
Abstract
Ebstein’s anomaly is a rare type of congenital heart defect characterized by a malformation of the tricuspid valve and the right side of the heart. This case study presents a well-documented case of Ebstein’s anomaly that was diagnosed prenatally using sonography. The ability of prenatal sonography to detect and accurately diagnosis this case allowed for a change in the management of the pregnancy to properly evaluate the condition and prepare for treatment. In addition, information regarding Ebstein’s anomaly is reviewed and specifically addresses etiology, symptoms, diagnosis, treatment, characteristic sonographic appearance, and common differential diagnoses.
Introduction
Congenital heart defects (CHDs) are the most common type of birth defect and are a leading cause of birth-defect associated morbidity and mortality. 1 The prevalence of CHDs is approximately 80 per 1000 live births. 2 Ebstein’s anomaly is a type of CHD characterized by a malformation of the tricuspid valve and the right side of the heart. 1 Ebstein’s is a rare CHD, accounting for less than 1% of all CHDs, with an estimated prevalence of approximately 0.5 per 10 000 live births. 3 A variety of cardiac abnormalities are associated with Ebstein’s anomaly, including atrial septal defect, conduction system abnormalities, patent foramen ovale, pulmonary stenosis or atresia, and ventricular septal defect. 1 Echocardiography is the diagnostic test of choice to definitively diagnosis Ebstein’s anomaly. However, other modalities such as chest radiograph, electrocardiogram (ECG), and prenatal sonography are often the initial tests to detect the cardiac abnormality, leading to further evaluation.
Case Report
A 34-year-old G2P1 Caucasian female presented to a high-risk fetal medicine facility at 20 weeks 1 day gestation based on last menstrual period (LMP) for a routine morphology examination. A dating and nuchal translucency (NT) sonogram was previously performed at 12 weeks 0 days and confirmed accurate gestational age by LMP. The NT measured 1.58 mm, which was within normal limits. No abnormalities were noted. The patient had a history of a twin cesarean delivery with no other complications.
The morphology sonogram was performed using a Philips iU22 ultrasound machine with a 5 MHz curvilinear array transducer (Philips Ultrasound, Bothell, WA, USA). The sonogram showed a borderline thickened nuchal fold measuring 5.34 mm as well as a complex cardiac defect. The abdominal situs was normal. The heart was correctly located in the left chest but displayed severe levocardia, as the apex of the heart was pointed too far leftward. In addition, the heart was also found to be enlarged, with a cardiac-to-thoracic ratio of .75 (Figure 1). Ventricular situs and orientation of the great vessels appeared normal. The right atrium was found to be abnormally enlarged with apical displacement of the anterior septal leaflet (Figure 2). Pulsed-wave Doppler evaluation demonstrated severe tricuspid valve regurgitation (Figure 3). No significant mitral valve regurgitation was noted. Ventricular systolic function was determined to be normal by M-mode investigation. The right and left ventricles appeared to be normal in size. The foramen ovale was visualized and confirmed to be normal, with the flap opening to the left atrium. The pulmonary artery appeared to be somewhat reduced in size, although antegrade flow was still seen (Figure 4). The ductus arteriosus flow appeared to be normal, demonstrating flow from the right ventricle to the descending aorta, allowing blood to bypass the nonfunctioning fetal lungs. At the time of the examination, there were no signs of fetal hydrops. Based on the sonographic findings, the perinatologist diagnosed these findings as consistent with Ebstein’s anomaly, with possible differential diagnosis of partial atrial ventricular canal defect. The patient was then referred to a pediatric cardiologist at the same facility, who agreed with the diagnosis of Ebstein’s anomaly.
