
Abstract
Summary
Cystic fibrosis (CF) is caused by a defect in the transmembrane conductance regulator (CFTR) protein that functions as a chloride channel. Dysfunction of the CFTR protein results in salty sweat, pancreatic insufficiency, intestinal obstruction, male infertility and severe pulmonary disease. In most patients with CF life expectancy is limited due to a progressive loss of functional lung tissue. Early in life a persistent neutrophylic inflammation can be demonstrated in the airways. The cause of this inflammation, the role of CFTR and the cause of lung morbidity by different CF-specific bacteria, mostly Pseudomonas aeruginosa, are not well understood. The lack of an appropriate animal model with multi-organ pathology having the characteristics of the human form of CF has hampered our understanding of the pathobiology and chronic lung infections of the disease for many years. This review summarizes the main characteristics of CF and focuses on several available animal models that have been frequently used in CF research. A better understanding of the chronic lung infection caused particularly by P. aeruginosa, the pathophysiology of lung inflammation and the pathogenesis of lung disease necessitates animal models to understand CF, and to develop and improve treatment.
Overview of human cystic fibrosis
First described by Fanconi in 1936 and further characterized by Andersen (1938), cystic fibrosis (CF) is the most common and fatal autosomal-recessive disorder in the Caucasian population. It affects about one in 3300 live births, and with a combination of therapies and diet results in a life expectancy of 30 years (Welsh et al. 2001). The CF transmembrane conductance regulator (CFTR) gene, mapped on human chromosome 7 and responsible for CF, encodes a cAMP-regulated chloride ion channel, the CFTR (Kerem et al. 1989, Riordan et al. 1989, Rommens et al. 1989), which acts as a chloride ion (Cl-) channel in the apical membrane of epithelial cells (Welsh 1996). The balance of transport in normal and in CF airways is summarized in Figure 1.
Transport in normal and cystic fibrosis (CF) airways. In a normal airway an appropriate balance between Na+ absorption (mediated by epithelial sodium channel [EnaC] in the apical membrane) and anion secretion (mediated by apical CF transmembrane conductance regulator [CFTR] and alternative anion channels) determines the volume of fluid on airway surfaces. This transport balance regulates the height of the periciliary liquid (PCL) in which cilia beat to permit effective mucus clearance. Mall et al. (2004) created a mouse overexpressing β-ENaC to augment sodium absorption. This reduced PCL depth impedes ciliary movement and mucus clearance whereas normally, CFTR downregulates ENaC activity. This process does not occur in CF because CFTR is absent or defective. In β-ENaC transgenic mice and perhaps in CF airways failure of mucus clearance promotes accumulation of airway secretory products, including glycosaminoglycans (GAGs) and pool of chemokines (e.g. interleukin-8), neutrophil proteases and growth factors that promote airway inflammation and mucus cell hyperplasia. Modified from Fizzell et al. (Frizzell & Pilewski 2004)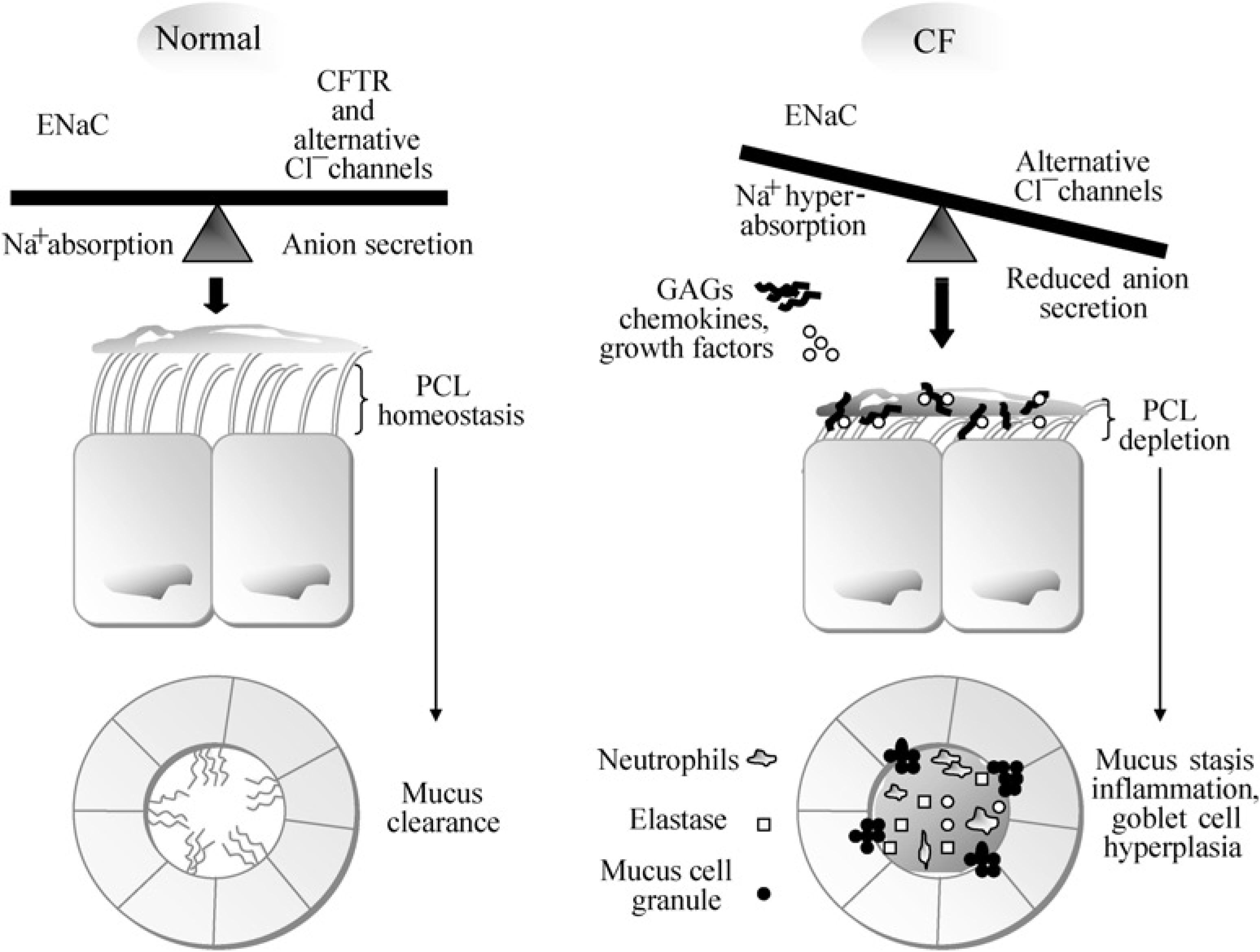
The ΔF508 mutation occurring in approximately 70% of human CF cases is a trinucleotide deletion in exon 10 resulting in the absence of a phenylalanine amino acid residue from nucleotide-binding domain 1. The CFTR protein is still produced, but is non-functional because it lacks this amino acid, is not properly processed in the endoplasmic reticulum and does not reach the apical cell membrane. Curiously, it has been shown that the protein can be functional when it reaches the plasma membrane. Many other mutations also cause CF; these include mutations that lead to dysfunctional or non-functional proteins, as well as complete ‘null’ mutations (Davidson & Dorin 2001). CFTR is expressed in many epithelial cells, including sweat duct, airway, pancreatic duct, intestine, biliary tree and vas deferens. The characteristic manifestations of CF are salty sweat, pancreatic insufficiency, intestinal obstruction, male infertility, biliary cirrhosis, congenital bilateral absence of the vas deferens and severe pulmonary disease characterized by recurrent bacterial infections and bronchiectasis. Lesions in CFTR can give rise to other clinical syndromes or vulnerabilities as well, but most clinicians will reserve the term CF for those who will ultimately develop progressive, fatal lung disease (Davis 2006).
