
Abstract
Horseshoe kidney in the presence of autosomal dominant polycystic kidney disease is a rare occurrence of two relatively common and unrelated renal findings. Visualization of multiple, bilateral cysts along with fusion of the kidneys by a midline isthmus can usually isolate these diagnoses. Accurate sonographic evaluation is essential in determining the degree of disease progression and possible complications associated with these diseases. Sonography is also useful in identifying extrarenal involvement and eliminating differential diagnoses.
Individually, horseshoe kidney (HSK) and autosomal dominant polycystic kidney disease (ADPKD) are not uncommon.1,2 However, the likelihood of an HSK to occur in conjunction with ADPKD is rare. The incidence of this dual diagnosis is estimated to be 1 in up to 8,000,000 with fewer than 20 cases documented at that time. 3 Many patients with ADPKD progress to renal failure by their sixth decade of life. 2 Therefore, serial medical imaging is important for aiding in prognosis and management. The sonographic evaluation of this rare incidence is made difficult by the combination of the large size of polycystic kidneys and the abnormal location of HSKs. Sonography remains the modality of choice when evaluating these types of kidneys, although magnetic resonance imaging (MRI) and computed tomography (CT) have been used in combination with sonography for further characterization.
Case Report
A 27-year-old woman with a history of ADPKD presented to an outpatient ultrasound department for a renal sonogram. The examination was performed to obtain an accurate evaluation of the size of the kidneys. Accurate measurements were needed in order for the patient to qualify to receive Jynarque, an FDA (United States Food and Drug Administration) approved medication used to slow the progression of ADPKD and maintain renal function. The patient had MRI 4 years prior to the ultrasound that reported the presence of multiple cysts of varying signal intensity and size, as well as the presence of an HSK. The MRI was completed in a T2-weighted phase in which the cysts appeared bright, or hyperintense, with respect to relative tissue (Figure 1). The isthmus was also visualized on MRI passing over the abdominal vessels anteriorly in the midline abdomen (Figure 2). No liver or pancreatic cysts were noted.
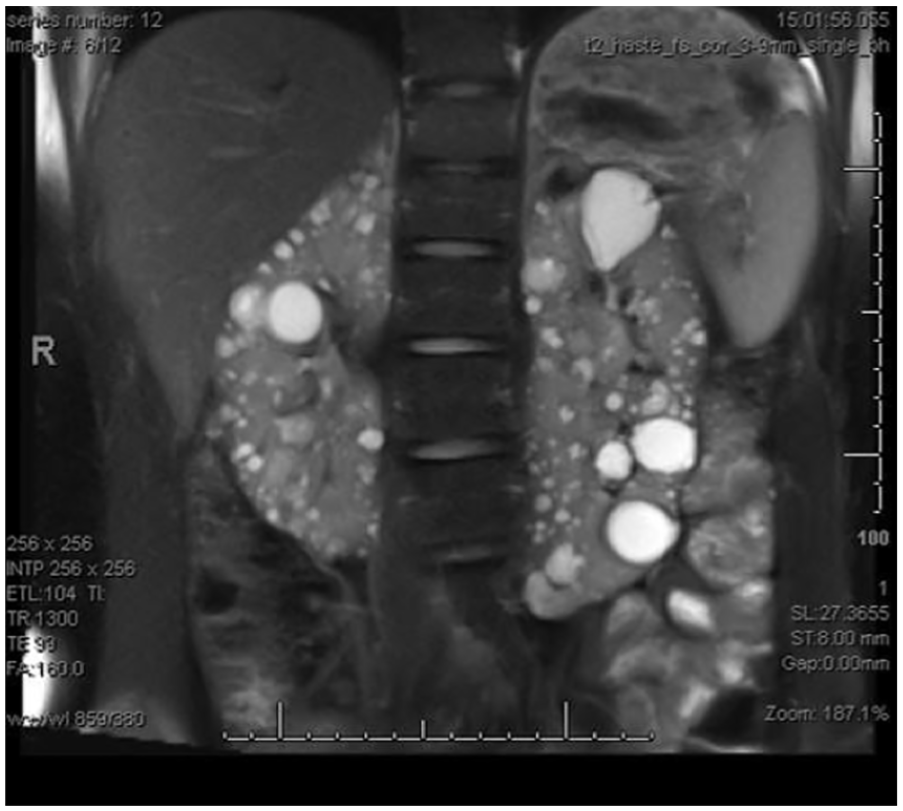
T2-weighted magnetic resonance imaging image showing multiple hyperintense cysts and bilateral enlargement of the kidneys.
