mice (Entpd5 -/-) resulted in 2 major histopathologic lesions: hepatopathy and aspermia. The hepatopathy was progressive and characterized by centrilobular hepatocyte hypertrophy, oval cell proliferation, bile staining of Kupffer cells, and hepatocyte degeneration with increasing incidence and severity of degenerative lesions, development of multiple foci of cellular alteration, and hepatocellular neoplasia with age. Greatly increased proliferation of hepatocytes in young adult as well as aged Entpd5 -/- mice was demonstrated by Ki67 immunohistochemistry and 5î-bromo-3î-deoxyuridine incorporation. Of 15 Entpd5 -/- mice between 44 and 69 weeks of age, all showed foci of cellular alteration in the liver, and at least 6 of 15 developed hepatocellular carcinoma (HCC), hepatocellular adenoma, or both. Significantly, none of these lesions were observed in 13 wild-type Entpd5 ++ littermates. These findings, combined with the historically low incidence (about 5%) of HCC in mice up to 2 years of age with the same genetic background, strongly suggest that loss of Entpd5 promotes hepatocellular neoplasia in mice. In humans, ENTPD5 has been found to be identical to the PCPH proto-oncogene, and dysregulation of this gene has been demonstrated in some human cancers. This mouse model could contribute to the understanding of the influence of ENTPD5/PCPH on cellular proliferation and neoplasia.
Keywords
Introduction
Nucleotides (tri-, di-, and monophosphate [NTP, NDP, and NMP, respectively]) have diverse essential functions in the cell. Not only do they compose the building blocks of DNA and RNA, but they also function as energy sources, in protein folding, and in cell signaling. Nucleotides mediate intercellular signal transmission, at least in part, through G-protein–coupled P2 cell membrane receptors (P2X n , P2Y n , and P2T), 6, 7, 12 which have been shown to modulate cell metabolism, adhesion, activation, development, proliferation, differentiation, and apoptosis (reviewed in Robson et al.). 27 The levels of extracytoplasmic (extracellular and intraorganellar) nucleotides are modulated by the degradative activity of ectonucleoside triphosphate phosphohydrolases (ENTPDs). ENTPD family members include enzymes that are membrane bound, soluble within the endoplasmic reticulum (ER) and Golgi apparatus, secreted, or in a combination of these states. 27
ENTPD5 (also known as ER-UDPase) is a soluble enzyme that has been localized in the ER of rat hepatocytes, where it is believed to drive protein glycosylation with uridine diphosphate (UDP)–sugars. One of these pathways is a repetitive glucosylation/deglucosylation that assists in folding complex proteins. 31 In the ER, unfolded proteins bind and are monoglucosylated by UDP-glucose : glycoprotein glucosyltransferase (GT), causing them to bind calnexin or calreticulin (ER lectin/chaperones), which promotes protein folding. The folded protein is released by hydrolysis of the bound glucose; however, because several rounds of glucosylation/deglucosylation might be necessary to properly fold complex proteins, each round generating a molecule of UDP, several molecules of UDP could be generated in the process. Other glycosylation processes also use UDP-sugars, generating UDP as an end product. ENTPD5 hydrolyzes UDP to uridine monophosphate (UMP), which is then transported from the ER lumen to the cytoplasm in exchange for nucleotide sugars; this activity prevents end product inhibition and promotes the importation of reactants, thus driving glycosylation forward.
ENTPD5 has also been identified as the PCPH proto-oncogene. 24 The PCPH oncogene is a truncated form 33 of ENTPD5 that shows activating interactions with Ras that were not found with the native protein. 25
We report the effects of ENTPD5 deficiency in the mouse. The primary feature of the gene knockout presented in this manuscript is a progressive hepatopathy characterized by centrilobular hepatocyte hypertrophy with increased hepatocyte turnover, leading to the development of focal hepatocellular alterations and neoplasia with time. Our findings suggest a previously unknown role for Entpd5 in the development of hepatocellular neoplasia in mice and represent the first report clearly demonstrating the development of hepatocellular tumors in response to mutation of this gene. This knockout mouse model could provide insight into the pathogenesis and treatment of human hepatocellular carcinoma.
Materials and Methods
Generation of mutant Entpd5−/− mice
Entpd5 null mice (Entpd5−/− ) were generated by homologous recombination as described. 36 Exons 7–10 of the Entpd5 gene (accession NM_001026214.1) were deleted and replaced with a selection cassette containing both LacZ and Neo (see Fig. 1A). The upstream and downstream arms of homology were generated by long-range polymerase chain reaction (PCR) with the use of primers Entpd5-2 [5′-CAGCTTGCCACAGAGGACAGCTTACAGTTCG-3′] and Entpd5-5 [5′-GGTGAATTCTTGAAGATCTCCTCTACCTAGAG-3′] (upstream arm), and primers Entpd5-7 [5′-CATCTCCAGTCTGGGCCTCAAGCGTATGTG-3′] and Entpd5-9 [5′-GCACCCTCATTCTCCTTCCCGTCTACTGGG-3′] (downstream arm). Correctly targeted 129S5/SvEvBrd embryonic stem cell clones were confirmed by Southern analysis (see Fig. 1B) and were microinjected into C57BL/6J-Tyr c-Brd blastocysts. Resulting chimeras were mated to C57BL/6J-Tyr c-Brd females to generate heterozygous mice (Entpd5+/− ). Animal genotypes were determined by quantitative Neo PCR 11 and were confirmed by gene-specific genotyping with the primer set Entpd5-12 [5′- GGGAGGTGAACTAAGACGTACAG-3′] and GT-IRES [5′- GCTAGACTAGTCTAGCTAGAGCGG-3′].
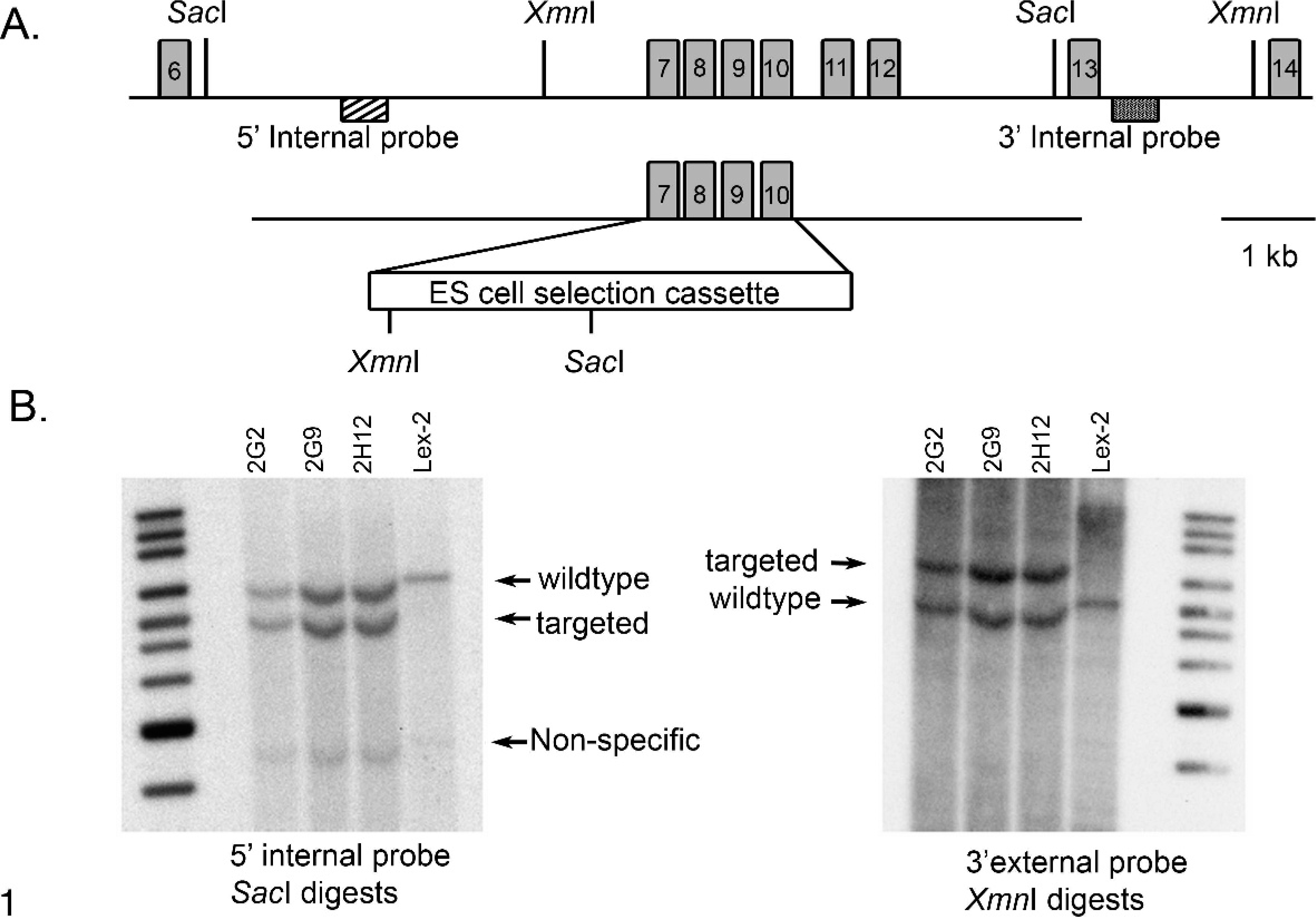
Construction of the Entpd5 targeting vector and Southern strategies for clone confirmation. Fig. 1A. A schematic representation of the Entpd5 locus is shown, including relevant restriction sites used for Southern hybridization analysis (SacI and XmnI). A homologous recombination vector targeting exons 7–10 of the Entpd5 locus was constructed with PCR-generated homology arms and a selection cassette containing a LacZ reporter gene, as well as a Neo gene conferring resistance to G418. Fig. 1B. Southern hybridization demonstrating proper targeting of the embryonic stem cell clones. Three correctly targeted clones were identified by Southern analysis with the use of both 5′ and 3′ probes (generated by genomic PCR). Clones 2G2 and 2G9 were injected into blastocysts, and clone 2G2 ultimately achieved germ line transmission. Lex-2 represents control 129S5/SvEvBrd embryonic stem cell DNA.
Mouse husbandry
Mice were housed in a barrier facility at 24°C on a fixed 12-hour light and 12-hour dark cycle and were fed rodent chow 5001 (Purina, St. Louis, MO) ad libitum. Procedures involving animals were conducted in conformity with the Institutional Animal Care and Use Committee guidelines that are in compliance with state and federal laws and standards outlined in the Guide for the Care and Use of Laboratory Animals. 23 Quarterly sentinel surveillance of the source colonies was conducted (Charles River Laboratories) and showed no evidence of infection by pathogenic rodent viruses, Mycoplasma sp. or Helicobacter sp.
The reference aging study was conducted on F1 mice generated as described above, to evaluate lesions in an unrelated gene knockout, but only results from the wild-type (Wt) control animals are presented. Mice were necropsied when reported for illness or at 2 years of age. Full gross and histologic examinations were performed on all mice.