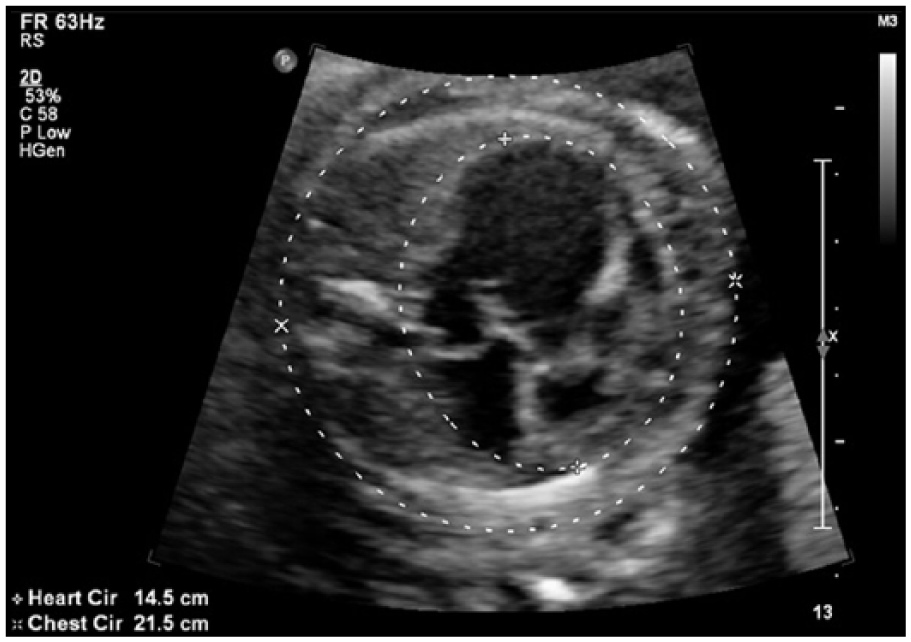
Transverse gray-scale sonogram of the fetal thorax showing an enlarged heart with a cardiac-to-thoracic ratio of .75.
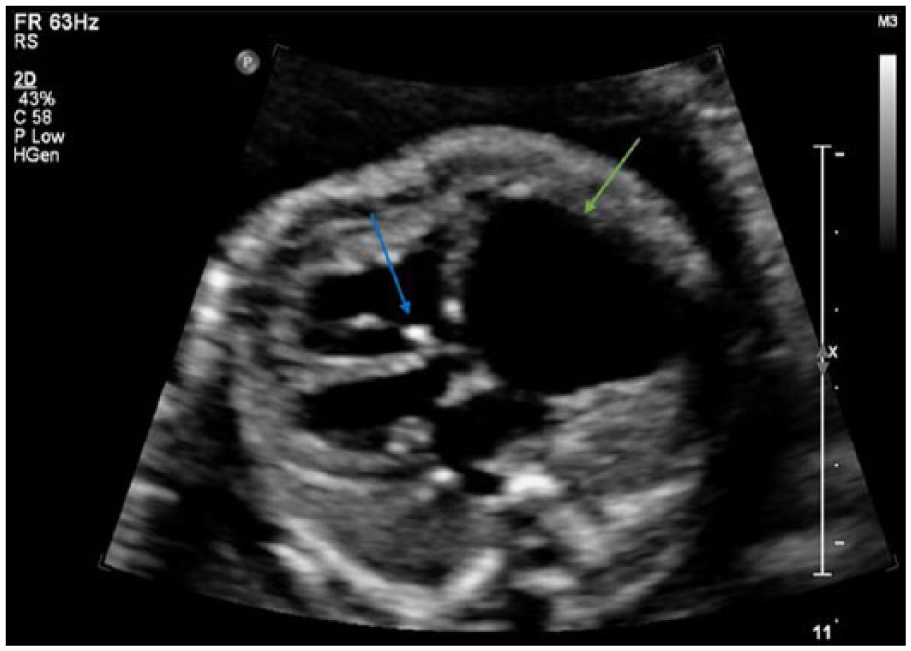
Transverse gray-scale sonogram of the fetal heart showing an abnormally enlarged right atrium (green arrow) with apical displacement of the anterior septal leaflet (blue arrow).