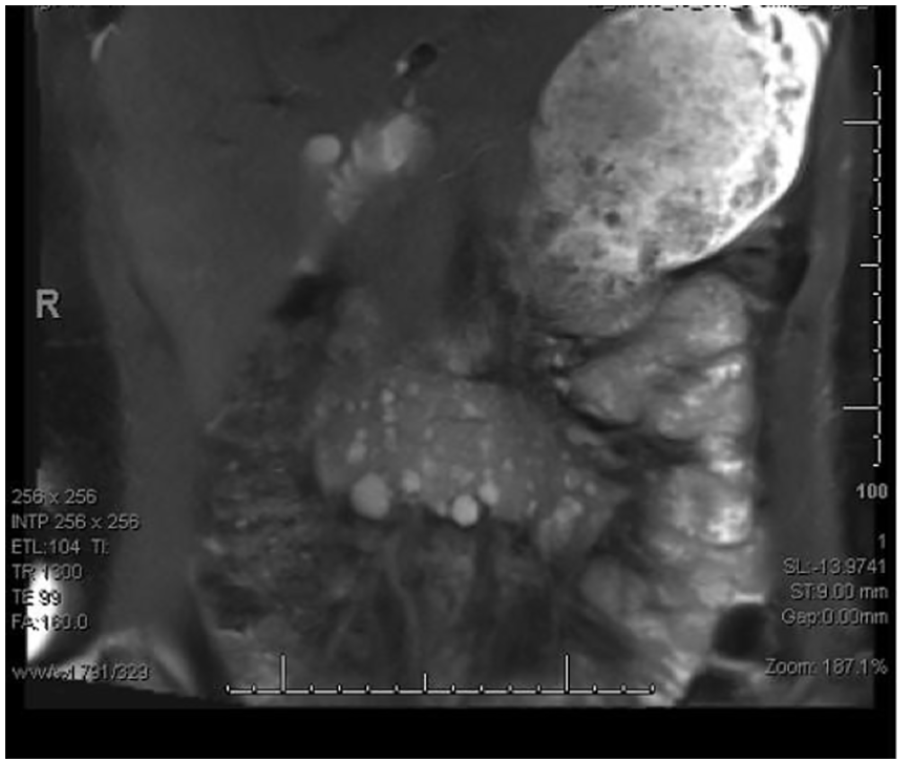
A magnetic resonance imaging image of midline isthmus connecting the inferior poles of the horseshoe kidney.
Despite the MRI findings, the patient was asymptomatic at the time of her sonogram and had a normal creatinine level of 0.61 mg/dL. Other relevant comorbid factors included early onset hypertension and a body mass index (BMI) of 35.5. Four of her immediate family members also had a history of ADPKD, one of which passed away from end stage renal failure.
The sonogram was completed using a Philips iU-22 ultrasound system (Philips Medical, Bothell, WA) with a C5-1-MHz curved linear transducer. Images revealed two large, medially rotated kidneys located high in the abdomen, inferolateral to the sternum and connected at the inferior poles by an isthmus. The isthmus was located anterior to the abdominal aorta with an AP diameter of 1.3 cm (Figure 3). The dimensions of the right and left kidneys measured 18.7 × 6.5 × 5.4 cm and 21.3 × 6.4 × 5.6 cm, respectively, displaying a left-sided dominance. Dual screen was used to obtain an accurate measurement of the left kidney (Figure 4). Each side of the HSK contained innumerable cysts of varying complexity and size that replaced much of the normal parenchymal tissue (Figure 5). The largest cyst on the right side was located on the superior pole and measured 3.3 × 3.6 × 3.5 cm, containing internal septations and peripheral blood flow seen with color Doppler. The largest cyst on the left side had a similar appearance to that on the right, measuring 2.6 × 2.6 × 2.3 cm with internal septations. This cyst contained a small calcification on the posterior aspect that revealed twinkle artifact with color Doppler. Due to the orientation, the inferior poles were difficult to visualize. There was no evidence of nephrolithiasis, hydronephrosis, or extrarenal cysts in the pancreas or the liver. A brief scan of the uterus was performed to rule out uterine anomalies, none of which were seen. This patient was lost to follow-up likely because she was asymptomatic with few complications.