Phenotypic screen
F2 generation Wt and homozygous knockout (Entpd−/− ) mice were subjected to a comprehensive battery of phenotyping screens, to include behavioral tests (such as circadian rhythm, open field, inverted screen, prepulse inhibition of the acoustic startle response, tail suspension, marble burying, and context trace conditioning), fundoscopy and angiography exams, blood pressure measurements, serum chemistries, complete blood count, urinalysis, quantitative magnetic resonance, CAT-scans, and Micro-CAT scans as previously described. 2, 37
Histopathology
Tissues were collected and immersed in 10% neutral buffered formalin for 48 hours, except for the eyes, which were removed and fixed by immersion in Davidson's fixative (Poly Scientific, Bay Shore, NY) overnight at room temperature. All tissues were embedded in paraffin, sectioned at 4 μm, mounted on positively charged glass slides (Superfrost Plus, Fisher Scientific, Pittsburgh, PA), and stained with HE for histopathologic examination. Tumors and other liver lesions were diagnosed histologically by 2 pathologists (R. Read and P. Vogel) using previously described diagnostic criteria. 18, 19
β-Galactosidase staining and LacZ histochemistry
As previously described,
35
anesthetized mice were perfused sequentially with β-galactosidase (β-Gal) fixative (0.2% glutaraldehyde, 1.5% paraformaldehyde, 2 mM MgCl2, 5 mM EGTA, 100 mM sodium phosphate [pH 7.3]), followed by 2 ml of β-Gal rinse (0.2% Nonidet-P40 [NP-40], 0.1% sodium deoxycholate, 2 mM MgCl2,100 mM sodium phosphate), and finally 10 ml of β-Gal stain (5 mM K3Fe[CN]6, 5 mM K4Fe[CN]6, 1 mg/ml 5-bromo-4-chloro-3-indolyl-
Immunohistochemistry
Immunohistochemistry for ubiquitin was performed with rabbit anti-ubiquitin (Dako Z0458) at 1 : 400 dilution followed by biotinylated goat anti-rabbit immunoglobulin G (IgG; Vector BA-1000) at 1 : 500 dilution and 3,3′-diaminobenzidine (DAB) chromogen. Immunohistochemistry for catalase was performed with rabbit anti-catalase (Abcam ab1877) at 1 : 500 dilution, followed by horseradish peroxidase (HRP)–conjugated goat anti-rabbit-IgG (Bethyl Laboratories A120-501P) and DAB chromogen. Immunohistochemistry for Ki-67 was performed with rat anti-mouse Ki-67 (Dako M7249) at 1 : 30 dilution and biotinylated rabbit anti-rat IgG (Vector BA-4001) at 1 : 400 dilution, and detection was performed with Vector Elite ABC Kit (PK-6100) and DAB chromogen with light hematoxylin counterstain. For bromodeoxyuridine (BrdU) labeling, mice were given 1 mg BrdU (BD Pharmingen, catalog No. 550891) intraperitoneally once, 5 days before necropsy. Tissues were processed and embedded in paraffin as described above. Sections were deparaffinized and stained with BrdU In Situ Detection Kit (BD Pharmingen, catalog No. 551321) according to the manufacturer's instructions with DAB chromogen and light hematoxylin counterstain.
Cytochrome P450 activity assessment
Portions of liver (about 1 g/piece) were collected and flash frozen in liquid N2 immediately after euthanasia of groups of 4 male and female Entpd−/− mice and groups of 2 age-matched Wt controls (45 to 55 weeks old). Hepatic microsomal fractions were prepared from all 12 samples. About 4 volumes of extraction buffer containing 100 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES, pH 7.2), 100 mM KCl, and 2 mM EDTA, as well as 1 mg/ml protease inhibitors, was added to each liver sample to yield an approximately 28% (w/v) homogenate. The liver tissues were homogenized with Mini Beadbeater. About 2 ml of homogenized mixture was transferred into a new collection tube. The samples were put into centrifugation at 9,000 rpm for 20 min. The supernatants were transferred into a new tube. Calcium chloride was added to a final concentration of 8 mM and mixed well, and the samples were subjected to centrifugation in a microfuge at 14,000 rpm for 60 min. Supernatants were discarded and the pellets were resuspended in the same extraction buffer with 8 mM CaCl2. The above step was repeated once. The microsomes were then resuspended into the same working buffer. The protein content was determined by BCA™ Protein Assay Kit (Pierce, Rockford, IL). Ethoxyresorufin O-dealkylation was used to assess CYP1A activity, warfarin 7-hydroxylation and omeprazole hydroxylation were used to assess CYP2C activity, dextromethorphan N-demethylation was used to assess CYP2D activity, and midazolam hydroxylation was used to assess CYP3A activity. A cocktail containing the 5 selective probe substrates was incubated with the mouse liver microsomes for 1 hour at room temperature. The reaction was terminated with the addition of an equal volume of methanol containing 5 μM verapamil. Each sample (10 μl) was injected onto an Agilent SB-C8 column using standard gradient conditions. Liquid chromatography–mass spectrometry (LC/MS/MS) was used to simultaneously measure the appearance of metabolites of each CYP-specific probe substrate. Differences between knockout (KO) and Wt mice were analyzed by 1-way analysis of variance (ANOVA) with Dunnett's post hoc test at P < .05.
Results
Mice homozygous for the deleted Entpd5 (Entpd5−/− ) were generated by breeding heterozygous parents. Homozygous Entpd5−/− mice were born in a normal (1 : 2 : 1) Mendelian ratio with their heterozygous and Wt littermates (actual ratio was 53−/− : 97+/− : 51+/+). Entpd5−/− mice appeared normal at birth but grew more slowly and were still stunted at 1 year of age (Fig. 2).
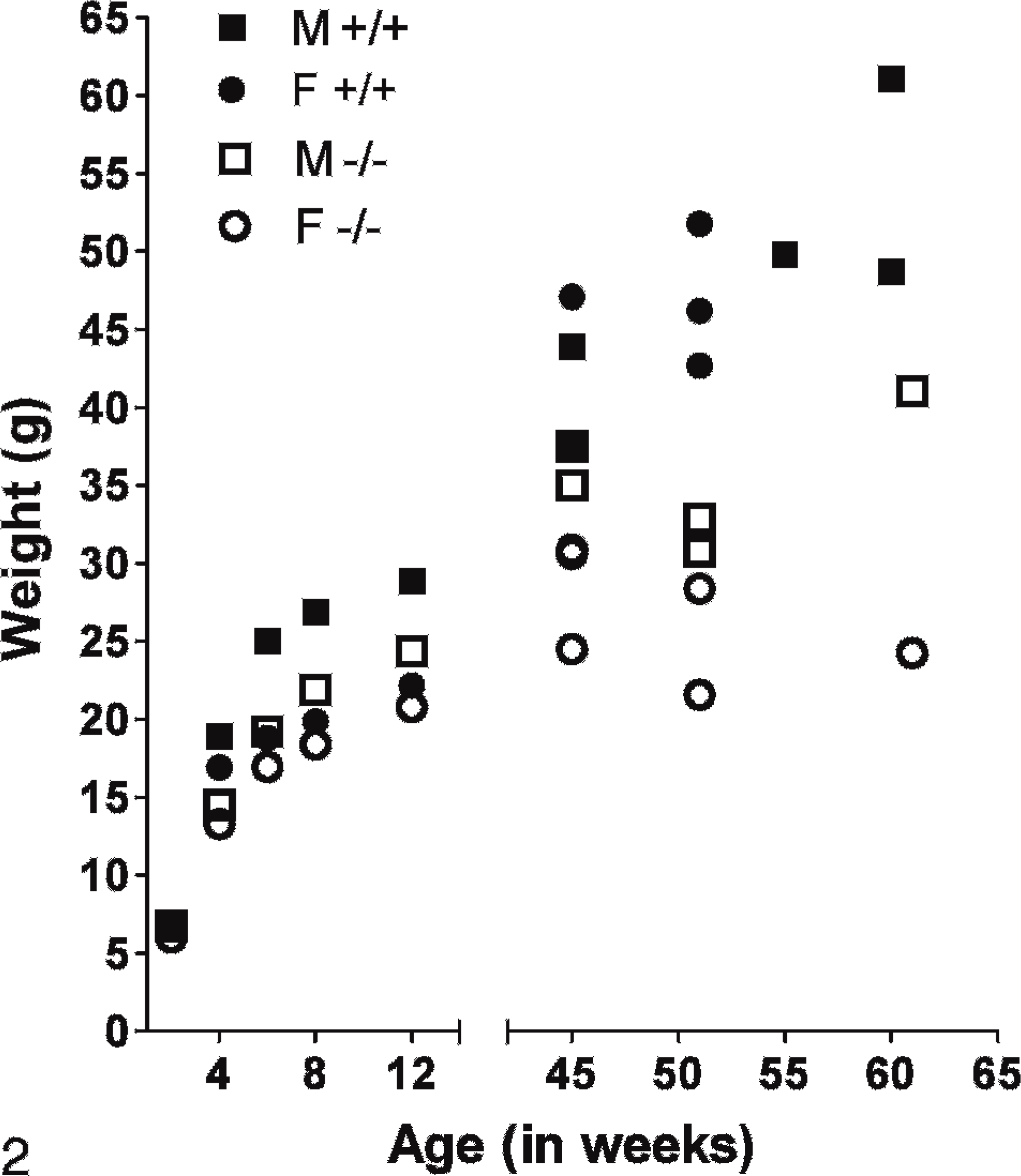
Growth curves of Entpd−/− mice and Wt littermates. Weights were taken at intervals for Entpd−/− and Wt Entpd+/+ progeny of heterozygous matings. Weight information at 2, 4, 6, 8, and 12 weeks represent the mean of 4 male or female Wt or 8 male or female Entpd−/− mice. Weights taken at 45–62 weeks of age are from individual mice. Growth of Entpd−/− mice is mildly depressed.
Serum chemistry showed increases in alanine aminotransferase (ALT), alkaline phosphatase, and bilirubin with decreases in serum albumin, glucose, cholesterol, and triglycerides (Table 1). Interestingly, the perturbations of hepatic indicators did not appear to worsen with increasing age. Additional hematologic, physiologic, and behavioral metrics were within normal limits of variation, except that breeding trials showed that Entpd−/− male mice were infertile.
Serum chemistry from Entpd -/- and Entpd +/+ mice. †
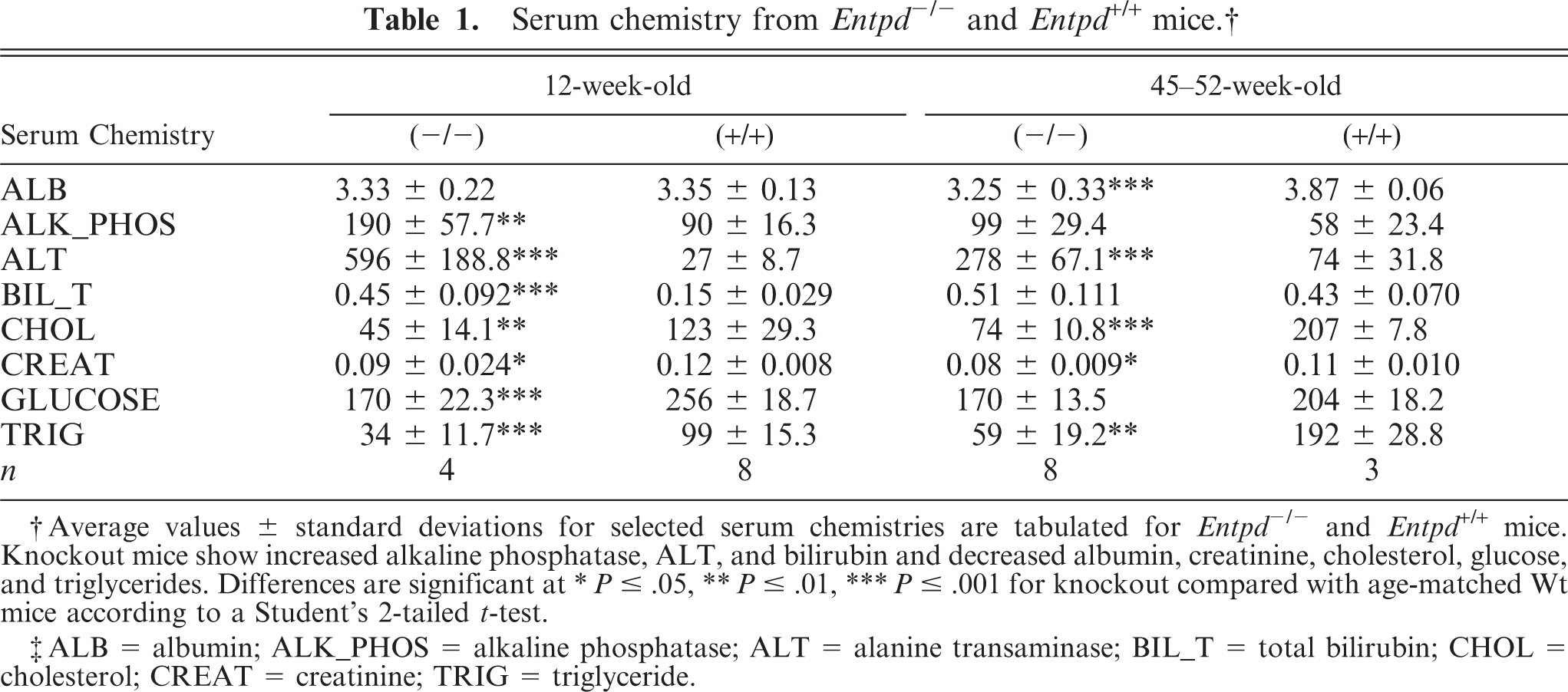
† Average values ± standard deviations for selected serum chemistries are tabulated for Entpd -/- and Entpd +/+ mice. Knockout mice show increased alkaline phosphatase, ALT, and bilirubin and decreased albumin, creatinine, cholesterol, glucose, and triglycerides. Differences are significant at
∗ P ≤ .05,
∗∗ P ≤ .01,
∗∗∗ P ≤ .001 for knockout compared with age-matched Wt mice according to a Student's 2-tailed t-test.