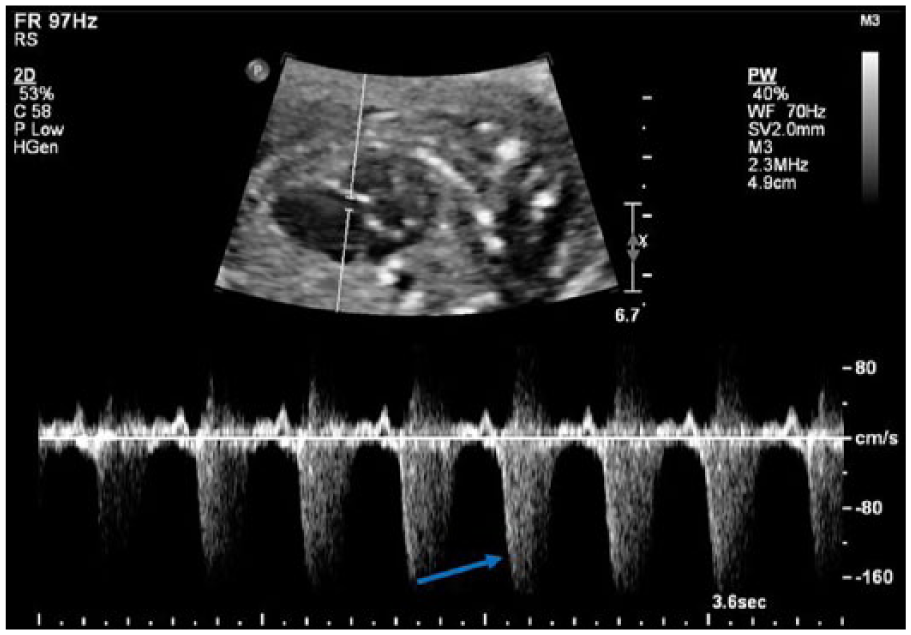
Pulsed-wave Doppler sonogram of the tricuspid valve showing severe tricuspid valve regurgitation (blue arrow).
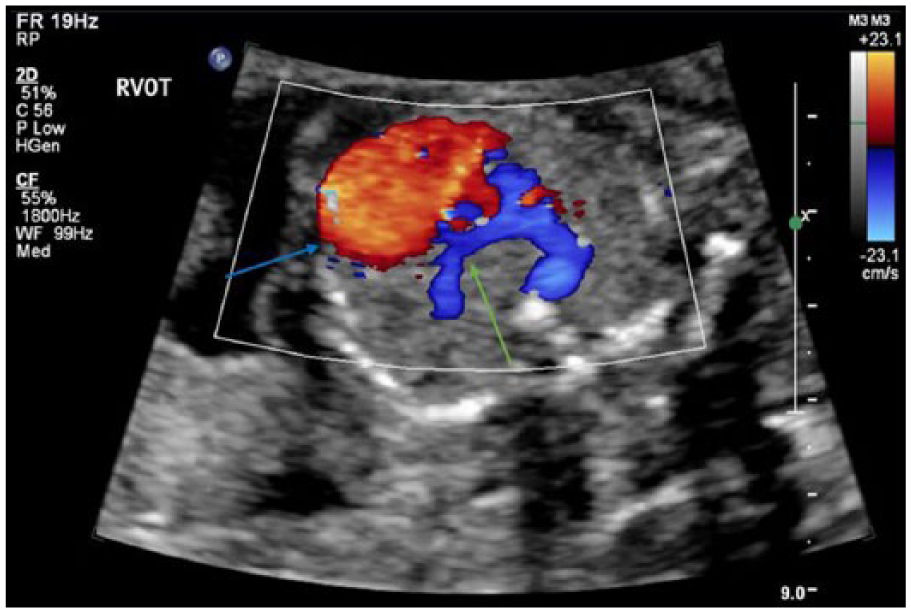
Transverse color Doppler sonogram showing an abnormally enlarged right atrium (blue arrow) and normal antegrade pulmonary artery flow (green arrow).
After receiving the diagnosis from the initial perinatologist, the patient chose to seek a second opinion to verify the diagnosis. Fetal echocardiography performed at a separate facility resulted in a slight discrepancy in diagnoses. The perinatologist at the second facility agreed with the initial diagnosis of Ebstein’s anomaly; however, the pediatric cardiologist at the second facility diagnosed the condition as tricuspid valve dysplasia. Due to the difficulty in definitively diagnosing the condition, and the possible differential diagnosis of partial atrial ventricular canal defect, which has a strong association with aneuploidy, an amniocentesis was recommended and performed. The amniocentesis revealed a normal fluorescence in situ hybridization (FISH) and microarray. The initial perinatologist recommended growth sonograms at 4-week intervals. Each growth sonogram included measurement of the Tei index (TI) of the left ventricle, which evaluated the overall left ventricular myocardial performance, evaluation of the ductus venosus to monitor for right ventricle failure, assessment of cardiac-to-thoracic ratio, and evaluation for hydrops. In addition, weekly biophysical profiles (BPPs) were recommended to begin at 32 weeks.