Lung infection
The lung is exposed to a constant barrage of inhaled harmful agents and microorganisms. Several layers of defences in the normal lung help prevent infection from inhaled or aspirated microorganisms. These include the mechanical filtering of particles that occur in the nasal airway, the trapping of particles in mucus and mucociliary clearance. Respiratory epithelial cells also secrete surfactant proteins, antimicrobial peptides and complement; all these secreted proteins are important in the innate mucosal immunity (Fleming & Allison 1922, Franken et al. 1989, Thompson et al. 1990, Reynolds 1997). In addition, alveolar macrophages, neutrophils, lymphocytes and circulating antibodies participate in the clearance of microorganisms from the lung, usually at the cost of some degree of inflammation (Reynolds 1997). A loss of some of these barriers frequently results in lung infection.
Lung disease in CF is characterized by chronic microbial colonization and repeated acute exacerbations of pulmonary infection caused mostly by a unique spectrum of bacterial pathogens (Gilligan 1991). Pathological studies indicate that, at birth in CF, the lung is normal and the only abnormality detected before infection is widening of the mouths of the submucosal glands, as if the glands were already impacted with mucus (Sturgess & Imrie 1982). In contrast, the pancreas and the gut are often damaged at birth. Like normal infants, patients with CF acquire viral infections which are not more frequent but are more likely to be symptomatic (Wang et al. 1984). Unlike normal infants, however, patients with CF develop bacterial infections early in life, which initially appear to clear with vigorous antibiotic therapy. Unfortunately, permanent colonization of the airways is established with age. It is likely that bacterial colonization occurs because reduced chloride secretion and increased sodium reabsorption in airway epithelium leads to reduced water content of secretions as well as reduced depth of periciliary fluid, which in turn leads to trapping of inhaled bacteria and slower clearance (Saiman 2004). These episodes precipitate progressive, irreversible, inflammatory lung damage. Bronchoalveolar lavage (BAL) fluid shows a predominantly neutrophil inflammation with elevated interleukin (IL)-8 and neutrophil elastase (Gutierrez et al. 2001) (Figure 1). This persistent inflammation is the major cause of progressive lung injury and destruction leading to a decrease in lung function.
In CF patients, infection of airways with Pseudomonas aeruginosa leads to a wide spectrum of pathological responses and lung injuries. One extreme is the acute nosocomial necrotizing pneumonia associated with a high incidence of morbidity and mortality. In spite of aggressive antibiotic therapy, P. aeruginosa infections lead to epithelial destruction and bacterial invasion of the pulmonary vasculature, bacteraemia and its sequellae. At the opposite extreme is persistent airway infections, where early onset inflammation characterized by neutrophilia and pro-inflammatory cytokines in BAL precedes chronic infection (Armstrong et al. 1995, Balough et al. 1995, Kent et al. 1997).
Development of appropriate animal models for understanding the pathophysiology of lung inflammation and the pathogenesis of lung disease in CF with infecting bacteria is needed to improve current therapy and develop future treatments (Heijerman 2005).
Spectrum of bacterial pathogens in CF lung
The major causes of morbidity and mortality in CF patients are respiratory infections. Early in life, a large spectrum of bacterial invaders is detected in CF lungs including Staphylococcus aureus, Haemophilus influenzae and the Burkholderia cepacia complex (BCC) (Tarran et al. 2001, Festini et al. 2006). Eventually, P. aeruginosa appears and is maintained permanently in the CF lung forming biofilm-containing cells with a mucoid phenotype. These events are marked by an acceleration in persistent lung infections and a decline of pulmonary functions (Bals et al. 2001). P. aeruginosa infections incite an exuberant, persistent neutrophilic inflammatory response; the combination of neutrophils and bacterial products ultimately destroy the lung airway wall. Moreover, there is an increase in the volume of glands and secretory cells in the epithelium where their secretions contribute to airway impaction. Bronchiectasis ensues and the growth of blood vessels predisposes to massive haemoptysis. With time, a modest degree of emphysema develops. Bronchial cysts can develop and reach the periphery of the lung predisposing to pneumothorax (Tomashefski et al. 1985). Bacterial infections persist and periodically exacerbate, requiring treatment. Despite intensive therapy, infection is difficult to eradicate.
Over time, bacteria highly resistant to antibiotics supplant the initial colonization. Atypical mycobacteria, yeast and fungi are common. Allergic bronchopulmonary aspergillosis occurs in patients with CF, and more than half of adolescents and adult patients with CF have Aspergillus fumigatus cultured from the sputum (Stevens et al. 2003). Antibiotic-resistant bacteria also invade the lung including Stenotrophomonas maltophilia and Achromobacter xylosoxidans, the pathogenic role of which is not yet entirely clear but appear to have little impact on the course of the disease (Goss et al. 2004). In 1980, the isolation of Mycobacterium spp. from the sputa of CF patients was reported (Boxerbaum 1980). Species implicated included M. tuberculosis, M. avium-intracellulare, M. kansasii, M. gordonae, M. chelonei and M. fortuitum (Hjelte et al. 1990, Kilby et al. 1992). The prevalence of mycobacterial infection among CF patients ranges from approximately 4% (Torrens et al. 1998) to almost 20% (Hjelte et al. 1990). However, the relative clinical importance of these infections is unclear. Alcaligenes xylosoxidans is another pathogen found in CF patient oropharyngeal cultures (Burns et al. 1998), but again its finding has unclear clinical significance. Many other pathogens are occasionally isolated from sputum cultures of CF patients, but their clinical importance is an open question. One common thread to all of these unusual pathogens is that they can colonize and infect CF lungs extensively damaged by years of chronic mucoid P. aeruginosa infections (Lyczak et al. 2002).
P. aeruginosa and BCC are Gram-negative bacteria widespread in the external environment (stagnant fresh water) and in specific reservoirs of hospital environments (e.g. washbasin drains). They are usually innocuous for immunocompetent subjects; however, in hospital environments they may turn into severe pathogens affecting patients with lowered immune defences (burns, immunodepressed and oncological patients; ventilated patients in intensive care). Although the mechanism is not fully understood, these bacteria have specific affinity for the lungs of CF patients and are well adapted to CF respiratory mucosa; their maintenance is facilitated by the chronic inflammatory condition that gradually builds up in the lungs. Initially, the presence of these bacteria is intermittent and clinically silent. Over time, P. aeruginosa is established permanently giving rise to episodes of acute infections (pneumonia), progressive clinical deterioration and worsening of the prognosis (Festini et al. 2006).