‡ ALB = albumin; ALK_PHOS = alkaline phosphatase; ALT = alanine transaminase; BIL_T = total bilirubin; CHOL = cholesterol; CREAT = creatinine; TRIG = triglyceride.
Pathologic evaluation of Entpd−/− mice at 14 and 20 weeks found no gross lesions; however, histologic examination demonstrated a distinct hepatopathy in both male and female mice and aspermia in male mice.
The hepatopathy in Entpd−/− mice at 14 and 20 weeks of age was characterized by diffuse moderate to prominent enlargement of centrilobular hepatocytes (Fig. 3). This contrasts with Wt mouse livers in which centrilobular hepatocytes might show mild enlargement because of glycogen or steatosis but are relatively uniform across hepatic lobules (Fig. 4). In older Entpd−/− mice (45 to 62 weeks old), hepatocellular alterations (hypertrophy, vacuolation, eosinophilia, basophilia) showed a more eccentric lobular orientation. Oval cell proliferation was also much more prominent in older mice (Fig. 5). The livers of older mice also contained occasional periportal infiltrates of lymphocytes with smaller numbers of histiocytes and granulocytes.
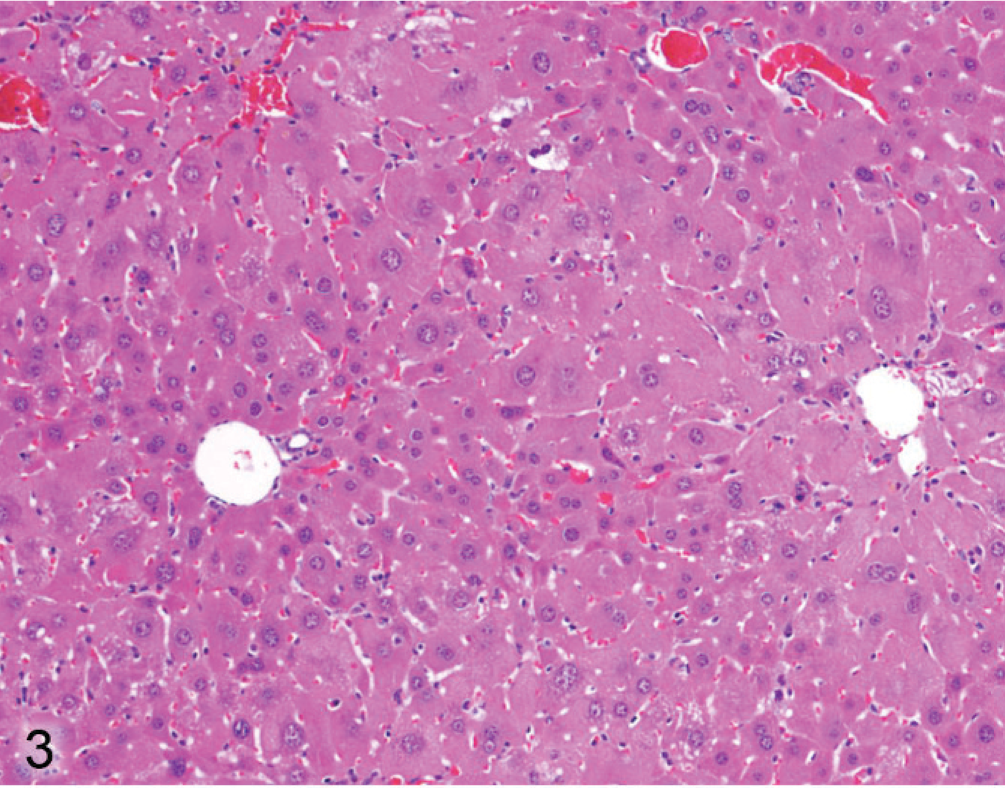
Liver; Entpd−/− mouse No. 118, 14.1 weeks old. Hepatocytes around the central vein (vessel on right) are larger than those around the portal tract (vessel on left). HE.
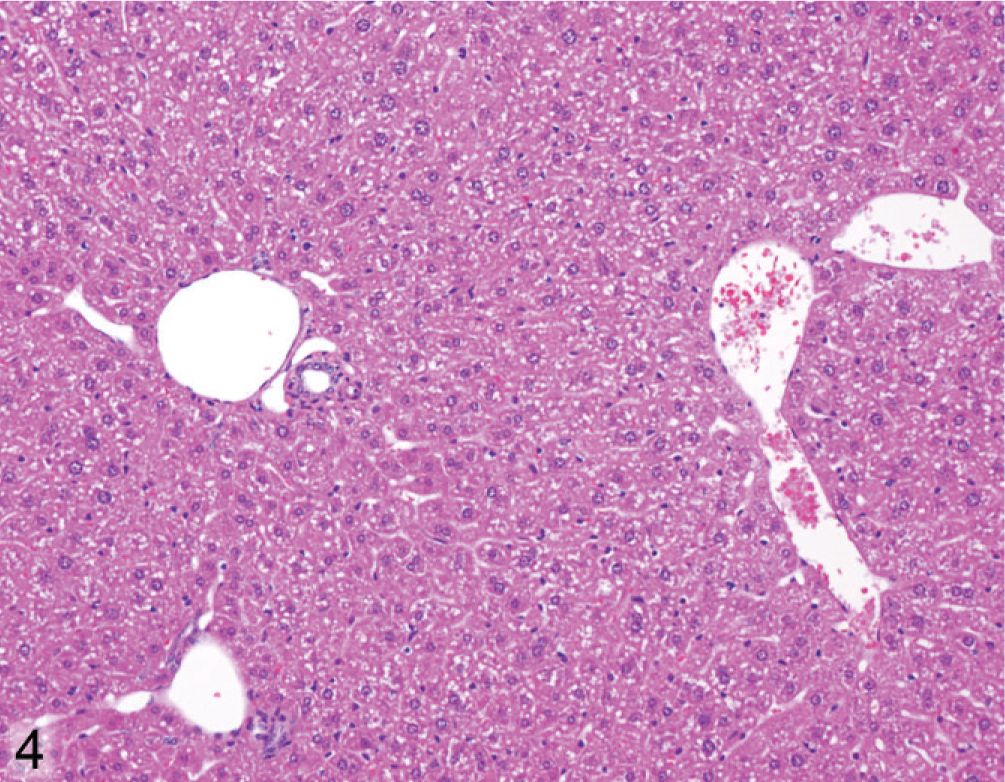
Liver; Wt mouse No. 126, 22.1 weeks old. Hepatocytes show relatively uniform size across lobule (portal tracts on left, central vein on right). HE.
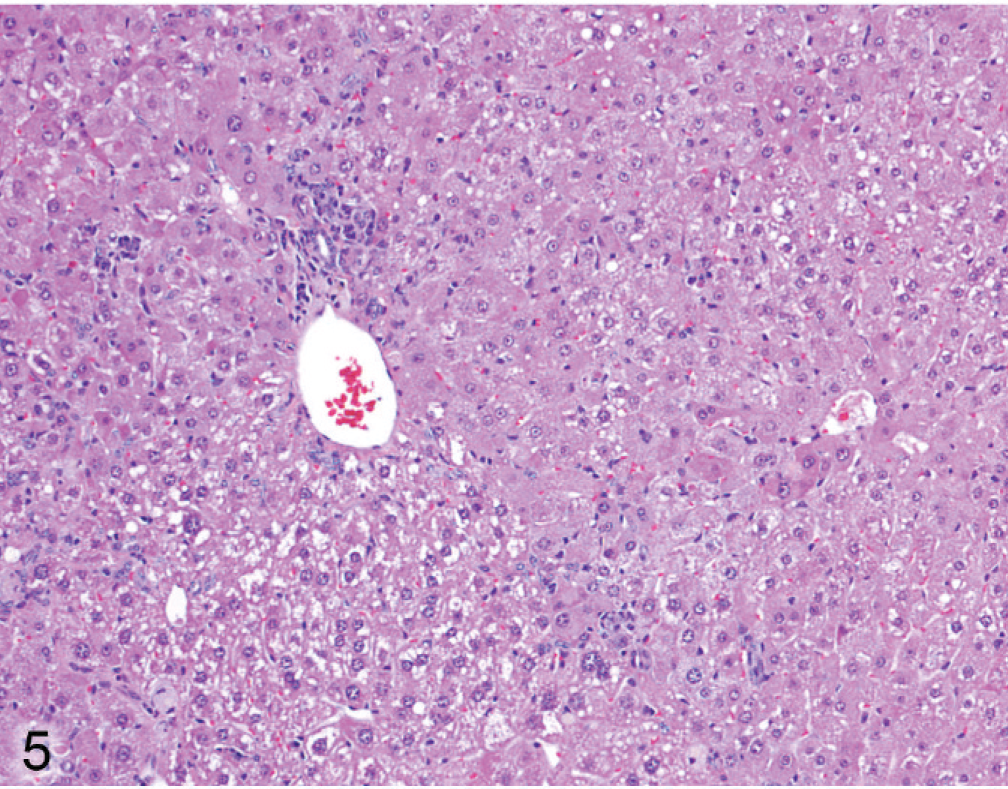
Liver; Entpd−/− mouse No. 286, 44.7 weeks old. Alteration in size and cytoplasm of hepatocytes is irregular with respect to hepatic lobules (portal tract on left, central vein on right). HE.
The enlarged centrilobular hepatocytes of the younger Entpd−/− mice showed increased amounts of coarsely variegated cytoplasm with clumps and stripes of basophilic material juxtaposed with areas of pale vacuolated or typical eosinophilic cytoplasm (Fig. 6). Young mice also showed increased numbers of mitotic figures in mature hepatocytes. In these mice (14 to 22 weeks old), widely scattered degenerating hepatocytes included many shrunken rounded hypereosinophilic globular bodies typical of apoptosis, as well as smaller numbers of enlarged pale degenerating hepatocytes (Fig. 7). Abundant yellow-green bile pigment was present in numerous Kupffer cells with rare canalicular bile casts, suggesting that bile staining of phagocytes was primarily due to phagocytosis of dying hepatocytes rather than a primary cholestatic process.
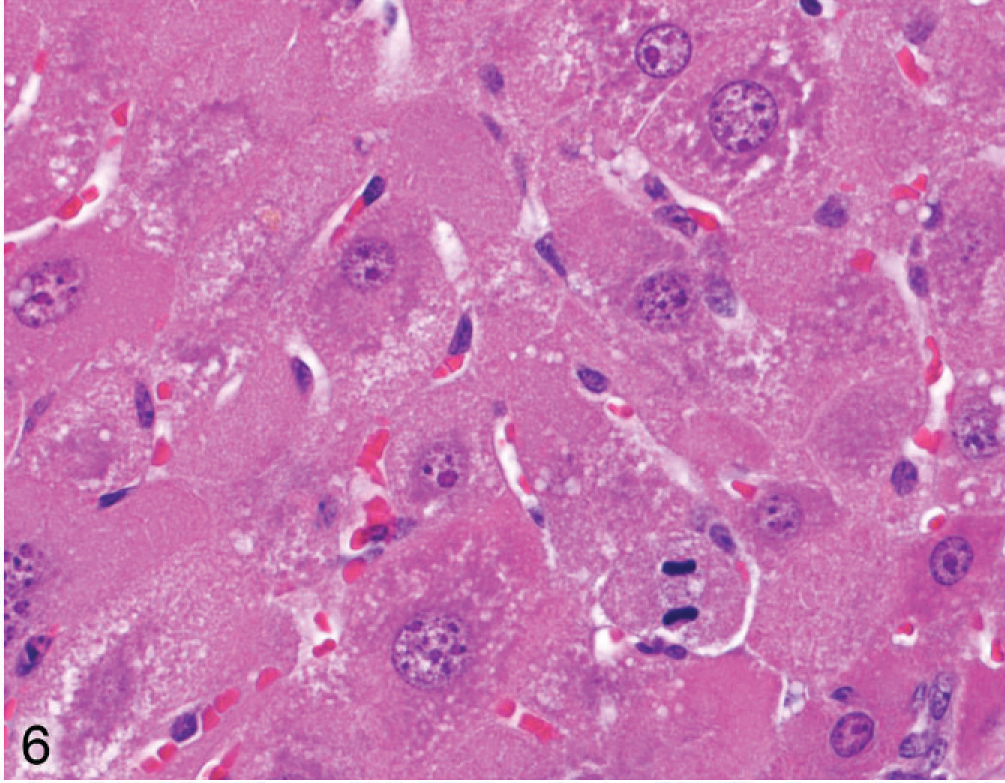
Liver; Entpd−/− mouse No. 131, 22.1 weeks old. Enlarged hepatocytes show coarse variegated cytoplasm and mitotic figures. HE.