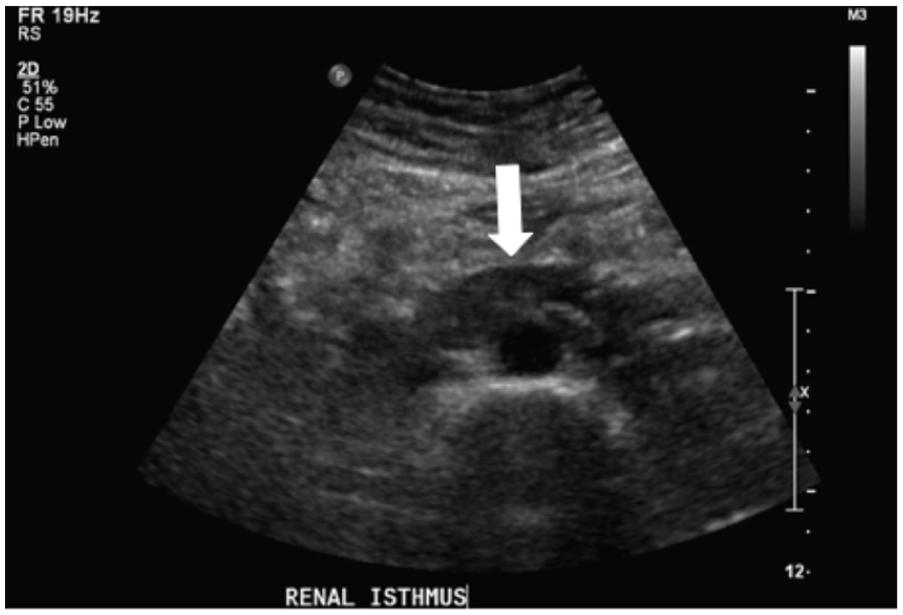
A transverse sonogram of the horseshoe kidney isthmus (arrow) located anterior to the abdominal aorta.
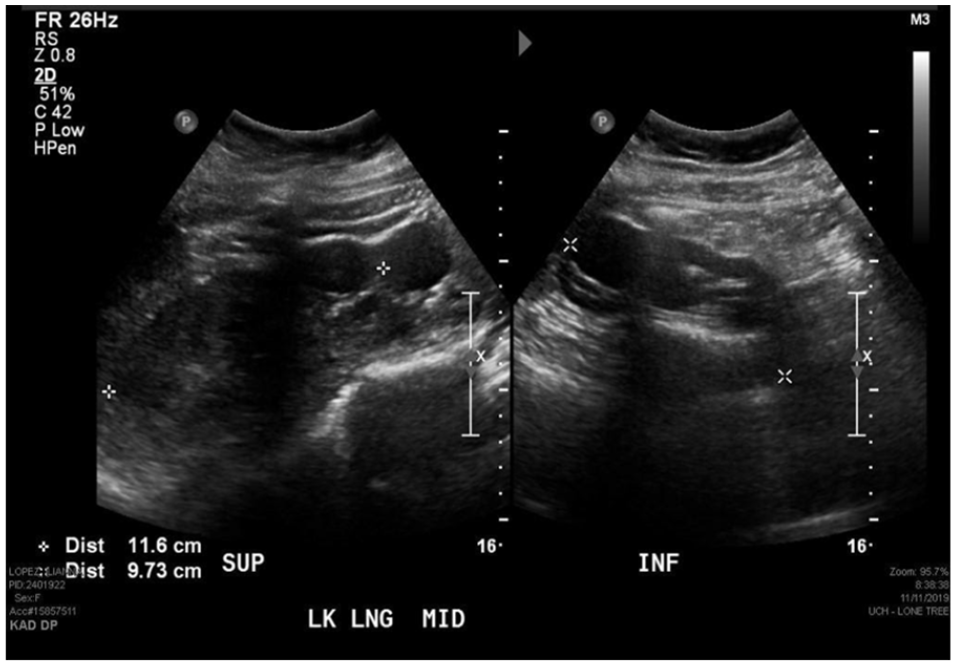
A sonogram of the left portion of the horseshoe kidney in a longitudinal plane using dual screen to obtain measurement.