P. aeruginosa is the most common and clinically important pathogen in patients with CF. The study of P. aeruginosa is complicated by phenotypic alteration that this organism undergoes over the course of chronic infection in the lungs of CF individuals. Although the initial infections are with planktonic strains, rapid deterioration of the lung usually occurs after transformation of P. aeruginosa into the mucoid form. It is not clear what the impact and significance of previous infections with other organisms and antibacterial chemotherapies primes for P. aeruginosa infections (Davidson & Rolfe 2001). Chronic colonization of the lungs with P. aeruginosa in patients with CF is associated with reduced lung function and life expectancy (Taccetti et al. 2005).
Pathogenesis of human CF lung disease
Although lung disease is the major clinical concern in CF and has been extensively studied, the mechanisms by which mutations in the CFTR gene result in the characteristic human pathology remains keenly debated. One current hypothesis emphasizes the role of CFTR in determining the volume and the ionic concentration of the airway surface liquid (ASL) lining in the lung epithelia (Wine 1999). Thus, CF might be the result of an abnormally low ASL volume that compromises mucociliary clearance mechanisms or a raised ASL salt content that inhibits salt-sensitive antimicrobial agents, such as beta-defensins and lysozyme. Other theories include increased bacterial adherence to CF epithelial cells (Saiman & Prince 1993) and decreased bacterial internalization (Pier et al. 1997). Hence, a better understanding of CF and, in particular, the initiation and maintenance of bacterial infections requires animal models of chronic lung infection.
Animal models for CF studies and chronic infections with P. aeruginosa
Traditional therapies in CF have been used to alleviate the pulmonary, gastrointestinal and pancreatic manifestations of CF disease (Ramsey 1996). The mainstays of current therapy are chest physiotherapy with postural drainage, aggressive antibiotic therapy and pancreatic enzyme supplementation. The use of an aggressive regimen incorporating these treatments has been responsible for the current improvements in survival of CF patients. However, novel approaches are required to achieve further increases in life expectancy and to improve quality of life. Exciting information gained from the human genome sequence, the CFTR locus, knockout (KO) and CF mice can only be exploited with relevant animal models (Zeitlin 1999). Animal models provide not only an important resource for the development and testing of new therapies, but also for studies into the initiation and maintenance of bacterial infections, particularly with P. aeruginosa.
In addition to animal models, there is a collection of numerous wild-type and CFTR-/- cell lines available which can be cultured as polarized or non-polarized. The in vitro culture of epithelial cells from the lungs of CF patients has permitted important studies in CF cell biology (Bals & Hiemstra 2004, Davidson et al. 2004, Ulrich & Doring 2004, Willems & Jorissen 2004, da Paula et al. 2005). However, many of these immortalized cell lines do not maintain proper cellular differentiation and typical epithelial cell properties. Primary cells can maintain their epithelial properties for several generations but are limited by the availability of tissues (e.g. nasal polyps) (Lau et al. 2005). Unfortunately, these cell lines lack the specific differentiation phenotypes and structural complexity of intact organs. Animal models are still essential for the study of CF pathogenesis, for studying bacterial chronic lung infections and for the evaluation of new therapeutics.
Table 1 presents a list of animal models that have been used for the analysis of P. aeruginosa virulence and its capacity in causing opportunistic infections. In this compilation, which is by no means comprehensive, priority is given to murine models of infection. The rationale for this selection is the increasing availability of transgenic mice with various defects relating to CF and lung disease, which represents a constantly expanding and invaluable resource as an animal model of first choice.
Animal models for Pseudomonas aeruginosa infection studies
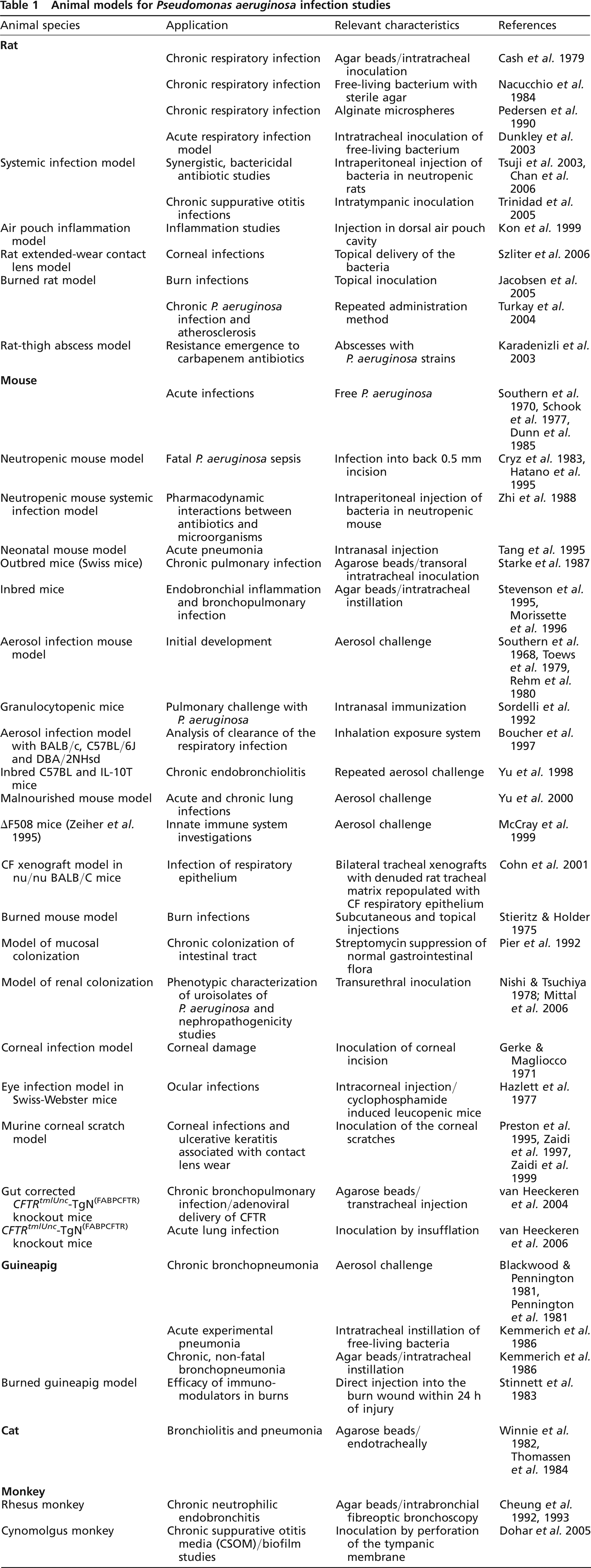
Despite vigorous screening of non-human primates, natural animal models of CF have not yet been identified (Glavac et al. 2000). Because of this limitation, a collection of so-called animal models have been developed in the laboratory. As depicted in Figure 2, numerous in vivo and in vitro infection models have been used to study P. aeruginosa virulence. Acute pneumonia models are useful to study non-CF-related pneumonia and may be of interest for the initial phase of lung infection during P. aeruginosa colonization of CF airways. As infection progresses into the chronic stage in CF lungs, P. aeruginosa switches into a biofilm mode of growth coupled with a mucoidy phenotype; the B-band O-antigen of its lipopolysaccharide (LPS) undergoes changes and bacterial cells lose flagellar motility. The introduction of free-living and mobile P. aeruginosa cells into the lung provides a model of acute lung infection; there is either rapid clearance of the organism or acute sepsis and death (Southern et al. 1970, Schook et al. 1977, Dunn et al. 1985). Some investigators have employed single or repeated aerosol administration of bacteria in attempts to reproduce the acquisition and initial symptoms of infection caused by P. aeruginosa as seen in CF patients (Yu et al. 1998, McCray et al. 1999). Because P. aeruginosa is an opportunistic pathogen, chronic infections are obtained more readily by using P. aeruginosa cells impregnated in agar or alginate beads. Cash et al. (1979) originally devised a model of chronic bronchopulmonary infection in rats using P. aeruginosa cells embedded in agar beads. Starke et al. (1987) modified the system and adapted the embedded bacteria for use in mice. Nacucchio et al. (1984), however, have indicated that free-living P. aeruginosa in the presence of sterile agarose beads is sufficient to cause histopathological changes that resemble those seen using P. aeruginosa-laden agar beads in rats. Alternative use of seaweed alginate beads in a chronic bronchopulmonary infection model was developed by Pedersen et al. (1990).