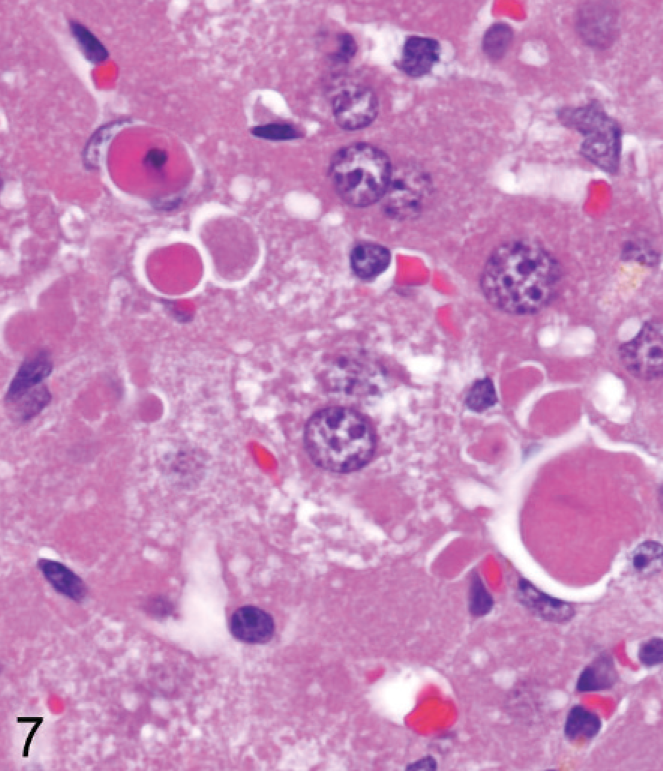
Liver; Entpd−/− mouse No. 118, 14 weeks old. Degenerating hepatocytes in liver from young adult knockout mice include many shrunken hypereosinophilic hepatocytes typical of apoptosis. HE.
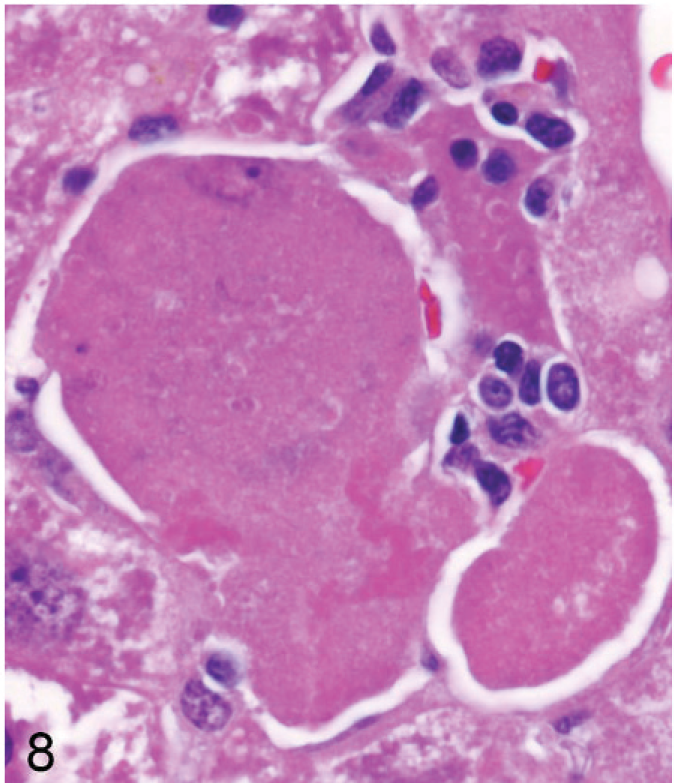
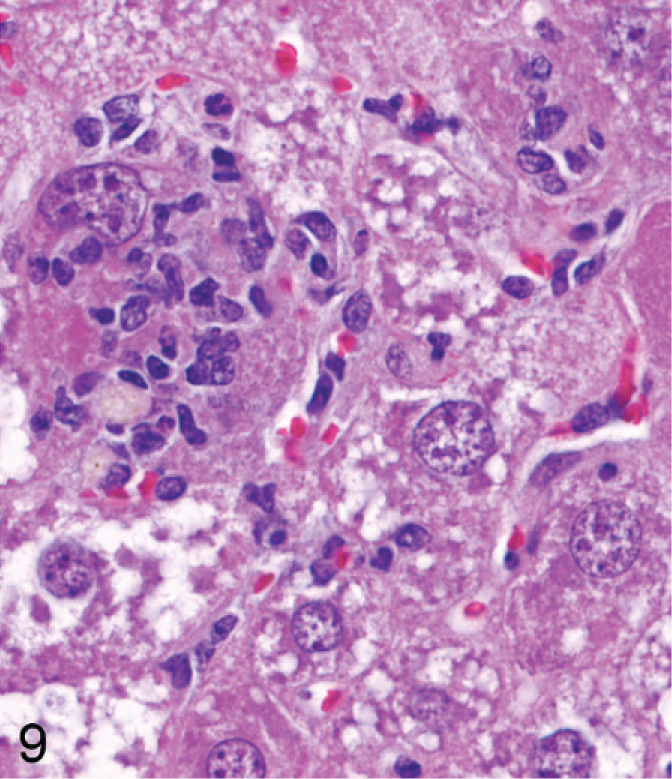
In the older mice (45–62 weeks old) most degenerating hepatocytes were greatly enlarged, pale, and rounded (Fig. 8). Some enlarged degenerating hepatocytes with intact nuclei were invaded by inflammatory cells, particularly in midzonal areas (Fig. 9). Bile pigment in phagocytes was slightly more prominent in older mice than in younger mice. Nodules of pale histiocytes, often containing fat vacuoles and bile pigment, were scattered in most livers of older Entpd−/− mice, and small strands of oval cells extended frequently from portal tracts.
Liver; Entpd−/− mouse No. 264, 45.3 weeks old. Degenerating hepatocytes in older knockout mice were often greatly enlarged. HE.
Liver; Entpd−/− mouse No. 264, 45.3 weeks old. The enlarged degenerating hepatocyte with intact nucleus is invaded by inflammatory cells. HE.
Using immunohistochemical staining of liver for catalase, an indicator of peroxisome proliferation, the older Entpd−/− mice showed patchy areas of increased staining because of marked variations in staining intensity of individual hepatocytes and multifocal groups of hepatocytes, but the increased staining did not show a regular lobular orientation (Fig. 10). In contrast, Wt control mice showed no significant variation in catalase staining intensity across the liver (Fig. 11). Immunohistochemistry for ubiquitin in older KO mice demonstrated a similar irregular patchy increase in staining, with individual cells showing greatly increased cytoplasmic staining (Fig. 12) that was not seen in Wt mice of the same age (Fig. 13). Enhanced ubiquitin staining in hepatocytes of older KO mice often showed densely staining granules and elongated cytoplasmic structures, most likely representing aggregates of denatured and ubiquitinated proteins (Fig. 14).
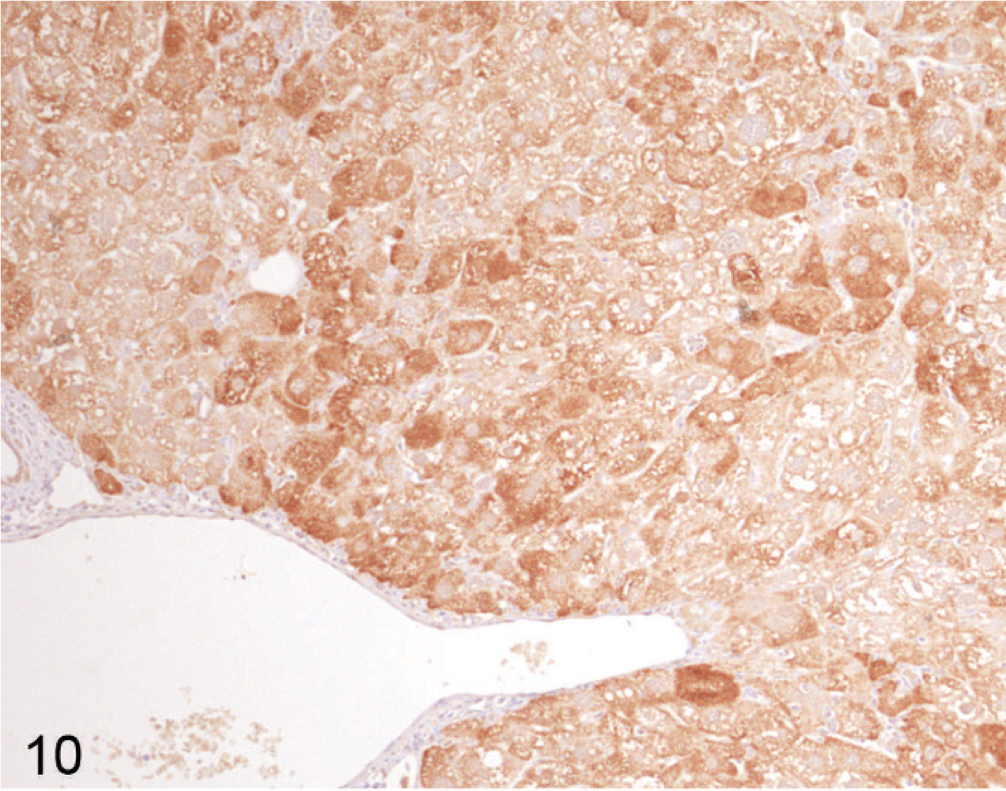
Liver; Entpd−/− mouse No. 264, 45.3 weeks old. Catalase stain for peroxisomes shows dense staining in a patchy pattern without uniform lobular orientation, highlighting focal and regional alterations in gene expression patterns. Immunohistochemistry (IHC) for catalase, DAB chromogen, hematoxylin counterstain.
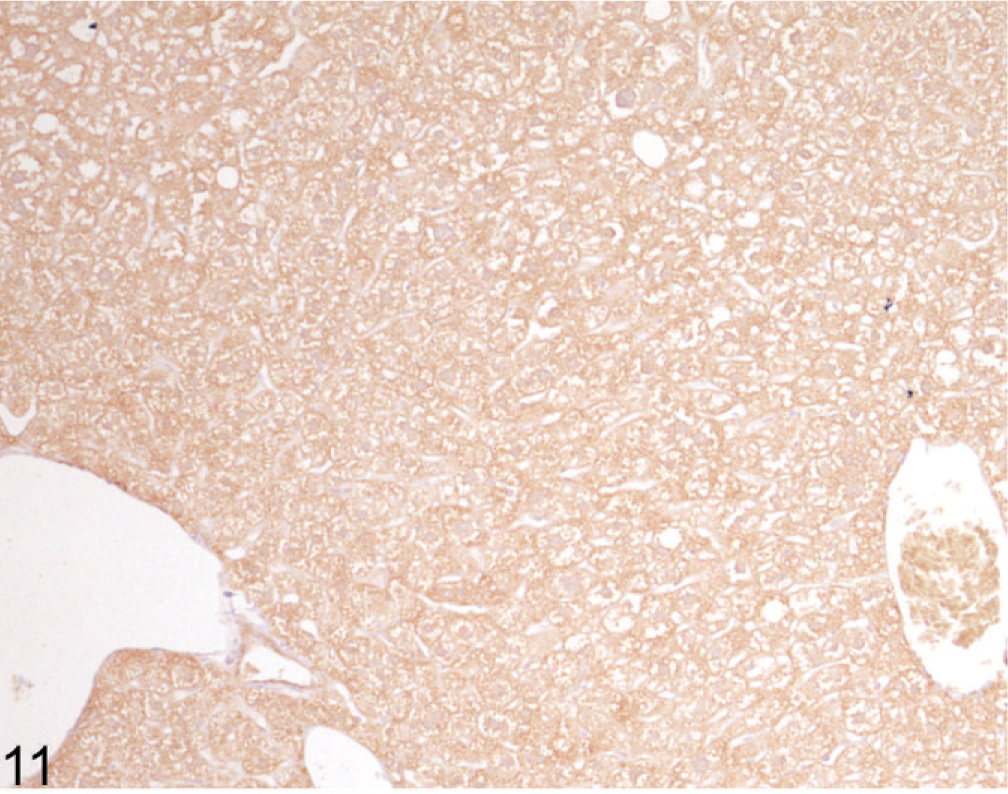
Liver; Wt mouse No. 282, 44.7 weeks old. Catalase stain for peroxisomes shows uniform expression in Wt mice. IHC for catalase, DAB chromogen, hematoxylin counterstain.