At 34 weeks, the patient presented for a weekly BPP examination. Fetal movement, breathing, and tone were all observed sonographically. The amniotic fluid index (AFI) measured 31.93 cm, with the largest pocket measuring 10.0 cm, diagnostic of polyhydramnios. An intermittent cardiac arrhythmia was also noted, consisting of premature atrial contractions (Figure 5). The nonstress test (NST) was nonreactive with a negative contraction stress test. Therefore, the fetus received a final score of 8 out of 10. A repeat BPP/NST was recommended for the following day.
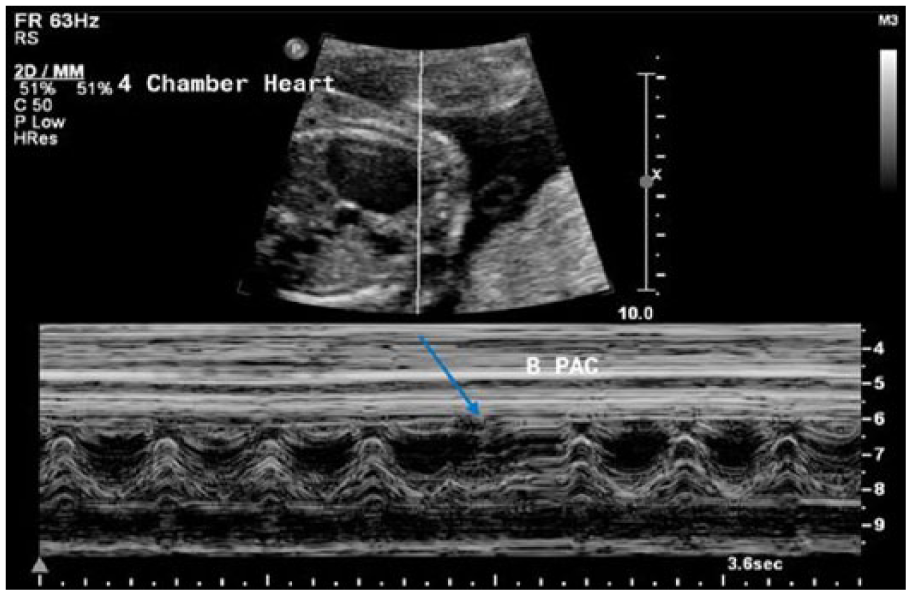
Duplex sonogram showing M-mode investigation through the gray-scale fetal heart. A fetal cardiac arrhythmia, consisting of premature atrial contractions, is identified by the blue arrow.
The next day, a repeat BPP with growth was performed. Fetal movement and tone were observed, and fetal breathing was not demonstrated. The AFI measured 30.22 cm, which was consistent with past examinations. Ultimately, the fetus received a score of 6 out of 8. Fetal biometry indicated an estimated fetal weight of 5 pounds 15 ounces (79th percentile) at 34 weeks 1 day. In addition, fetal ascites and concurrent pericardial effusion were visualized, which were indicative of fetal hydrops and heart failure (Figures 6 and 7). Based on these findings, the NST was not performed and the patient was immediately transported to the hospital for prenatal care and anticipated delivery.
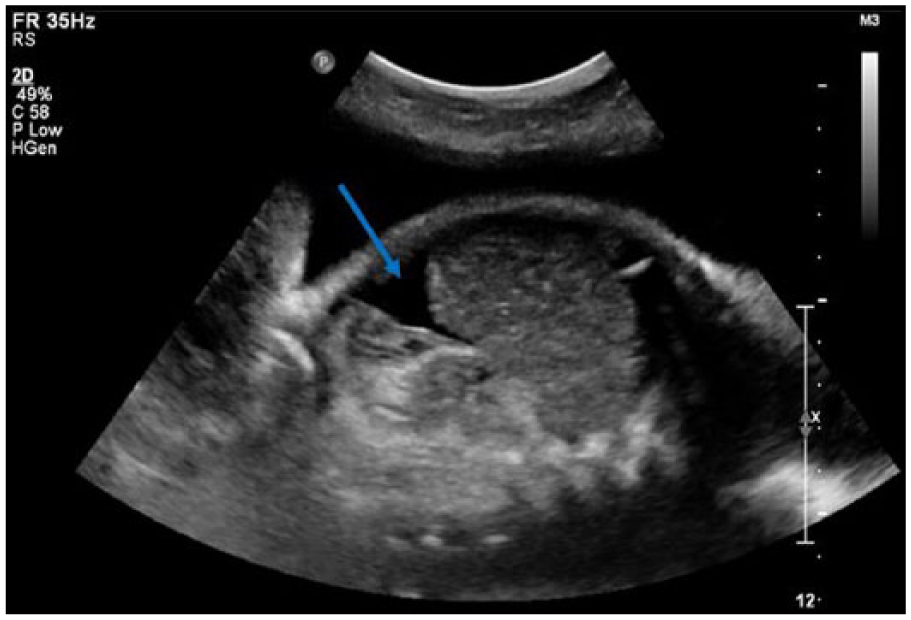
Longitudinal gray-scale sonogram demonstrating fetal ascites in the abdominal cavity (blue arrow).