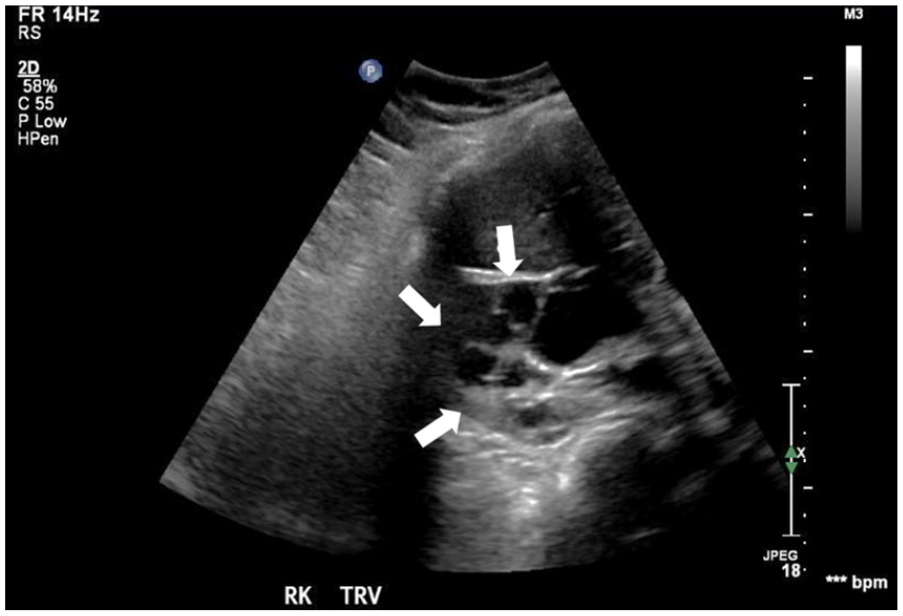
A transverse sonographic image of the right kidney (arrows) showing multiple cysts within the renal parenchyma.
Discussion
The kidneys are paired retroperitoneal organs located at the level of L1-L3. The superior poles of the normal kidneys are rotated slightly more medially than the inferior poles. The length of the kidneys ranges from 9 to 12 cm, with volumes from 120 to 170 g, but may vary with BMI, age, and gender. The cortical thickness should measure between 7 and 10 mm in a healthy kidney. 4 Embryologically, the kidneys begin to develop by the fourth week of gestation in the sacral region of the embryo. Between Weeks 6 and 9 of gestation, they begin to ascend and rotate to their permanent position in the abdomen. 5 In most cases, sonography is the ideal modality for the evaluation of the kidneys due to their anatomic position and accessibility.
HSK is a congenital renal fusion anomaly with an incidence of 1 in 500, making it one of the most common congenital renal anomalies. 3 It is twice as common in males than in females. 6 HSK occurs when the inferior (90% of cases) or superior poles of the embryologic kidneys fuse prior to ascending to their permanent position. 1 The connection between the left and right kidney is called the isthmus and is the most important characteristic to visualize when diagnosing HSK. The isthmus is typically seen lying anterior to the abdominal aorta and inferior vena cava in a midline transverse view.5,7 In 80% of cases, the isthmus is composed of functioning renal tissue, the remaining being composed of fibrous connective tissue. 1 The majority of HSK are left-side dominant, meaning the left kidney is larger and contains the majority of the isthmus. 1 A hydroureter may also be present and some cases have shown the ureters to be found coursing anterior to the isthmus. 5 The isthmus may also receive an independent blood supply originating from branches of the abdominal aorta. 8
There is no confirmed genetic cause for this anomaly although certain etiological factors such as maternal teratogen exposure, alcohol consumption, and uncontrolled hyperglycemia may contribute to the development. 1 This renal anomaly is also associated with a number of chromosomal abnormalities including trisomy’s 13, 18, 21, and Turner’s syndrome. 8 Due to its embryologic origin, HSK is also often associated with uterine anomalies such as bicornuate and septate uterus, as well as testicular anomalies such as cryptorchidism. Up to one third of patients with this anomaly are asymptomatic so it is typically an incidental finding. 8 However, patients with HSK are more susceptible to complications such as nephrolithiasis, infections, and pelviureteric obstruction due to anomalous anatomy and malposition of the ureters. 5 One study proposed that 36% of patients with HSK will acquire nephrolithiasis throughout their lifetime. 1 Ureteropelvic junction obstruction (UPJ) is one of the most common findings associated with HSK secondary to the abnormal insertion of the ureters into the renal pelvis. UPJ obstruction often causes reflux and delayed bladder emptying, posing an increased risk for ascending infections. 5
Treatment is often not necessary because HSK is considered a benign condition and when isolated, patients are typically unaffected. Intervention may be required with coexisting nephrolithiasis in which lithotripsy or laparoscopic removal is used. 1 The mortality rate increases when HSK is associated with other severe chromosomal anomalies.