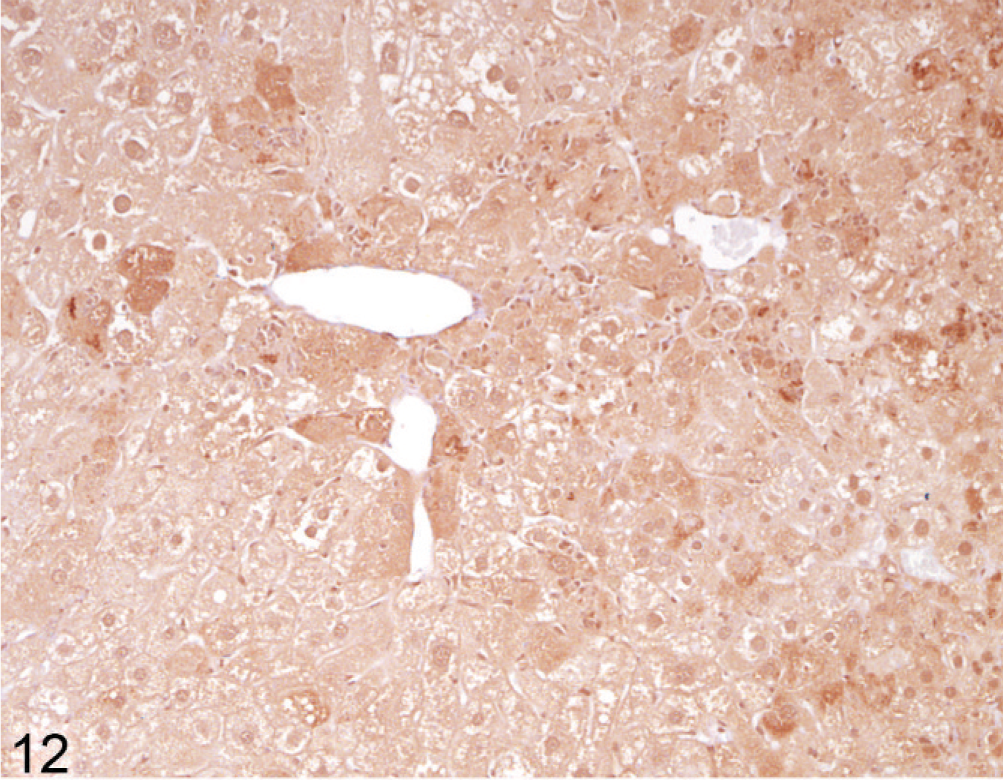
Liver; Entpd−/− mouse No. 264, 45.3 weeks old. Ubiquitin stain shows increased granular stain in many individual hepatocytes. IHC for ubiquitin, DAB chromogen, hematoxylin counterstain.
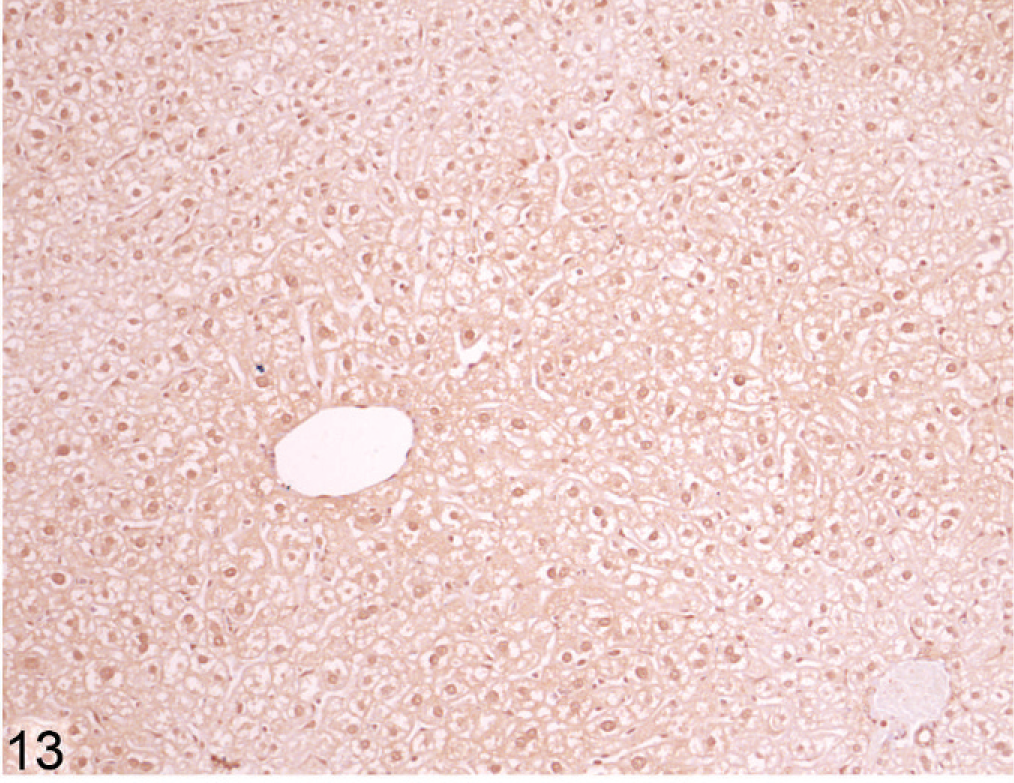
Liver; Wt mouse No. 261, 45.4 weeks old. Ubiquitin stain on Wt liver does not show strong enhancement of staining in individual hepatocytes. IHC for ubiquitin, DAB chromogen, hematoxylin counterstain.
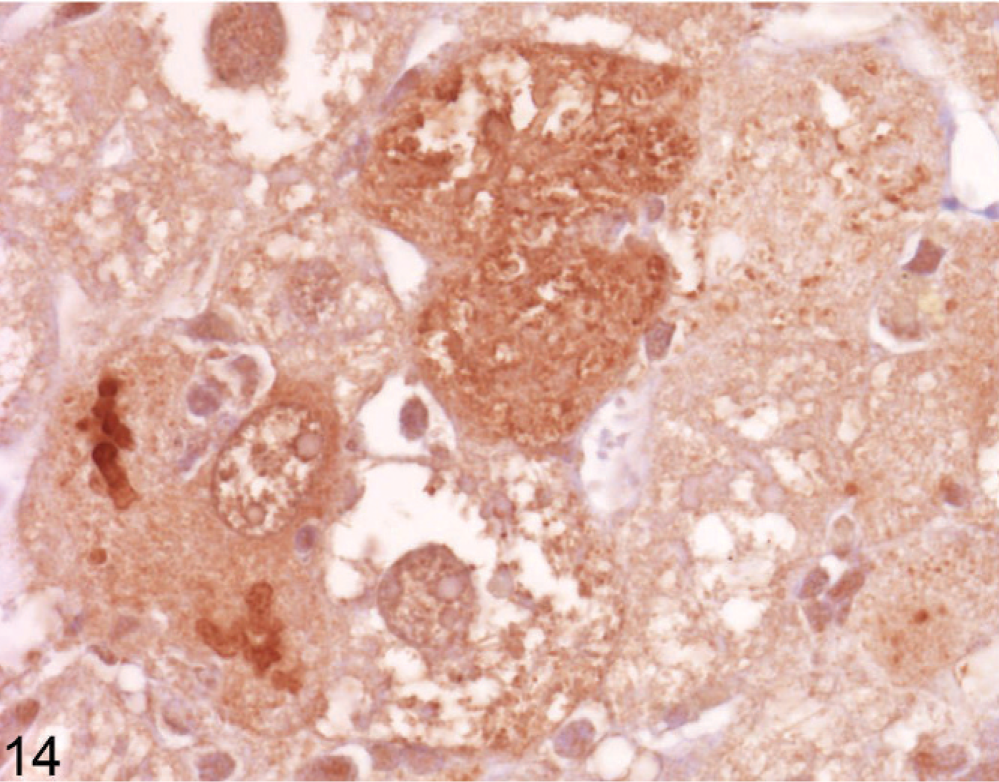
Liver; Entpd−/− mouse No. 264, 45.3 weeks old. Higher power view of ubiquitin stain in Fig. 12 shows variation in granular cytoplasmic stain and densely staining linear bodies suggestive of ubiquitinated protein aggregates. IHC for ubiquitin, DAB chromogen, hematoxylin counterstain.
Because gene inactivation was performed with a targeting vector containing β-Gal reporter gene under the control of the native promoter, we stained for reporter gene expression in the Entpd−/− mouse. X-gal staining for reporter gene expression showed an irregular patchy pattern in the liver, with considerable variation in staining intensity of individual hepatocytes (Fig. 15); close inspection showed reporter gene staining of linear cytoplasmic structures, consistent with secretion of the gene product into the ER (Fig. 15 inset). X-gal staining also showed strong specific diffuse cytoplasmic staining of mature epithelial cells in the sebaceous glands, the subcomissural organ (modified ependymal cells) of the brain, the proximal convoluted renal tubules (but not distal convoluted or straight tubules or glomeruli), and villus and luminal surface epithelia in the small and large intestines (data not shown).
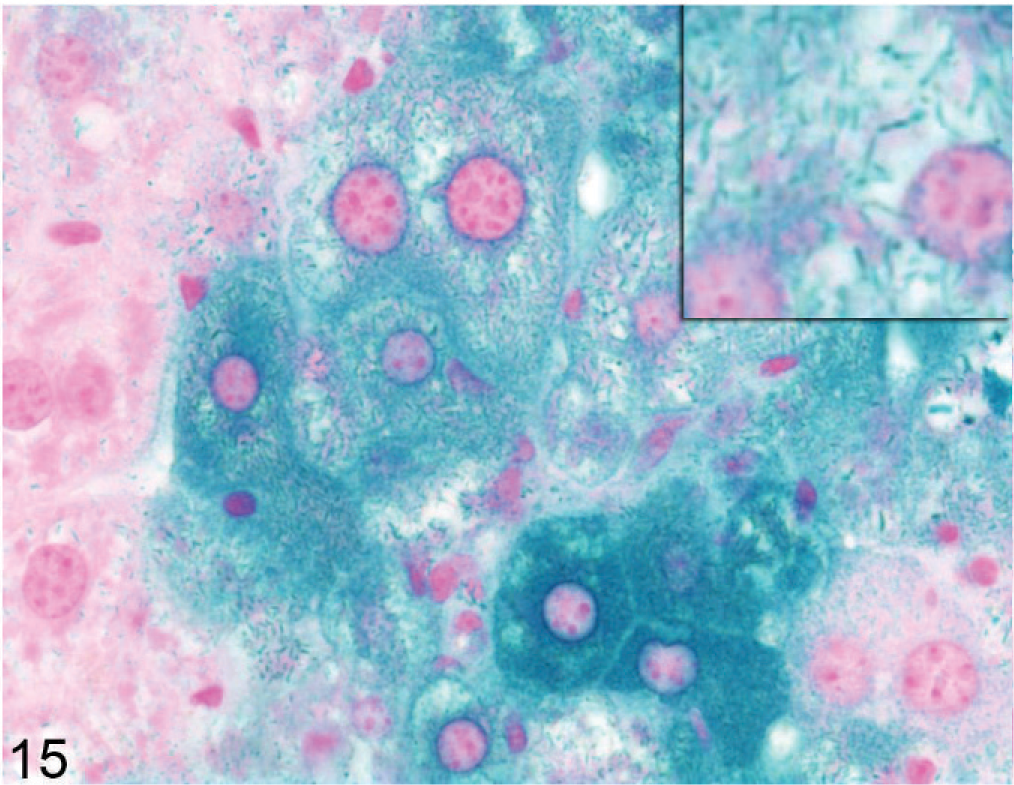
Liver; Entpd−/− mouse No. 290, 43.3 weeks old. X-gal stain shows variable staining (reporter gene expression) in individual hepatocytes; staining of linear cytoplasmic structures is consistent with localization in the ER (inset). X-gal stain.