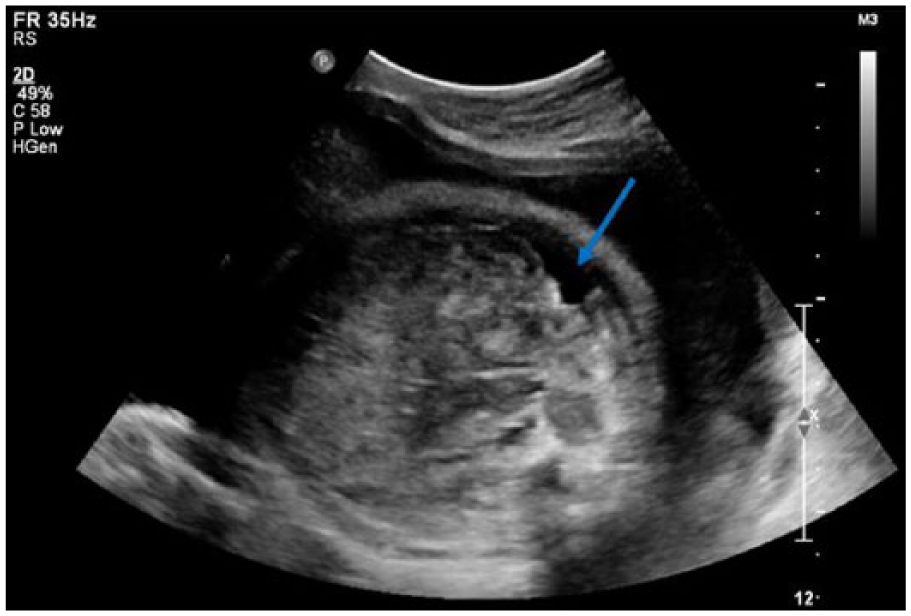
Transverse gray-scale sonogram showing fetal ascites in the abdominal cavity (blue arrow).
The patient delivered by a planned cesarean section at 37 weeks 2 days. Upon delivery, they were able to convert the neonate’s arrhythmia with one shock. The baby’s oxygen saturation measured in the 70s, so she was placed on a normal ventilator that kept her oxygen in the upper 80s. She was also administered arterial infusion of prostaglandins to maintain the patency of the ductus arteriosus, due to pulmonary artery dysfunction. The neonate was found to have pulmonary atresia, which occurs when a solid sheet of connective tissue grows and closes off the pulmonary artery valve, therefore not allowing any blood to flow from the heart to the lungs to become oxygenated. Maintaining the patency of the ductus arteriosus allows some of the oxygen-poor blood to travel to the lungs, where it can become oxygenated to provide for the body. To ensure that the neonate was receiving enough oxygen, she was placed on extracorporeal membrane oxygenation (ECMO), which allows blood to bypass the heart and lungs, while a machine adds oxygen to the blood and removes carbon dioxide.
Surgery was planned and performed a few days following birth, in which the tricuspid valve was repaired, the atrial size was reduced, and a replacement pulmonary valve was placed. Following surgery, the baby developed an aortic valve thrombosis that was restricting the valve opening. Heparin, an anticoagulant, was administered to prevent the clot from growing and prevent future clots. In addition, an electroencephalogram monitored the baby for brain bleeds as a result of treatment. Her heart began to show no pulsatility; doctors decided that the pulmonary valve replacement was causing too much resistance on blood flow and therefore affecting the heart’s pumping ability. Another surgery was scheduled and performed to adjust the original pulmonary valve replacement. After the surgery, some pulsatility was regained and the baby was taken off ECMO a few days later. At this time, there is no additional follow-up information available.