HSK cannot be diagnosed clinically; therefore imaging plays an important role. CT, MRI, and sonography are the recommended methods for the diagnosis of HSK and the associated complications.6,9 CT and MRI are especially accurate in detecting the isthmus and anatomic position of the kidneys. On sonography, the anatomic location of the isthmus of HSK may be hard to appreciate depending on body habitus. The inferior poles of HSKs are often suboptimally visualized because of their midline fusion and can result in an inaccurate renal length measurement. 7 Understanding the sonographic appearance of this anomaly is important as the isthmus may be mistaken for a midline mass or may be missed altogether. 8 Due to the operator dependent nature of sonography, diagnostic accuracy is variable. 5
Identifying the vascularity of an HSK and the location of the isthmus is important in the setting of abdominal surgery as to avoid serious iatrogenic injury. Multiple imaging modalities are often used to fully evaluate the anatomy of an HSK prior to abdominal and vascular surgeries. 6 Crossed fused renal ectopia is a congenital renal anomaly that may be confused for HSK. However, the kidneys are often fused via the superior pole of the normally located kidney and the inferior pole of the ectopic kidney. Crossed fused kidneys are differentiated from HSK because of their more inferiorly located position in the abdomen. In addition, they are differentiated by their position relative to the spine, both kidneys will be located on the same side of the spine. 10
ADPKD is an adult-onset inherited disease resulting from the gene mutations PKD1 (85%–95%) located on chromosome 16 or PKD2 (10%–15%) located on chromosome 4. Both mutations lead to the formation of multiple cysts within the renal parenchyma and diffuse enlargement of both kidneys.9–11 It is the most common inherited kidney disease with an incidence between 1/400 and 1/1000. 9 The biggest risk factor for developing this disease is a positive family history. However, 10% to 15% of ADPKD cases do not have a positive family history which can be explained by new mutations, mosaicism, or an unknown family history. Patients with the PKD1 mutation are more likely to undergo more rapid progression and a more severe course of the disease than those with the PKD2 mutation. 11
ADPKD is one of the leading causes of end stage renal failure, responsible for up to 10% of patients on dialysis. 12 The rapid development of large cysts in the renal cortex and medulla leads to destruction and derangement of the nephrons, ultimately leading to kidney failure around the fifth or sixth decade of life. 10 ADPKD is likely to have extrarenal manifestations as well, with cysts often developing in the liver, seminal vesicles, pancreas, arachnoid membrane, and spinal meninges.13,14
Patients with ADPKD are susceptible to a wide range of complications including hypertension, nephrolithiasis, obstruction, and urinary tract infections. It is estimated that up to 35% of patients with ADPKD have reported nephrolithiasis. 2 Common symptoms attributed to these complications include chronic pain, fever, gross hematuria, and sleep disturbances all of which may decrease patients’ quality of life. 14 Hypertension is the most common complication associated with ADPKD, developing in 50% to 70% of patients at an early age.2,14 Elevated blood pressure is correlated with the abnormal activation of the renin-angiotensin-aldosterone system (RAAS) that is usually controlled by healthy kidneys. Patients with ADPKD and hypertension show larger kidney volumes and decreased renal function compared with patients with ADPKD and normal blood pressure. 2 Therefore, it is crucial to start hypertension treatment early in the disease, to reduce renal damage and complications. 14 Other associated health conditions that are thought to be correlated with ADPKD mutations are mitral valve prolapse, aortic aneurysms, inguinal hernias, and diverticulitis. 11
Clinically, this disease is monitored with serial serum creatinine levels and glomerular filtration rates (GFRs). 9 Elevated creatinine and a decreased GFR are linked to advancing disease; however, many patients’ lab values remain normal for many decades before there is a loss of kidney function.9,14 Renal function begins to decline when the vast majority of nephrons have been destroyed. 14 Until recently, the only treatment option for patients with ADPKD was lifestyle modification and management of symptoms. These include remaining well hydrated, restricting dietary sodium and caffeine intake, avoiding smoking, and increasing physical activity. The FDA has recently approved a pharmacological treatment of ADPKD called Tolvaptan (Jynarque), a highly selective vasopressin V2 receptor antagonist, which has been shown to slow the decline of kidney function. 15 This medication is indicated in adults at risk of rapidly progressing ADPKD. However, patients taking this drug are at an increased risk of hepatotoxicity, hypovolemia, and hypernatremia. Therefore, the risks and benefits of taking this medication must be weighed. 15 For those with disease that advances to end stage renal failure, renal transplantation is the preferred treatment. Commonly, the volume of the diseased native kidneys decreases significantly after transplantation; because of this, native kidney nephrectomy along with transplantation is usually only indicated if patients have severe complications or if additional space within the abdomen needs to be made. 2
Sonography is the imaging modality of choice when it comes to the diagnosis, evaluation, and monitoring of ADPKD. 11 Gray-scale and Doppler imaging aid in characterizing cysts from other solid lesions, differentiating simple from complex cysts, and determining internal versus peripheral vascularity. 16 Sonography is accurate in detecting cysts as small as 5 mm. 17 Sonographically, the cysts should appear well-circumscribed, smooth, thin-walled, and have posterior enhancement. The size of the cysts is variable and can range from less than 1 cm up to 5 cm in diameter. 18 One challenge of using sonography to image polycystic kidneys is accurately documenting their size. It is often difficult to obtain the entire length of the kidney in one image. 11 The use of dual-screen and/or extended field of view may be helpful for documenting accurate kidney measurements.