Proliferation of hepatocytes was determined by counting individual or pairs of adjacent positively stained hepatocyte nuclei labeled by Ki67 immunohistochemistry on liver sections from 14- to 22-week old mice and by BrdU labeling in 51 to 61-week-old mice. Significantly increased proliferation of hepatocytes in both young and older Entpd−/− mice was found compared with age-matched Wt mice (Table 2; Figs. 16, 17).
Proliferation labeling index (LI) of hepatocytes. †
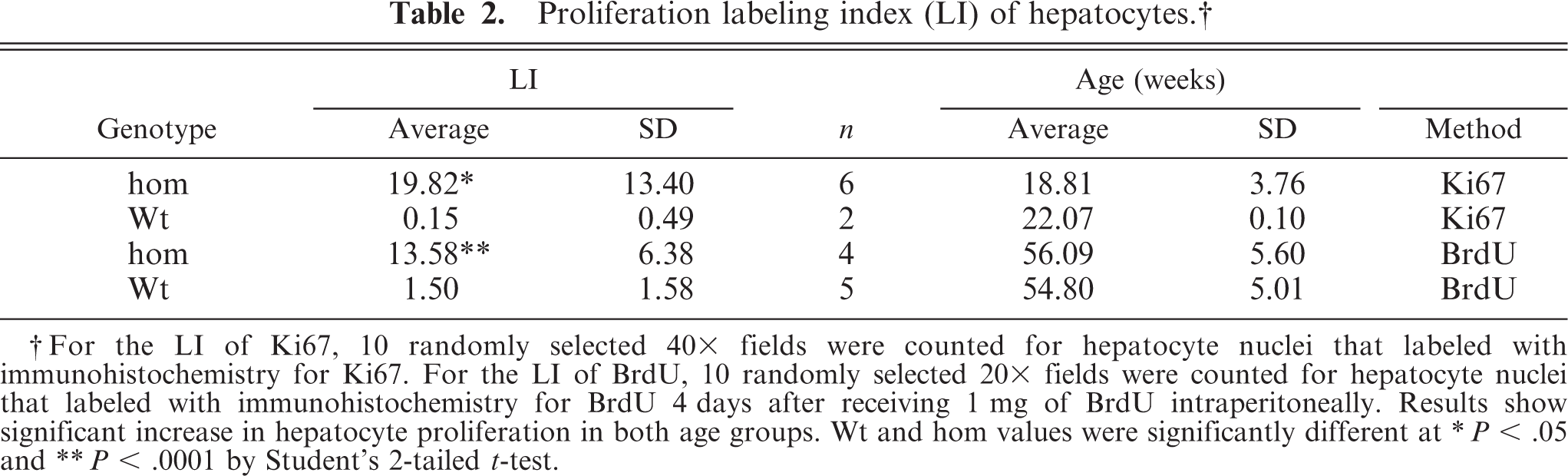
† For the LI of Ki67, 10 randomly selected 40x fields were counted for hepatocyte nuclei that labeled with immunohistochemistry for Ki67. For the LI of BrdU, 10 randomly selected 20x fields were counted for hepatocyte nuclei that labeled with immunohistochemistry for BrdU 4 days after receiving 1 mg of BrdU intraperitoneally. Results show significant increase in hepatocyte proliferation in both age groups. Wt and hom values were significantly different at
∗ P < .05 and
∗∗ P < .0001 by Student's 2-tailed t-test.
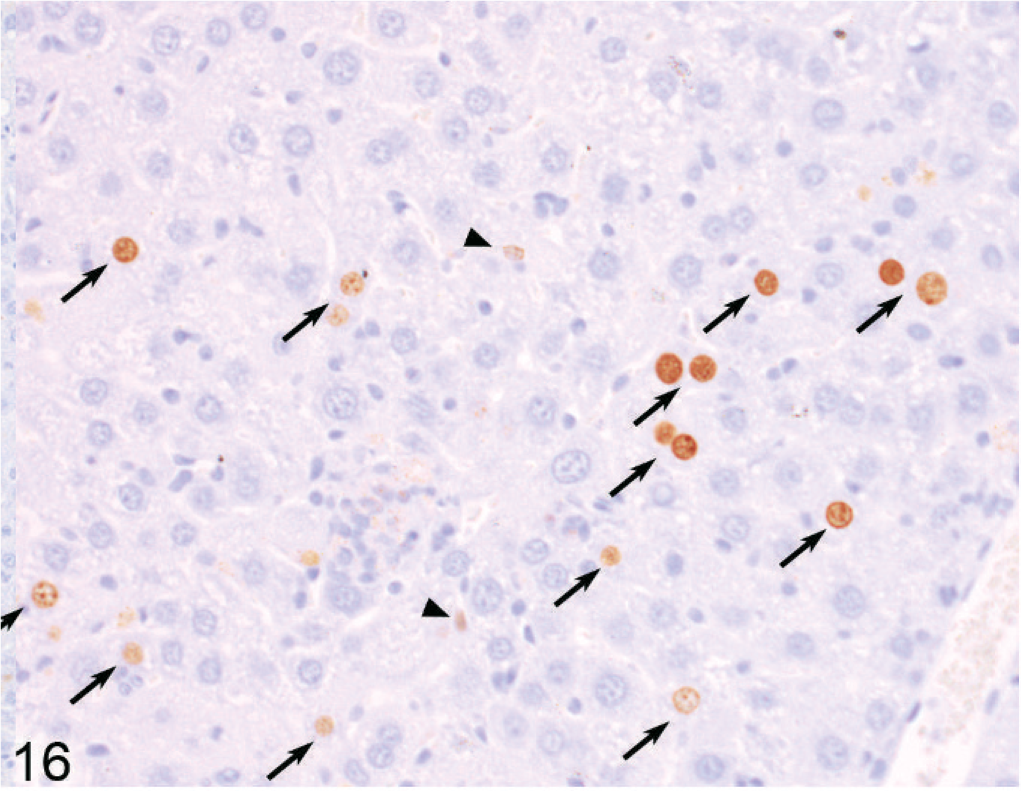
Liver; Entpd−/− mouse No. 185, 60.9 weeks old. BrdU-labeled hepatocyte nuclei (indicating DNA synthesis during exposure to BrdU) are common in the liver of knockout mice. Arrows indicate counted BrdU-positive individual or pairs of hepatocyte nuclei, and arrowheads indicate BrdU-positive nonhepatocyte nuclei (not counted). IHC for BrdU, DAB chromogen, hematoxylin counterstain.
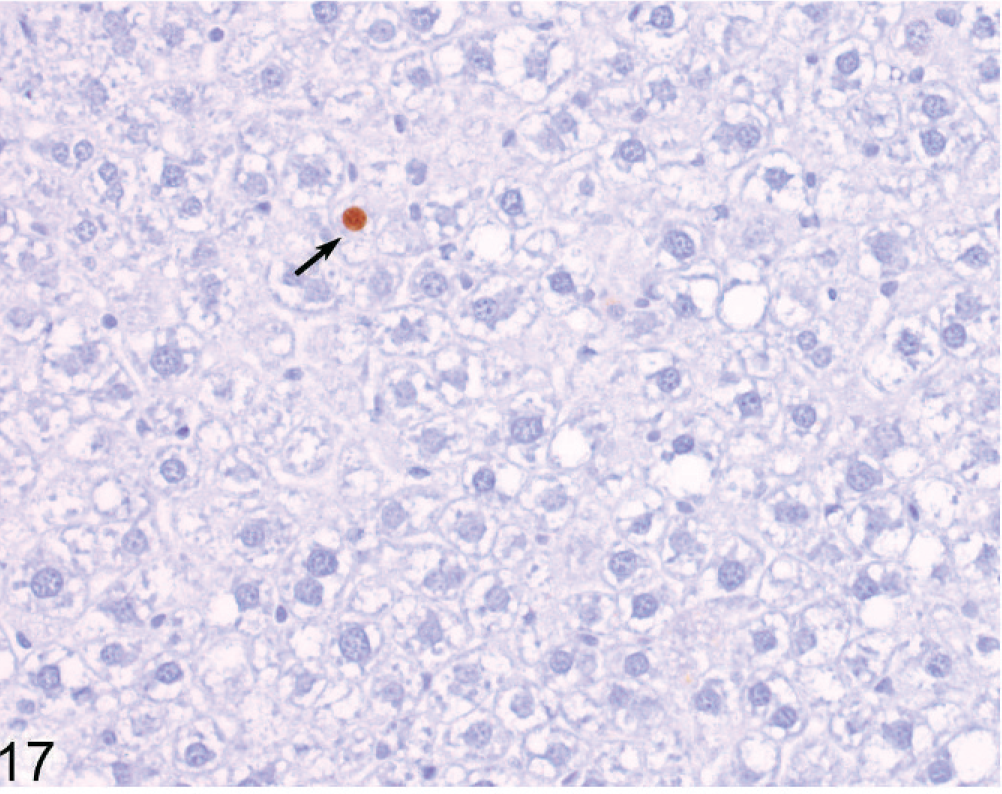
Liver; Wt mouse No. 252, 51 weeks old. BrdU-labeled hepatocyte nuclei are uncommon in the liver of Wt mice. IHC for BrdU, DAB chromogen, hematoxylin counterstain.
Aspermia caused by late spermatogenic arrest or defective spermiation was evident in the reproductive organs of all male Entpd−/− mice examined. Histologic examination of testes and epididymides from Entpd−/− mice showed seemingly normal spermatogenesis and maturation through the elongated spermatid stage in seminiferous tubules, but markedly decreased numbers of tail structures within tubular lumina and an absence of normal spermatozoa in the epididymis (not shown). Epididymal tubules instead contained small numbers of degenerating round cells, a few tail structures, and, very rarely, individual eosinophilic necrotic spermatozoa.
Hepatocellular neoplasia
Older Entpd5−/− mice with Wt (Entpd5+/+ ) littermate controls were examined from 44 to 69 weeks of age. The livers in all 15 of the older Entpd5−/− mice examined histologically showed prominent hepatopathy with at least 1 focus of cellular alteration, which are often regarded as preneoplastic lesions. In contrast, none of the liver sections examined from 13 age-matched control mice showed a focus of cellular alteration (P < .00001, Fisher's 2-tailed exact test). Out of 15 aged KO mice, liver sections from 2 could not be evaluated because of extensive neoplasia, 1 contained a single focus of cellular alteration, 3 showed multiple foci of cellular alteration, and the remaining 9 showed multiple foci that formed an irregular patchwork of coalescing foci (Fig. 18) of different types (Fig. 19).
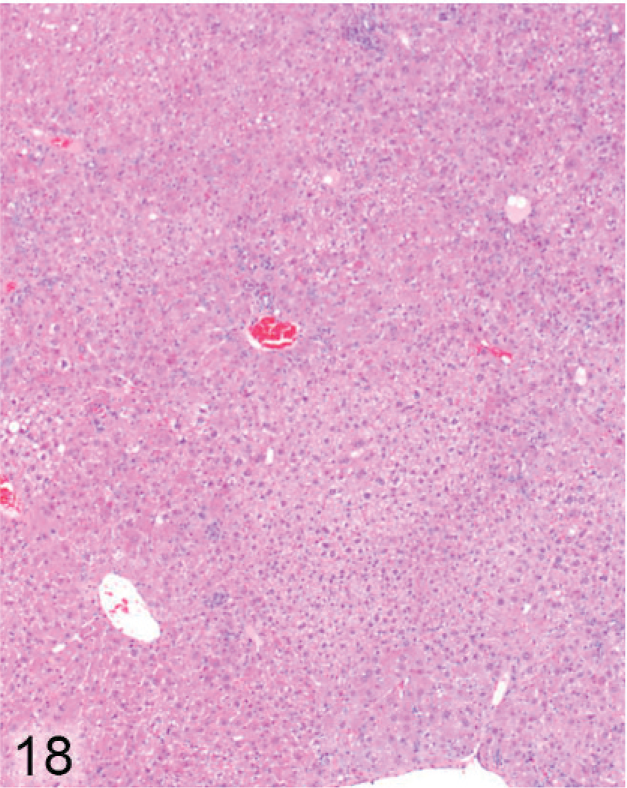
Liver; Entpd−/− mouse No. 286, 44.7 weeks old. Liver from older mouse shows a patchwork of focal cellular alteration. HE.