Discussion
Ebstein’s anomaly develops during the first weeks of intrauterine life as the tricuspid valve fails to develop normally. During week 7, the atrioventricular valve leaflets begin to develop from the myocardial tissue of the ventricles by a process called delamination. By the end of week 12, the tricuspid valve is fully formed. 1 In a normal heart, the tricuspid valve has three leaflets: anterior, posterior, and septal. 4 Ebstein’s anomaly is a malformation of the tricuspid valve and right ventricle characterized by (1) adherence of the septal and posterior leaflets to the underlying myocardium (failure of delamination, namely, splitting of the tissue by detachment of the inner layer during embryologic development); (2) downward (apical) displacement of the functional annulus (septal > posterior > anterior); (3) dilation of the “atrialized” portion of the right ventricle, with various degrees of hypertrophy and thinning of the wall; (4) redundancy, fenestrations, and tethering of the anterior leaflet; and (5) dilation of the right atrioventricular junction (true tricuspid annulus).3,4 The spectrum of malformation in Ebstein’s anomaly may range from only minimal displacement of the septal and posterior leaflets to an imperforate membrane or muscular shelf between the inlet and the trabecular zones of the right ventricle. 5 Many patients with Ebstein’s anomaly have other structural cardiac anomalies and abnormalities of the cardiac conduction system, such as atrial septal defect, patent foramen ovale, pulmonary stenosis or atresia, and ventricular septal defects. 1
Most cases of Ebstein’s anomaly are sporadic and the exact mechanism for the anomaly is not entirely understood. Case-control studies suggest genetic, reproductive, and environmental risk factors; for example, the anomaly is more common in twins, in those with a family history of congenital heart disease, and in those with maternal exposure to benzodiazepines. 4 There is also a weak link between maternal lithium therapy and the presence of Ebstein’s anomaly, although this rarely occurs. 4
The hemodynamic variations and clinical presentation of Ebstein’s anomaly depend on the age at presentation, anatomic severity, hemodynamics, and degree of right-to-left interatrial shunting. However, the cardinal symptoms of Ebstein’s anomaly are cyanosis, right-side heart failure, arrhythmias, and sudden cardiac death. 6 Neonates with Ebstein’s anomaly may present with cyanosis, congestive heart failure caused by tricuspid valve regurgitation, and marked cardiomegaly. 7 Symptomatic children with Ebstein’s may have progressive right-side heart failure, but most will reach adolescence and adulthood. Adults and children older than 10 years often present with arrhythmias, although they can also present with progressive cyanosis, decreased exercise tolerance, fatigue, or right-side heart failure. 8
Echocardiography, or sonography of the heart, is the diagnostic test of choice for Ebstein’s anomaly, due to its ability to accurately evaluate the tricuspid valve leaflets and the size and function of the cardiac chambers. 5 Sonographic findings of Ebstein’s anomaly include apical displacement of the septal leaflet of the tricuspid valve, which is the principal feature of the anomaly. In addition, marked enlargement of the right atrium and atrialization of the right ventricle are seen. Due to the malformation of the valve, tricuspid valve regurgitation is often noted, although the site and degree are variable. The feasibility of valve repair can also be assessed with echocardiography.