Schematic representation of experimental systems available to study Pseudomonas aeruginosa virulence. In vivo, in vitro and model host systems used to study P. aeruginosa virulence. Modified from Lau et al. (2005)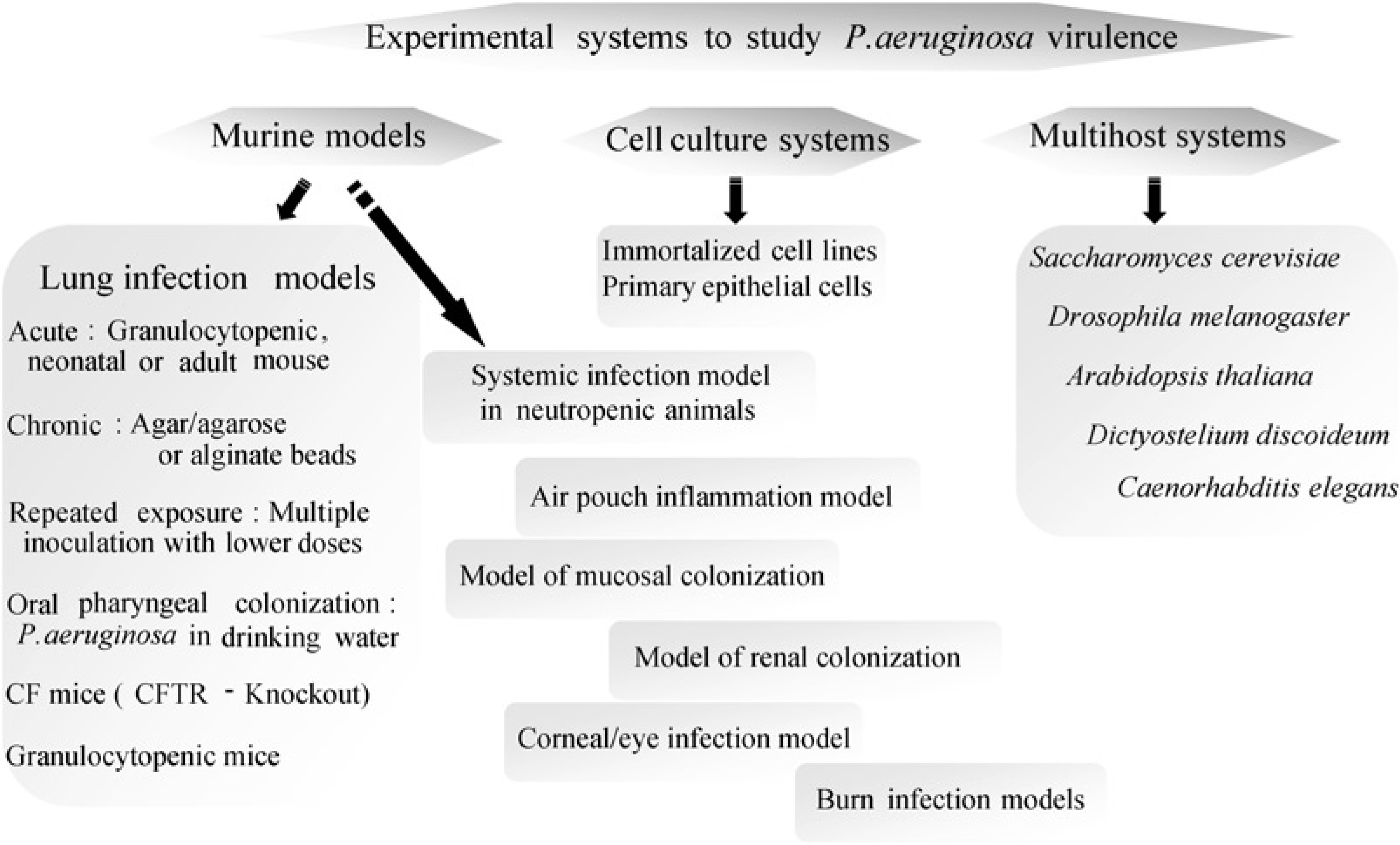
Since the CF gene was cloned in 1989, several mouse models have been created using the sequence for gene targeting within embryonic stem (ES) cells. The current mouse models encompass several phenotypes of CF and include mice with a complete KO of CFTR function, with residual CFTR function, and with precise mutations corresponding to those found in human CF. All CF mice demonstrate the characteristic CF ion-transport defect, show some evidence of intestinal disease and large variations in survival. Genetic background has also been shown to affect the intestinal phenotype of CF mice. This has allowed identification of a ‘genetic modifier’ locus of CF in humans (Davidson & Dorin 2001).
Lung disease in human CF is the major cause of morbidity and death in early adulthood. This condition is not entirely reproduced in CF mice but repeated exposures of the lung to clinical pathogens reveals a significantly abnormal pathogen-related response in the residual-function of the mouse lung. CF mice have been successfully used to investigate the safety and efficacy of various pharmacological and gene-therapy protocols. As new cloning techniques and gene constructions become available, these CF models can be refined to mimic human CF (Davidson & Dorin 2001).
Furthermore, many variants of animal models of infection were developed for studies of P. aeruginosa virulence in urinary, otitis and corneal infections, ulcerative keratitis associated with contact lens wear and infected burn wounds (see Table 1).
Several hosts have been used extensively to identify novel virulence factors from P. aeruginosa by using genome-wide mutagenesis screens (Figure 2). Several determinants were subsequently confirmed to be required for the initiation and maintenance of P. aeruginosa in the lung. In addition, these animal models of infection have been used to identify and define the molecular mechanisms of bacterial virulence. These model systems represent convenient methods to screen for conserved P. aeruginosa virulence determinants essential for infecting different hosts; these findings still require confirmation in mammalian systems (Lau et al. 2005).