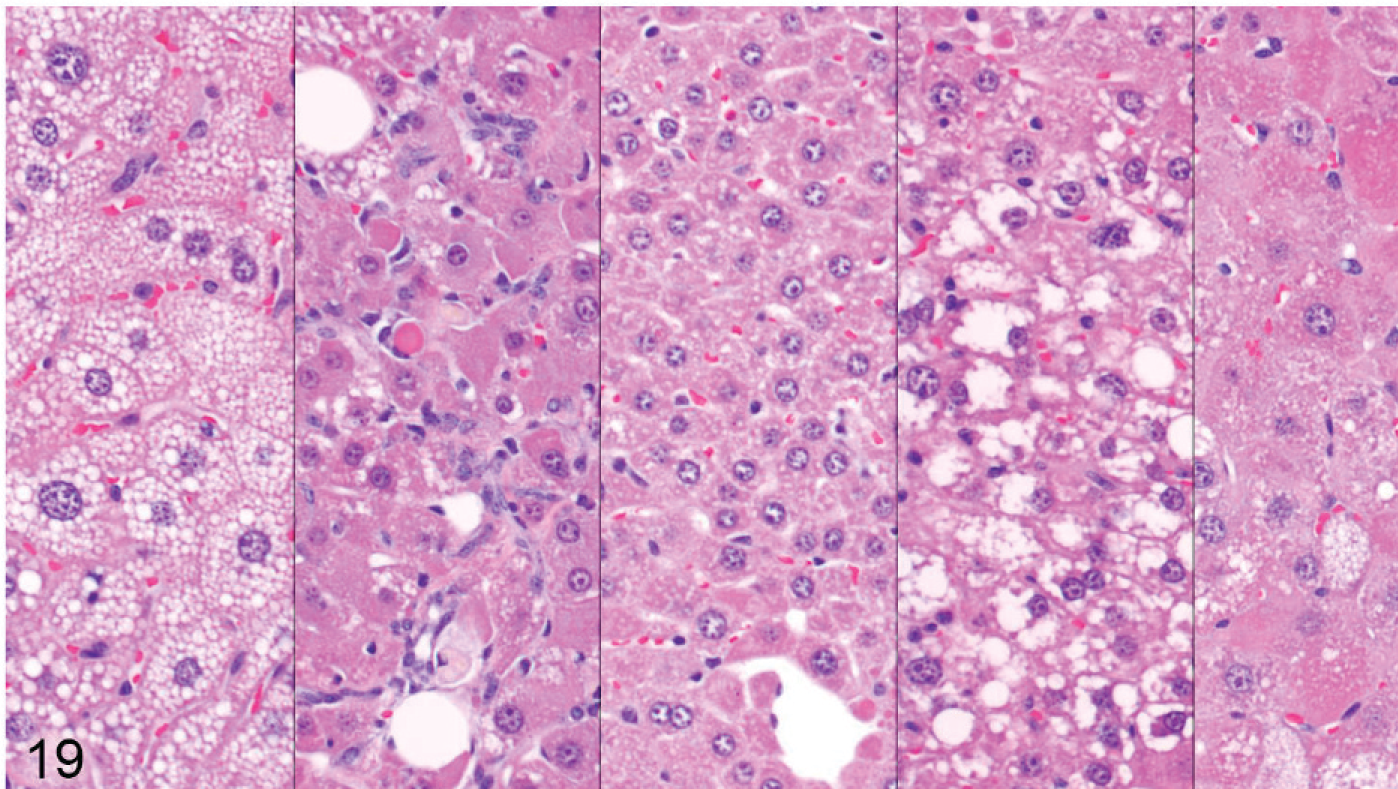
Liver; Entpd−/− mouse No. 286, 44.7 weeks old. Higher power views of 5 areas from the liver section seen in Fig. 20. Five panels show distinct morphologies of hepatocytes in different foci. HE.
In about half of the cases (n = 8), only a single section of liver was available because the remaining liver was used for enzyme analysis; in the other animals, histologic evaluation of the liver was limited to 3 routine sections, although sections of liver masses were added if apparent on gross exam. In one group, 3 of 8 Entpd5−/− mice showed gross liver masses at necropsy, but one of these was lost to histologic analysis and therefore is not counted as a neoplasm-bearing liver. Out of 15 Entpd5−/− livers evaluated histologically, 5 showed HCC (2 multicentric and 1 with an additional adenoma), and an additional liver showed hepatocellular adenoma. Of the HCC, 1 showed mixed morphology, having areas of well-differentiated hepatocytes that blended into areas of poorly differentiated pleomorphic and often elongated cells (Figs. 20–22). In summary, of KO mice (44–69 weeks old), 6 of 15 (40%) Entpd5−/− mice showed HCC, hepatocellular adenoma, or both, whereas none of the 13 Wt controls showed neoplasia (P = 0.018, Fisher's 2-tailed exact test). If the lost liver mass was assumed to be neoplastic, 7 of 15 KO livers compared with none of 13 controls with neoplasia would increase the statistical significance of this finding (P = .0069, Fisher's 2-tailed exact test). Increased confidence that Entpd5−/− mice have a significant increase in hepatic neoplasms was provided by results of a recent aging study that can serve as a reference population in estimating the incidence of HCC in mice having the same genetic background. In this study, out of 82 Wt mice (52 male, 30 female; age range 36–105 weeks, average 87 weeks old), 3 showed HCC (males aged 73, 87, and 104 weeks) and 1 had hepatocellular adenoma (male age 52 weeks). The mice with HCC in this reference population were older than similarly affected Entpd5−/− mice. In the reference study, HCC was identified only in male mice (average 79 weeks of age), whereas hepatocellular neoplasia was found both in male and female Entpd5−/− mice, with an average age of 54 weeks. These findings are summarized in Table 3.
Ages of mice with hepatocellular neoplasia. ∗

∗ The average age (weeks) and number of mice (n) in each group and age of mice (weeks) with liver neoplasms found in older Entpd -/- mice (male KO, female KO) and Wt controls (male Wt, female Wt) described in this study, as well as in a reference study of aging F1 Wt mice of the same genetic background (ref male Wt, ref female Wt).
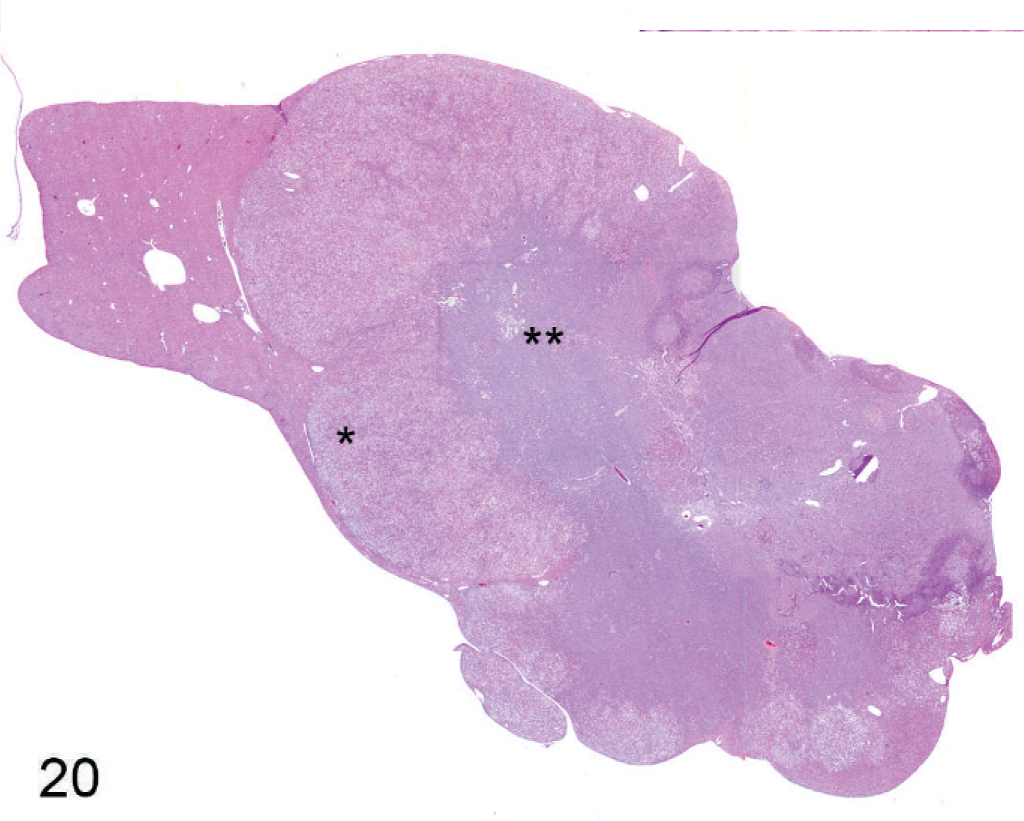
Liver; Entpd−/− mouse No. 259, 45.3 weeks old. Low-power view of histologic section of hepatocellular carcinoma showing 2 distinct populations of neoplastic cells. Figures 21 (∗) and 22 (∗∗) are taken from areas marked with asterisks. HE.
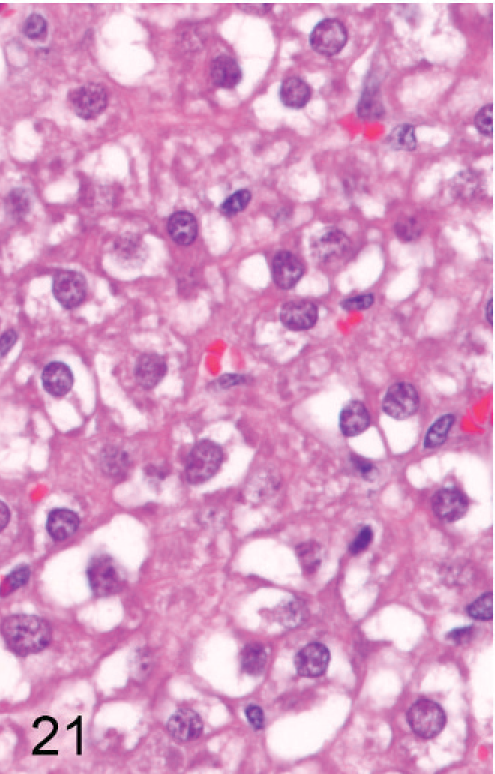
Liver; Entpd−/− mouse No. 259, 45.3 weeks old. High-power view of tissue in Fig. 20 marked with a single asterisk (∗) showing the population of well-differentiated neoplastic hepatocytes. HE.
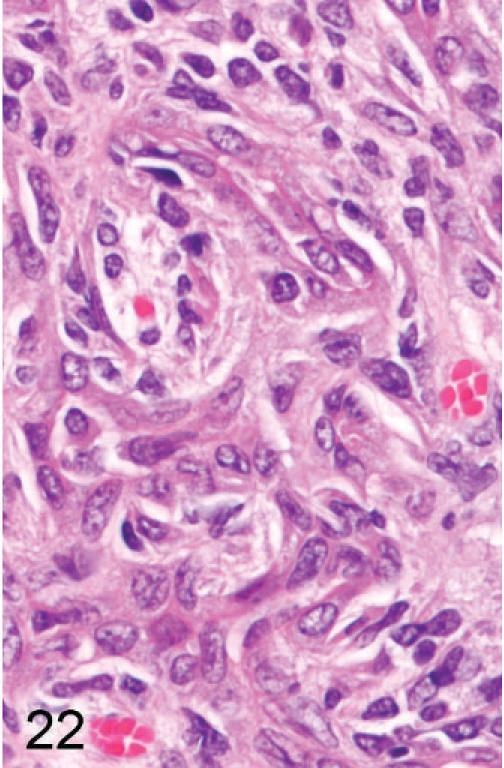
Liver; Entpd−/− mouse No. 259, 45.3 weeks old. High-power view of tissue in Fig. 20 marked with a double asterisk (∗∗) showing pleomorphic elongated neoplastic cells with occasional tubular structures and few cells recognizable as hepatocytes. HE.