Criteria has been established for sonographic diagnosis of ADPKD in patents with the PKD1 mutation, known as Ravine’s criteria. 19 In patients under the age of 30 with a positive family history, the presence of two cysts in a single kidney is sufficient for diagnosis of ADPKD. Those in this same age group with a negative family history must have at least five total renal cysts for diagnosis. Those between the ages of 30 and 59 with a positive family history must have at least two cysts in each kidney or at least five with a negative family history. Finally, those over the age of 60 with a positive family history may be diagnosed if there are at least three cysts in each kidney or eight total cysts with a negative family history. Sensitivity of this criteria in patients over the age of 30 with the PKD1 mutation is nearly 100%, the specificity of patients with PKD2 is much lower.11,19 ADPKD can be ruled out completely in patients over the age of 40 with two or fewer cysts regardless of family history. 11 There are currently no established criteria for MRI or CT diagnosis of ADPKD. 17
Due to the fact that there are many cystic disease processes of the kidneys, many differential diagnoses should be considered when renal cysts are visualized. Benign and simple renal cysts have an increased prevalence with age; approximately 50% of men and 35% of women in their sixth decade of life have simple cysts that are not associated with genetic mutations. 12 Acquired renal cystic disease is differentiated from ADPKD by normal or small kidney size as opposed to large, and is only found in patients with end stage renal disease. 12 Multicystic dysplastic kidney disease is a congenital disease in which the entire renal parenchyma is replaced by cysts. If the condition occurs bilaterally, it is incompatible with life. 7 It is important to use clinical information and patient history when performing a sonogram on a patient with renal cysts to aid in making an appropriate diagnosis to guide treatment options.
The coexistence of HSK and ADPKD is extremely rare with an estimated prevalence of 1 in 134,000 to 1 in 8,000,000; only 20 patients with ADPKD with coexisting HSK have been reported at that time. 3 There does not appear to be any genetic association between this congenital anomaly and the inherited disease. 20 ADPKD will affect the entirety of the HSK which may differentiate it from other diseases like multicystic dysplastic kidney which is unilateral. The risk for progression to end stage renal failure in ADPKD is not thought to increase with the presence of HSK, and the indication for nephrectomy or transplant is unchanged. However, this double diagnosis may warrant closer surveillance and more frequent imaging. 21 The co-occurrence of these pathologies makes imaging difficult because of the abnormal positioning and large size of the kidneys. Little is written in the literature about the imaging techniques of polycystic HSK due to its rarity. The sonographic appearance of each disease process should be considered when scanning a patient with these conditions.
Conclusion
Sonography is a fast, inexpensive, and safe imaging modality making it very useful in the evaluation and diagnosis of patients with ADPKD with HSK. A positive family history of ADPKD with visualization of multiple large bilateral cysts and the presence of a midline isthmus connecting the inferior poles of the kidneys is pathognomonic for the diagnosis of polycystic HSK. Considering that patients with ADPKD and HSK are at high risk of developing renal failure, nephrolithiasis, and infections, serial sonograms are useful for monitoring disease progression.
Footnotes
Declaration of Conflicting Interests
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author received no financial support for the research, authorship, and/or publication of this article.