Rat model of chronic lung infection
One of the first model of chronic bronchopulmonary infection in the rat was designed by Cash using P. aeruginosa embedded in agar beads (Cash et al. 1979). The use of agar or agarose beads (Klinger et al. 1983) is essential for initiating and creating a chronic lung infection. The purpose of embedding P. aeruginosa within beads is to retain the bacteria physically in the airways so that it mimics the bacterial biofilm present in the CF lung (Hoiby et al. 2001). Such retention presumably avoids physical elimination and leads to a persistent stimulation of host defences typical of CF. There is now evidence that injection of P. aeruginosa into agar beads for a chronic lung infection model is more representative of CF than administration of free-living P. aeruginosa alone. Surgical implant of the beads bypasses the primary host defences and does not take into account initial bacterial colonization. Although bacteria can migrate from the agarose beads in vivo, growth is slow within the beads; this phenomenon is comparable with what has been observed in biofilms and in the planktonic state (Hodgson et al. 1995). Although models using free-living bacteria alone may mimic some features of CF, inoculating animals with P. aeruginosa-laden agarose beads typically resembles the chronic lung infection of CF including histopathology, elevation in lung neutrophils and accumulation of cytokines. The ‘embedded bacteria model’ is particularly challenging since there is extensive neutrophil influx in response to the P. aeruginosa-laden beads, which may cause airway obstruction; effective gas exchange is not always optimal and a small percentage of animals may die. Given its simplicity and efficacy, this very first model of chronic bronchopulmonary infection with P. aeruginosa using agarose beads is still frequently used today (Cantin & Woods 1999, Amano et al. 2000, Omri et al. 2002, Duan et al. 2003, Potvin et al. 2003, Lehoux et al. 2004, Boyer et al. 2005, Joly et al. 2005, Woods et al. 2005, Zhang et al. 2005).
The use of alginate in several studies as a substitute for agar beads was attempted because it mimics natural conditions in CF lungs where bacteria are embedded with alginate in confluent biofilms (Wu et al. 2001, Ciofu et al. 2002, Wu et al. 2004, Song et al. 2005). The alginate bead model closely resembles the later stages of CF lung infection and offers the theoretical advantage of using a substance which is chemically similar to the alginate produced in vivo by P. aeruginosa (Pedersen et al. 1990).
Mouse models of P. aeruginosa chronic lung infection
The initial agar bead model of chronic bronchopulmonary infection with mucoid P. aeruginosa was modified for use in mice by Starke et al. (1987) and has frequently been used to study CF lung disease, for bacterial pathogenesis (Gosselin et al. 1995, Morissette et al. 1995, Stevenson et al. 1995, Morissette et al. 1996, Moser et al. 1997, Gosselin et al. 1998, Sapru et al. 1999, Tam et al. 1999, McMorran et al. 2001, Dagenais et al. 2005), and for evaluation of potential new treatments (Pier et al. 1990, Staczek et al. 1998, van Heeckeren et al. 1998, Chmiel et al. 1999, Wilmott et al. 2000).
Several CF mouse models have been generated using gene targeting to disrupt the murine CFTR locus by homologous recombination (Dorin et al. 1992, Snouwaert et al. 1992, O'Neal et al. 1993, Ratcliff et al. 1993, Hasty et al. 1995, Rozmahel et al. 1996) and by introducing specific human mutations into the equivalent mouse loci, including ΔF508 (Colledge et al. 1995, van Doorninck et al. 1995, Zeiher et al. 1995, French et al. 1996) and the G551D mutation (Delaney et al. 1996, Dickinson et al. 2000). Although intestinal disease manifestations are prominent in these animals, there is little evidence of lung disease in CF mice maintained under normal housing conditions. It has been proposed that the low incidence of lung infection in CF mice includes the presence of alternative Cl- transport pathways (Clarke et al. 1994, Grubb et al. 1994) and the presence of ‘modifier genes’ (Rozmahel et al. 1996).
CF mice models and lung disease
Murine CF gene
The murine homologue of the human CFTR gene is found on mouse chromosome 6 and was isolated in 1991 (Tata et al. 1991). The murine CFTR gene spans approximately 152 kb with all 27 exons being highly similar to the human homologue (Ellsworth et al. 2000). The murine CFTR protein is very similar to the human (78% identity) and the majority of known CF mutations occur in well-conserved regions suggesting conservation of function across species. The predicted mouse protein has a phenylalanine residue corresponding to the phenylalanine deleted in the common ΔF508 mutation, flanked by a 28 amino acid region that is identical to human CFTR. Although human and murine CFTR have many properties in common, some important differences in function have been described (Lansdell et al. 1998).
CF mice
The first mouse models of CF were created using gene targeting in ES cells to disrupt the murine homologue of the CFTR gene. This could be done with a technique pioneered by Thomas and Capecchi (Thomas & Capecchi 1987, Ledermann 2000) which uses homologous recombination in mouse to target a mutation to a specific site in the chromosome. The resulting ES cells are injected into blastocysts to produce chimeric mice and subsequently a mutant strain through germ-line transmission of the mutant allele (Scholte et al. 2004).
The initial CF KO and residual CF function mouse models contained mutations that resulted in a complete loss of function by using a ‘replacement strategy’ to produce an interruption of the CFTR gene and create absolute nulls and no CFTR protein production. Examples of these mouse strains are summarized in Table 2 (Snouwaert et al. 1992, Ratcliff et al. 1993, Hasty et al. 1995, Rozmahel et al. 1996). In addition, models using an ‘insertional strategy’ into the target gene without loss of genomic sequence were also described (Dorin et al. 1992, O'Neal et al. 1993). These insertional mutants retain the potential for reversion to wild-type and the production of normal CFTR mRNA (messenger RNA) by various mechanisms. This is particularly evident in the CFTRtm1Hgu mouse (Dorin et al. 1994), resulting in the description of this mutation as ‘residual in function’. The low level of normal CFTR is most likely responsible for the significant phenotype differences observed between the CFTRtm1Hgu mouse and the absolute nulls. It should be noted that human heterozygotes are also predicted to retain some level of normal CFTR function (Sheppard et al. 1993). These individuals are still diagnosed as having CF and although pancreatic sufficient, can develop severe lung disease. Thus, despite their residual-function, the insertional mutant mice still mimics CF and are important models (Davidson & Dorin 2001).
Cystic fibrosis (CF) mouse models

FABP: fatty acid-binding protein, ENaC: epithelial Na+ channels. References are in parentheses
While these CF mouse models have been fundamental to CF research, there was no clear evidence that these laboratory mutations would accurately replicate the phenotype of specific clinical mutations, in which dysfunctional or non-functional CFTR proteins are produced. To address this question, a collection of recombinant CF mouse models was generated containing clinically relevant mutations in CFTR by introducing specific human mutations into the equivalent mouse loci including ΔF508 (Colledge et al. 1995, van Doorninck et al. 1995, Zeiher et al. 1995, French et al. 1996), G551D (substitution of a glycine with an aspartic acid) (Delaney et al. 1996) and G480C (missense mutation) (Dickinson et al. 2000) (Table 2). These CF mouse models were created using either a replacement gene-targeting strategy or a double homologous recombination ‘hit and run’ procedure (Hasty et al. 1991). The latter technique produces a mutated exon without selection of marker genes or plasmid sequences in the intronic structure of the gene. The presence of such intronic ‘debris’, which arises during replacement gene targeting could be responsible for the observed transcriptional interference.