Analysis of liver showed increased activity of the cytochrome P450 enzymes CYP1A and CYP3A. CYP1A2 activity was statistically significantly elevated from 1.5 pmol product/min/mg microsome preparation in Wt mice to 2.8 pmol in Entpd−/− mice (Table 4). In contrast, CYP1A2 activity did not differ significantly between males and females of Entpd−/− mice or between males and females of Wt mice. CYP3A11 activity in the male and female Wt mouse liver samples was 100 pmol product/min/mg microsome preparation. In male KO mice, CYP3A11 increased significantly to 300 pmol product/min/mg microsome preparation, but CYP3A activity in the female KO liver preparations was only ∼20% greater than in Wt controls and not statistically significant. Neither CYP2C nor 2D activity differed between Wt and KO mice.
Liver CYP activities in KO and Wt mice. ∗
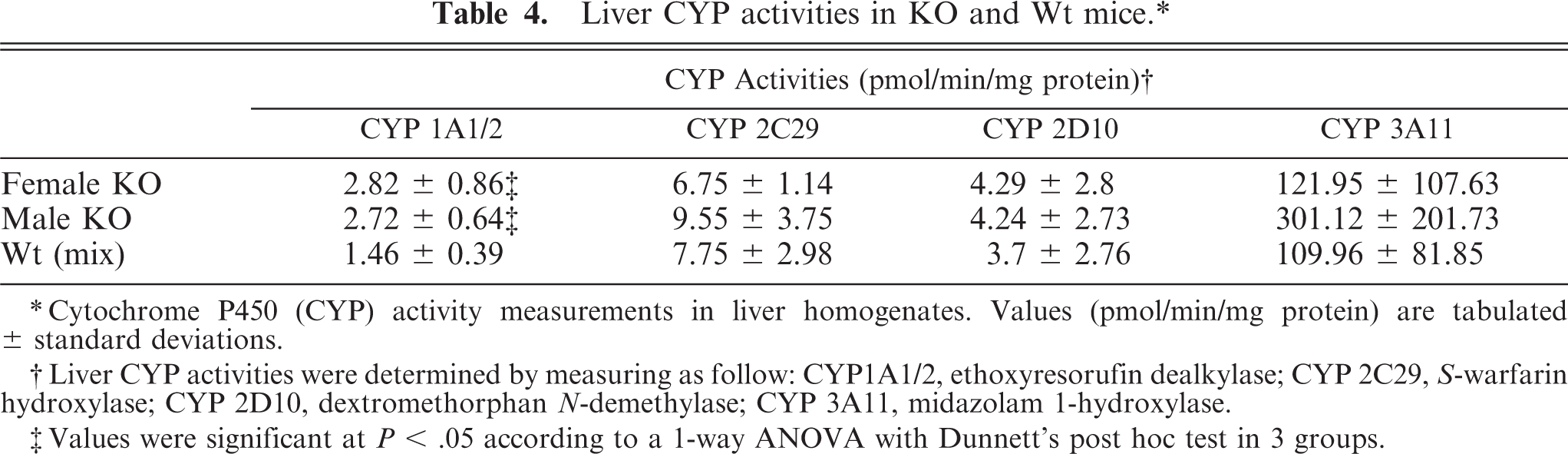
∗ Cytochrome P450 (CYP) activity measurements in liver homogenates. Values (pmol/min/mg protein) are tabulated ± standard deviations.
† Liver CYP activities were determined by measuring as follow: CYP1A1/2, ethoxyresorufin dealkylase; CYP 2C29, S-warfarin hydroxylase; CYP 2D10, dextromethorphan N-demethylase; CYP 3A11, midazolam 1-hydroxylase.
‡ Values were significant at P < .05 according to a 1-way ANOVA with Dunnett's post hoc test in 3 groups.
Discussion
We have shown that Entpd5−/− mice displayed mildly stunted growth, male infertility, and a proliferative hepatopathy with a high incidence of hepatocellular neoplasia. Hematology and most other biometrics were unremarkable, but serum chemistry and histopathologic evaluation revealed a slowly progressive hepatopathy characterized by greatly increased hepatocyte turnover (proliferation and degeneration), oval cell proliferation, centrilobular hepatocyte hypertrophy that progresses to include foci of hepatocellular alteration and an increased prevalence of hepatocellular neoplasia. The increased transaminase activities and bilirubin in the serum were not unexpected given the hepatocellular damage and bile pigment staining that was apparent microscopically. The decreased concentrations of albumin, glucose, triglyceride, and cholesterol in Entpd5−/− mice also suggest that overall hepatic function in these mice was compromised. Interestingly, with the exception of albumin, these serum markers of liver function did not worsen with age, suggesting adaptation by the liver. The cytologic alteration of hepatocytes progresses from a uniform diffuse centrilobular hepatocyte hypertrophy to a patchwork of hepatocytes displaying various morphologic changes, including neoplastic transformation. The patterns of focal and regional morphologic alterations that accumulate in older mice appear similar to the dramatic regional and cell-to-cell variation in gene expression demonstrated with anti-catalase staining. Furthermore, degeneration and death of hepatocytes changed with age from more typical apoptosis characterized by cell shrinkage in young Entpd5−/− mice, to cell death characterized by greatly swollen hepatocytes that were invaded by inflammatory cells in older animals; the change in the morphology of hepatocyte degeneration and death could be due to adaptation by acquiring resistance to apoptosis, which could also contribute to oncogenesis.
It has been demonstrated that ENTPD5 hydrolyzes NDP to NMP, so it is possible that increased hepatocellular turnover in Entpd5−/− mice is due to loss of the enzyme's degradation of NDP in the ER or in the extracellular environment. Loss of ENTPD5 from the ER could lead to inhibition of protein glycosylation mediated by end product (UDP) inhibition in that compartment. One consequence might be the accumulation of misfolded protein in the ER and Golgi, 31 triggering the ER stress response and apoptosis (reviewed by Szegezdi et al. 30 ). Consistent with this notion, in a related study, the unfolded protein response (ER stress) mediated through ire-1 was shown to up-regulate ER-UDPase expression in Caenorhabditis elegans. 32 Because unfolded proteins are ubiquitinated for targeted degradation in the 26S proteasome complex, our finding of ubiquitinated material accumulating in the cytoplasm of Entpd5−/− hepatocytes might signal an overload of this degradative pathway. The loss of these overloaded hepatocytes might be responsible for the observed increase in hepatocyte turnover in these Entpd5−/− mice, with the resulting hepatocellular hyperplasia serving to promote neoplasia.
Alternatively, the increased rate of hepatocyte turnover might result from dysfunctional extracellular nucleotide signaling. ENTPD5 has been shown to be secreted from COS-7 cells transfected with an expression vector containing the native leader peptide. 20 Because this protein is highly expressed in the liver, secreted ENTPD5 might well modulate NDP levels, either locally or throughout the body. Support for this hypothesis is found in the demonstration that modulation of ENTPD2 in cultured portal fibroblasts affects the rate of proliferation of cocultured (human cholangiocarcinoma) bile ductular epithelial cells, 13 suggesting that liver cell proliferation can be altered through ENTPD5 modulation of extracellular nucleotide signaling. The presence of hepatocyte and oval cell proliferation in the Entpd−/− mouse livers could therefore be a direct consequence of decreased ENTPD5 degradation of extracellular NDP as opposed to a response to hepatic injury.
The incidence of liver tumors was considerably increased in this small study of Entpd−/− mice. The development of hepatocellular neoplasms in 6 of 15 (40%) aged Entpd−/− mice is significant given that none of the 13 age-matched Wt control littermates from this cohort developed hepatocellular neoplasia, as well as the low incidence in older mice of the same genetic background. The incidence of hepatocellular neoplasia in the reference study of our C57Bl/6:129 mice up to 2 years of age was lower than that reported in untreated and chamber control B6C3F1 mice from most NTP carcinogenicity studies 18 but showed a similar predilection for male over female mice. The finding of 3 HCC and 1 hepatocellular adenoma in 10 Entpd−/− females (tumor-bearing females averaged 57.5weeks old) is significant compared with the absence of hepatocellular neoplasia in 9 cohort female Wt and 30 genetically similar although considerably older female Wt mice in the reference study. Larger controlled studies should confirm these findings.
The presence of the various morphologic hepatocellular changes and the cytochrome P450 induction we observed are similar to those previously associated with nongenotoxic carcinogenesis. 8, 16 Although the specific mechanism of CYP induction is not clear, it seems likely that the changes reflect hepatocyte responses or adaptation to cellular stress caused by inactivation of Entpd. Furthermore, it seems most likely that CYP induction is heterogeneous, with intercellular variation similar to that seen in the expression of catalase.
Hepatocellular carcinoma is the third most frequent cause of death by cancer in humans. 22 Hepatocytes normally remain in the G0 phase of the cell cycle and are mitotically inactive; however, numerous factors that induce hepatocyte proliferation can promote neoplastic transformation. 10 For example, chronic hepatocellular proliferation induced by chronic inflammation in patients with hepatitis C infections or alcoholic cirrhosis increase the incidence of liver neoplasia. Some specific genetic manipulations in mice have been shown to increase rates of hepatocellular neoplasia. Hepatocytes in mice lacking the Mdr2 gene cannot transport phospholipids into the bile, resulting in continuous injury to hepatocytes, inflammation, and hepatocellular proliferation that can progress to hepatocellular carcinoma by 18 months of age. 15, 29 Similarly, mice lacking peroxisomal fatty acyl-CoA oxidase develop chronic inflammation, hepatocellular regeneration, and then hepatocellular tumors by 15 months of age. 9 Increased hepatocellular proliferation and eventual neoplastic transformation is also seen in transgenic mice overexpressing TGF-α, 17 c-myc, 21 or SV40 large T antigen. 1
A potential role for ENTPD5 in the development of cancer has been shown in other studies. Expression of the normal ENTPD5 protein is deregulated or lost in some human cancers 3, 4 and in a wide variety of malignant cells, 28 consistent with a role as a tumor suppressor. In addition, a recent study of human hepatocellular carcinomas identifying putative tumor suppressor genes showed loss of heterozygosity in the chromosomal region 14q24.3 to 14q32.2, the same area in which the human ENTPD5 gene is located. 5, 14
Interestingly, overexpression of ENTPD5 was associated with expression of protein kinase C-delta and invasiveness in human prostate cancer. 34 A possible explanation for this finding is that unlike the full-length form of the enzyme, the truncated form was capable of cooperating with the Ras oncoprotein, thus producing a prolonged stimulation of ERK1 phosphorylation. 25 The transforming activity of the PCPH oncogene (a truncated form of ENTPD5) has also been attributed to reduction of intracellular ATP, possibly modulating the stress-response pathway mediated by JNK (or SAPK). 26 This highlights the possibility that the interrupted gene product, the residual N-terminal 154 amino acids of the Entpd5 gene joined with the selection cassette, might have an effect in addition to interruption of enzymatic activity of ENTPD5.
Neoplastic transformation of hepatocytes to HCC is thought to necessarily involve multiple molecular mechanisms, resulting from the accumulation of several genetic and epigenetic alterations. Many of the specific target genes and molecular pathways leading to neoplastic transformation of hepatocytes remain to be determined, but identification of these genes and pathways will accelerate the development of novel diagnostic and therapeutic strategies. Our findings suggest that loss of ENTPD5 activity results in increased turnover of hepatocytes and promotes hepatocellular oncogenesis. Therefore, Entpd−/− mice might provide a useful model in studying mechanisms involved in pathogenesis and progression of hepatocellular carcinoma. Such mice could also provide a model for testing new therapies and for rodent bioassays in drug and chemical safety assessments intended to predict the carcinogenic potential of test compounds.
Footnotes
Acknowledgements
Sincere thanks to Joe Shaw for his expert editorial assistance, to Ling Li and Jianghong Jiang for cytochrome P450 activity measurements, and to Ryan Vance and Kathy Henze for their excellent histology support.