In addition, Ebstein’s anomaly can lead to severe tricuspid valve regurgitation and cardiac dysfunction in utero, resulting in fetal cardiomegaly, heart failure, pulmonary hypoplasia, hydrops, and tachyarrhythmias. 9 Severe cardiac enlargement can inhibit lung growth by longstanding compression leading to severe pulmonary hypoplasia, which may complicate neonatal resuscitation. Fetal survival is strongly linked to the ability of the fetal heart to compensatorily increase the left ventricular volume flow. Therefore, the size of the fossa ovalis allowing the required increase of transatrial right-to-left shunt and a sufficient left ventricular diastolic and systolic function are mandatory in fetuses with Ebstein’s anomaly, to avoid occurrence of congestive heart failure followed by hydrops and in utero fetal death. 10 Fetal hydrops refers to the pathologic accumulation of fluid in greater than or equal to two fetal compartments, including the pleural or pericardial spaces, abdominal cavity, integument, or placenta. The mechanism of the development of hydrops in the fetus is thought to be a combination of increased hydrostatic pressure, decreased oncotic pressure, and in some, lymphatic obstruction. 11
Although echocardiography is used to definitively diagnosis Ebstein’s anomaly, other diagnostic tests such as a chest radiograph, ECG, and prenatal sonography can precede the echocardiogram, indicating the need for one. Chest radiographs in symptomatic patients with Ebstein’s anomaly will often demonstrate significant enlargement of the right atrium and cardiomegaly, creating the appearance of a globe-shaped, “wall-to-wall” heart that fills up the chest cavity. 1 In addition, most patients with Ebstein’s anomaly will have an abnormal ECG. Abnormal ECG findings most commonly seen with the anomaly are unusually tall, broad, P waves. 1 Fetuses with Ebstein’s anomaly can also be diagnosed in utero, as this case was, by routine prenatal sonograms.
The treatment for Ebstein’s anomaly can vary depending on the degree of tricuspid valve abnormality. An individual with mild Ebstein’s anomaly-related cardiac abnormalities can live into late adulthood without ever developing symptoms or having only mild symptoms related to the anomaly. In general, the earlier in life the symptoms develop, the more severe the patient’s symptoms. 1 Those with mild cases can be monitored by a cardiologist for signs of worsening, such as increasing heart size, right ventricular hypertrophy, and arrhythmias through the use of ECG, echocardiography, or chest radiograph. 1 Most neonates with the symptomatic Ebstein’s anomaly will not survive without surgical intervention in infancy. 12 Surgery can be performed to correct the underlying tricuspid valve and right ventricular abnormalities and correct any associated intercardiac defects. In addition, palliative procedures can be performed in the early days of life as a bridge to more definitive surgical treatment later, and surgery can correct any arrhythmias caused by the anomaly. Complete repair of Ebstein’s anomaly in symptomatic neonates has been shown to be feasible, with studies showing good early (78.1%) and late (74% ± 8%) survival rates and excellent functional status. 13 There are various surgical approaches that may be taken to treat the anomaly. The tricuspid valve can be repaired or replaced, with the preference being replacement due to increased durability. The atrialized portion of the right ventricle can be resected surgically, and the markedly dilated, thin-walled right atrium can be resected as well. 14
Similar to Ebstein’s anomaly, tricuspid valve dysplasia, which was presented as the diagnosis by another facility, involves a variation in tricuspid valve malformation. Tricuspid valve dysplasia is defined as an anomaly that encompasses a heterogeneous group of malformations involving abnormalities of the tricuspid valve, where the normal anatomic insertion of the tricuspid valve leaflet is maintained at the level of the tricuspid valve annulus, unlike Ebstein’s anomaly, where the septal and posterior leaflets are apically displaced. 15 In addition, tricuspid valve dysplasia has a very similar sonographic appearance, demonstrating an enlarged right atrium and tricuspid valve regurgitation, although the leaflets are not apically displaced and the valve is often abnormally thickened and irregular. In this case, the sonogram did demonstrate apical displacement of the septal and posterior leaflets of the tricuspid valve, which is why the initial diagnosis of Ebstein’s anomaly was suggested.
Conclusion
Ebstein’s anomaly is a rare type of congenital heart defect characterized by malformation of the tricuspid valve and the right side of the heart. Although echocardiography is the diagnostic modality of choice, prenatal sonography can offer early diagnosis of Ebstein’s anomaly, allowing for proper management and treatment of the condition. Because treatment can vary depending on the degree of malformation, with individuals presenting at earlier ages and needing more intervention, it is imperative that sonographers familiarize themselves with the principal sonographic findings of Ebstein’s anomaly, so cases can be accurately diagnosed prenatally. This case demonstrates the vital importance of the role that sonography plays in the accurate diagnosis of Ebstein’s anomaly, which drastically changed the management of the patient.
Footnotes
Acknowledgements
The authors thank Amy Bildner, RDMS, RDCS, RVT, Shelby Schoengarth, RDMS, RVT, Kelli Stein, RDMS, Kristy Helms, RDMS, RVT, and Erin Perry, RDMS, RVT, for their assistance, leadership, and guidance.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.