Finally, a transgenic KO model expressing a human CFTR with the G542X mutation under the control of the intestinal fatty-acid-binding protein (FABP) gene promoter has been generated and used to study the effect of aminoglycosides on suppression of this CFTR premature stop mutation (Du et al. 2002). These mouse models with specific CF mutations provide a clinically relevant in vivo system permitting the preclinical testing of compounds that emerge from large-scale screening programmes and mutation-specific therapeutic approaches. Thus, it is crucial that the possible consequences of the techniques used to generate the different mouse models are taken into account in interpretation of the phenotypes observed.
Phenotypes of CF mice
Animal models of human diseases must be carefully characterized and demonstrate appropriate pathology to justify their use in the understanding of the phenotype in pathogenesis of the disease and in the design and testing of novel therapies. One possible source of concern is the variations observed in phenotype from identical genotypic abnormalities between different species. The existence of numerous different CF mouse models provides the opportunity to study the contribution of a variety of factors causing a disease within the same species (Davidson & Dorin 2001).
Different CF mouse models presented in Table 2 include tight and leaky CFTR KOs as well as a number of specific CFTR mutations. In general, all models show a CF-like phenotype, in particular, a severely reduced cAMP-induced chloride conductance in various epithelial cells (small intestine and colon, gall bladder, airways). Pathology and mortality in CF mice is dominated by intestinal disease which is characterized by goblet cell hyperplasia and mucus accumulation in crypts. This is associated with reduced body weight and mortality due to intestinal plugging. In contrast to pancreas function in CF patients, the pancreas functions and the histology appear normal in CF mice. Under standard laboratory conditions, CF mice do not suffer from spontaneous lung disease (chronic inflammation caused by opportunistic pathogens and fibrotic regeneration) as is observed in most CF patients. However, several publications have claimed that when challenged with bacterial pathogens, CF mice are less capable of clearance and suffer from more pronounced inflammation. Davidson et al. (1995) demonstrated that with repeated bacterial challenges, there is clear evidence of decreased clearance of inhaled bacteria and persistent inflammatory disease. Van Heeckeren et al. (1997) found that CFTR null mice showed more inflammation and morbidity when challenged with agarose beads of P. aeruginosa. Kent et al. (1997) found that an inbred congenic strain of CFTR null mice spontaneously developed lung disease; unlike CF in humans the disease was primarily alveolar. Thus, with exposure to large bacterial loads or alteration of the genetic background, the CF mouse may develop lung disease (McCray et al. 1999). Yu et al. (2000) hypothesize that this effect could be secondary to the malnutrition caused by the CFTR defect and not necessarily by a dysfunction of pulmonary CFTR.
The mouse models differ from human CF with respect to the severity of the phenotypes resembling CF and which depends upon the type of mutations, the levels of mRNA expression and the genetic background of the mouse strain. The genetic background possibly relates to different levels of activity of ‘modifier genes’ affecting the CF mutations (Rozmahel et al. 1996). All CF mouse models available were originally produced in a mixed genetic background containing chromosomes from the mutant embryo stem cell line and the recipient blastocyst (FVB or C57BL/6). It is important to generate CFTR mutant mouse colonies that are backcrossed to several different mouse backgrounds. This would reduce experimental variability and allow a systematic analysis of ‘modifier genes’ in CF mouse models.
Survival of CF mice
Intestinal pathology and mortality appear to be the predominant hallmark of CFTR mutations in CF mice. The survival rates reported in the initial characterization of the different mouse models varied from <5% survival to maturity in CFTRtm1Unc nulls and CFTRtm1Cam mice, to approximately 90% in CFTRtm1Hgu mice and normal survival in CFTRtm2Hgu and CFTRtm1Eur mice. The remaining 10% of CFTRtm1Hgu animals died of intestinal obstruction at weaning. Interestingly, this proportion is similar to what has been observed for meconium ileus in human CF. The low-level production of normal CFTR has been proposed to be the explanation for the significantly greater survival rates in the CFTRtm1Hgu mice (Davidson & Dorin 2001).
Pathophysiology of airway epithelium in CF mice
The most significant impact of CF in affected individuals is progressive pulmonary disease, which is a major cause of morbidity and mortality. The lung disease is not evident at birth but develops over repeated acute exacerbations of pulmonary infections with the appearance of a spectrum of bacterial pathogens, chronic microbial colonization, tissue damaging inflammation and irreversible deterioration of lung function (Govan & Nelson 1992). Chronic pulmonary infection with P. aeruginosa is the primary concern with the characteristic transition of this organism to mucoid phenotype clearly correlating with poor prognosis and clinical decline (Parad et al. 1999). However, it remains unclear whether CFTR dysfunction results directly in an increased predisposition to infection with this organism or a broader susceptibility resulting in repeated infections with organisms such as S. aureus or respiratory syncytial virus and inappropriate inflammatory responses generating a lung environment that favours subsequent infections with P. aeruginosa (Scholte et al. 2004).
Initial characterization of the mouse models provided little indication of gross pulmonary abnormalities. These observations were initially surprising and raised concerns for these models. However, an expectation of mucus plugging, neutrophil accumulation and bronchiectasis in these mutant animals can now be considered to be unrealistic. This is particularly true if bacterial interaction is required to initiate a cycle of infection and inflammation because most of these mutant animals were maintained in semi-sterile to sterile facilities. Furthermore, the development of characteristic lung histopathology in CF individuals is a gradual process that occurs over years not weeks. The initial assessment of CF mouse was made after only a few weeks up to a few months. Indeed, in the case of null mice the vast majority died of gastrointestinal complications before a systematic assessment could be made. In this respect, the prolonged survival of the CFTRtm1Hgu mice is particularly valuable (Davidson & Dorin 2001).
As reviewed by Scholte et al. (2004), in the apparent absence of infection a variety of interesting observations have been made. These include excessive inflammation in CFTRtm1Hgu mice (Zahm et al. 1997), abnormal mucociliary clearance in the CFTRtm1Hgu (Zahm et al. 1997) and CFTRtm1Unc mice (Cowley et al. 1997), an increase in goblet cells with a decreased in volume of ASL in the nasal epithelium of CFTRtm1Unc mice (Tarran et al. 2001), a more distal extension of submucosal glands in CFTRtm1Hgu and CFTRtm1G551D mice (Borthwick et al. 1999), hypersensitivity of bone marrow-derived macrophages from CFTRtm1G551D mice to bacterial LPS (Thomas et al. 2000), and abnormalities in intracellular nitric oxide syntethase expression in CFTRtm1Unc and CFTRtm1Kth mice (Kelley & Drumm 1998, Steagall et al. 2000). Pulmonary abnormalities have been described in CFTRtm1Unc mice bred to congenicity using the C57BL/6 strain (Kent et al. 1997). In addition to obvious environmental influences such as infectious agents, data obtained suggest complex interplay between mutations and strain type (and thus independently segregating ‘modifier genes’) (Scholte et al. 2004).
Despite these promising observations, the most problematic phenotype to establish in CF mouse has been the most critical: pulmonary chronic infection with P. aeruginosa.
Initial studies using the CFTRtm1Unc, CFTRtm1Hgu and CFTRtm1Kth mice did not permit identification of abnormalities in response to infections with this pathogen (Cressman et al. 1998, McCray et al. 1999), significantly contrasting with CF lung infections in humans. Recent studies have demonstrated a defect in the epithelial cell ingestion of P. aeruginosa, with greater bacterial lung burden after 4.5 h in CFTRtm1Kth mice (Schroeder et al. 2001), oropharyngeal colonization, evidence of pulmonary spread and mucoid transformation in CFTRtm1Unc-TgN(FABPCFTR) mice following oral infection (Coleman et al. 2003). In addition, following infection with P. aeruginosa, defective airway epithelial cell apoptosis required for pulmonary clearance of this organism (Grassme et al. 2000) has been demonstrated in CFTRtm1Kth and CFTRtm1G551D mice (Cannon et al. 2003). These observations suggest that careful optimization may yet permit the development of an animal model of pulmonary infection with motile P. aeruginosa. These mouse strains may pave the way for an understanding of bacterial colonization, transformation to the mucoid phenotype and fibrotic lung damage having all the characteristics of human CF (Davidson et al. 2003).
To date, clear evidence of persistent infection and gross differences in response to P. aeruginosa have only been achieved using the agar bead model to mimic chronic colonization, both in CFTRtm1Unc (Heeckeren et al. 1997, Gosselin et al. 1998) and in CFTRtm1G551D mice (McMorran et al. 2001). These studies used agar to prevent normal clearance of the bacteria revealing mouse phenotype differences in survival and in cytokine response. Less pronounced differences in bacterial proliferation and lung pathology were found. Although the use of this technique may be effective in the study of the host response to established infection by superimposing bacterial retention on an otherwise unaffected lung, it seems less likely to be informative on the initiation and development of early stages of infection and in lung disease. Indeed, if predisposition to P. aeruginosa infection in CF is secondary to previous cycles of infection with other organisms and subsequent inflammatory damage, or due to the consequence of antibacterial chemotherapy, difficulty in modelling this infection in the CF mouse reared and maintained in sterile conditions may not be surprising. It may be that pulmonary exposure to P. aeruginosa following or in conjunction with repeated exposure to S. aureus could be more effective in triggering mouse lung disease (Scholte et al. 2004).
Despite some tantalizing similarities between CF lung disease in humans and in CF mouse models, the suitability of these animal models remains controversial, and significant differences are evident. Nevertheless, CF mouse models clearly demonstrate a range of abnormal pulmonary phenotypes as a result of the CFTR mutation. Although these species differences could prevent CF mouse models from accurately reproducing all aspects of CF lung disease in humans, they might prove as illuminating as the similarities. Rather than exclusively pursuing the development of all aspects of classical human CF lung disease in mouse models, studies should specifically address the effects of CFTR mutation upon the lung pathophysiology of mice. The consequence of CFTR mutations in the mouse lung can be addressed and the underlying mechanisms evaluated. By recognizing the key similarities and differences, CF mouse models may provide useful in vivo systems for the analysis of specific aspects of CF lung disease and for testing the validity of new hypotheses in CF disease (Davidson & Dorin 2001).
Mice with CF lung disease
The insertional KOs of the CFTR gene in mice promised to provide a long-awaited CF animal model and to better define the role of defective CFTR in lung disease. However, CFTR null mice did not live up to their promises, as animals failed to develop lung pathology that completely mimics the human disease (Grubb & Boucher 1999). Almost 15 years after the discovery of the CFTR gene, researchers have finally created a mouse with lung pathology similar to human CF. Rather than disturbing the function of CFTR, Mall et al. (2004) generated mice that absorb excess sodium in the airways. These animals show key abnormalities typical of CF including airway obstruction with dehydrated mucus. This CF model promises to answer important questions about the development of lung pathology and the origin of the inflammation that leads to lung destruction. In the future, these animals should be useful for evaluating new therapeutic interventions and new treatments (Frizzell & Pilewski 2004).
To enhance sodium absorption in mouse airways, Mall et al. expressed individual epithelial sodium channel (ENaC) subunits using a promoter specific for expression in airway cells. Transgenic mice overexpressing β-ENaC showed a 3-4-fold increase in sodium ion currents across their excised tracheas. There was no fetal mortality and at birth the airways were histologically normal. Further analysis revealed that β-ENaC mice showed characteristics of early lung disease in humans with CF. Several weeks after birth transgenic mouse had reduced periciliary liquid (PCL) depth in their lower airways obstructed with drier mucus. This mucus cleared slowly and adhered to airway surfaces. Despite the maintenance of mice in sterile facilities, mucus stasis led also to neutrophilic inflammation and increased pro-inflammatory cytokines in airway liquids. The concentrations of cytokines were not increased in lung homogenates or in the media from airway cells in primary culture. These results suggested that their production was a result of mouse development and not a primary consequence of enhanced ENaC expression. When challenged with bacteria, the airways of these mice were unable to effectively clear the organism, a situation analogous in the lungs of CF patients. This study addressed three important connections between ion transport and lung pathogenesis in CF. First, it showed that ion transport defects and the corresponding PCL depletion underlies obstructive lung disease in vivo. Second, it indicated that CF pathogenesis can arise from ion transport alterations in small, non-glandular airways and argues against earlier concepts that submucosal glands contributed to the early stages of lung pathogenesis. Finally, it showed that the inflammatory response is initiated primarily by aberrant ion transport and PCL depletion instead of bacterial infection (Frizzell & Pilewski 2004).
CF long-living mice
Almost at the same time that an animal model was developed with the characteristics of increased airway epithelial Na+ and giving a CF-like lung disease in mice (Mall et al. 2004), Durie et al. (2004) demonstrated for the first time that long-lived congenic C57BL/6J CFTR-/- mice developed the hallmarks of CF-like disease in all organs, a situation reminiscent of the pathology found in human CF. This is in striking contrast to previous observations of other CF murine models bred in a mixed genetic background (Dorin et al. 1992, Snouwaert et al. 1992, Colledge et al. 1995, Zeiher et al. 1995, Delaney et al. 1996, Dickinson et al. 2000) which showed severe intestinal disease but mild or no pathological changes in other organs affected by CF in humans (see Table 2 for details). In most published studies, the CF mice were sacrificed at an early age while progressive age-related changes in CF-like pathology became evident only after three months of age. Congenic C57BL/6J CFTR CFTR-/- and CFTR+/+ mice were maintained from weaning on a liquid diet, sacrificed at different time-points between one and 24 months of age, and their organs evaluated. The lungs of the CFTR-/- animals showed patchy alveolar overdistention, interstitial thickening and fibrosis with progression up to six months of age. The proximal and distal airway surface was encased with mucus-like material but lacked overt evidence of chronic bacterial infections or inflammation. All CFTR-/- animals showed progressive liver disease with hepatosteatosis, focal cholangitis, inspissated secretions and bile duct proliferation. After one year of age there was progression to focal biliary cirrhosis. The intercalated, intralobular and interlobular ducts and acinar lumina of the exocrine pancreas and the parotid and submaxillary glands of the CFTR-/- animals were dilated and filled with inspissated material as well as mild inflammation and acinar cell drop out. Quantitative measurements of the pancreas showed significant acinar atrophy and increased acinar volume in comparison with age-matched CFTR+/+ littermates. The ileal lumen and crypts were filled with adherent fibrillar material. After three months of age, the vas deferens of the CFTR-/- animals could not be identified. None of these pathological changes were observed in the CFTR+/+ littermates fed the same liquid diet. To conclude, this congenic KO murine model of CF has most of the hallmarks of human CF disease making it a valuable model for the study of CF pathobiology and for targeted therapies. Nevertheless, to allow significant pathological changes to occur in CF affected organs, it is necessary to breed the animals to a much older age than had been done previously. To maintain viability, intestinal obstruction can be diminished by nourishing the animals on a liquid diet. Alternatively, intestinal complications can be prevented by breeding murine models expressing CFTR cDNA of the intestinal tract under the control of the rat intestinal FABP gene promoter (Zhou et al. 1994).
Other animal models
Other animals are also being used for different P. aeruginosa virulence studies and in general in the CF field. One important advantage of these models is that the lung function, size and architecture of these animals more closely resemble the human situation.
Short-term organ culture isolated from ferret tracheal segments was used to evaluate whether P. aeruginosa alginate stimulates secretion from mucous or serous cells in the ferret trachea exposed to alginate and using concentrations reported to be present in the CF airway. A sandwich enzyme-linked lectin assay was used to measure mucin secretion and spectrophotometry to measure lysozyme secretion from isolated ferret tracheal segments (Kishioka et al. 1999).
A cat model of chronic pulmonary infection with P. aeruginosa embedded in agarose beads was developed. This model of chronic inflammation and macrophage stimulation is similar to the chronic pneumonia of human CF and may be a useful approach to answer questions on the role of macrophage activation in chronic lung disease (Thomassen et al. 1984).
Until recently, the generation of gene-targeted animal models has primarily relied upon homologous recombination following direct introduction of transgenes into ES cells. While this technique has been successful in the mouse, it has proven significantly more difficult in larger species. To date, the most exciting and promising research in transgenesis involves the use of fetal and adult somatic cells to produce genetically identical animals through nuclear transplantation (Renard et al. 2002). Successful production of cloned animals derived from somatic cells was first demonstrated in sheep (Campbell et al. 1996, Wilmut et al. 1997) and has more recently been demonstrated in mice (Wakayama et al. 1998), cattle (Kato et al. 1998), goats (Baguisi et al. 1999), pigs (Polejaeva et al. 2000), cats (Shin et al. 2002), rabbits (Chesne et al. 2002) and mules (Woods et al. 2003). Transgenic calves (Cibelli et al. 1998), gene-targeted sheep (McCreath et al. 2000) and α-1,3-galactosyltransferase KO pigs (Dai et al. 2002, Lai et al. 2002) have also been obtained by nuclear transfer from somatic cells. These successes have made animal modelling using nuclear transfer in less-studied species more feasible. It will be interesting to see which of these will be useful for CF.
The role of submucosal glands which are only found in the proximal trachea in mice can be studied in sheep, pig or cat models (Hug & Bridges 2001, Joo et al. 2001). The key questions that need to be answered in these models include: is mucus secretion from submucosal glands altered in CF? If so, how is it altered and how does it contribute to CF lung disease? Answering these questions will require an understanding of how the products secreted from these glands interact in other regions of the lung. So far, only mice have been successfully used in modelling by homologous recombination and transgenesis. Despite intensive efforts, CF sheep (Williams et al. 2003) or ferrets (Li & Engelhardt 2003) have not been created but may become a valuable contribution to studies of CF in animals and chronic lung disease applicable to human diseases.
Concluding remarks
The development of animal models for CF studies, particularly with P. aeruginosa infections progressed from those in which bacteria are artificially embedded in an immobilizing agent, such as agar, agarose or seaweed alginate to achieve a chronic lung infection. Additional models used in acute infection are induced by aerosol exposure to bacteria or by intratracheal administration of bacteria alone in normal or in immunosufficient animals and those in which the CFTR gene was disrupted. A number of CF mouse models have been described to provide in vivo systems to assist in dissecting the pathogenesis of CF lung disease. Developing an appropriate animal model to study the pathophysiology of infectious and inflammatory lung disease particularly in CF mice has proven challenging. The ability to pursue less invasive techniques and to control genetic and environmental influences should permit the development of CF mouse models. Appropriately controlled and defined studies acknowledging the similarities and the differences between mice and humans have the potential to answer questions about the basic pathophysiological processes regulating ASL in healthy lungs and in the presence of dysfunctional CFTR. In this manner, the use of CF mouse models should help to dissect the pathogenesis of CF lung disease and define many of the critical components of development of permanent infections.
Murine models of chronic P. aeruginosa lung infection are particularly useful for defining the role of the CFTR gene as well as the background of ‘modifier genes’ in host susceptibility. Overall, studies from mouse models using genetically and immunologically well-defined inbred strains support the concept of the role of local inflammation both in protection and in pathogenesis of bronchopulmonary infections. Mouse models provide useful tools for identifying the network of important pro-inflammatory and anti-inflammatory cytokines and the complex interactions among these molecules in the genesis of chronic P. aeruginosa infection in the CF lung.
The challenges for future use of mouse models of CF initially lie in refining existing models to replicate human disease as accurately as possible and in understanding the mechanisms that underlie the development of the mutant phenotypes observed only in mice. The phenotypic variability observed in mouse models of CF as a result of different genetic backgrounds, specific mutations in CFTR and environmental influences is of great potential for the definition of genetic modifiers and to refine the mouse models of CF.
To date, the most important connections between ion transport and the pathogenesis of CF lung disease arise from mice in which increased airway epithelial Na+ absorption was demonstrated (Mall et al. 2004). Ion transport defects and corresponding PCL depletion underlie obstructive lung disease in vivo, the CF pathogenesis can arise from ion transport alterations in small, non-glandular airways and the inflammatory response is initiated primarily by aberrant ion transport and PCL depletion instead of bacterial infection.
Although much has been learned about CF since the initial description of the various animal models and from subsequent studies in which vaccines, therapeutics and prophylaxis strategies were studied in different animal models, the limitations of the various models must also be recognized. Available data do not yet support a conclusion that there is a single preferred model for CF studies with P. aeruginosa in which all interventions or preventive strategies must be evaluated.
Footnotes
Acknowledgements
Work in R C Levesque's laboratory is funded by the Canadian Institute for Health Research as part of the CIHR Genomics Program. R C Levesque is also funded as a Research Scholar of Exceptional Merit and I Kukavica-Ibrulj obtained a PhD student scholarship from Le Fonds de la Recherche en Santé du Québec (FRSQ).