
Abstract
Cystic pulmonary airway malformation, or Craig’s disease, is a rare congenital abnormality of lung development. Medical imaging, particularly Doppler ultrasound, is currently one of the recommended methods for prenatal diagnosis and for the monitoring of this condition. We report three cases of Craig’s disease diagnosed at ultrasound: one at 26 weeks of amenorrhea and two others at 30 weeks of amenorrhea. We discuss according to the literature the main ultrasound aspects and specify the place of other radiographic explorations during the monitoring of this condition.
Cystic pulmonary airway malformation, or Craig’s disease, is a rare congenital abnormality of lung development and accounts for a quarter of congenital malformations of the lung. 1 The condition is characterized by a bronchogenic cystic proliferation caused by an arrest of lung development occurring primarily at the approximate sixth week of pregnancy.1,2 Although familial cases are described, Craig’s disease does not appear to be related to ethnic or genetic predispositions. Its evolution is unpredictable, hence the necessity of rigorous postnatal supervision. The discovery of three cystic pulmonary airway malformations during routine sonographic screening during prenatal surveillance is reported below, with a second objective of identifying the role of medical imaging in the prenatal diagnosis and management of this condition.
Case Report 1
A 24-year-old woman with no previous significant medical or surgical history presented for routine sonographic evaluation in her 26th week of pregnancy. This second obstetric sonogram, conducted as part of the prenatal check of the second term, showed a fetus without any fetal distress, with note made of right lung macrocysts with diameters greater than 2 cm (Figures 1 and 2). Color Doppler evaluation highlighted pulmonary vascularization feeding those cystic formations with a spectrum typical of pulmonary arterial flow by pulsed spectral Doppler analysis. There was no aberrant vascularization seen deriving directly from the aorta, and there appeared to be normal lung segmentation. The results of other routine prenatal checks including rubella serology, toxoplasmosis, syphilis, human immunodeficiency virus (HIV), blood counts, hemoglobin, serum glucose, urea, and creatinine were all normal.
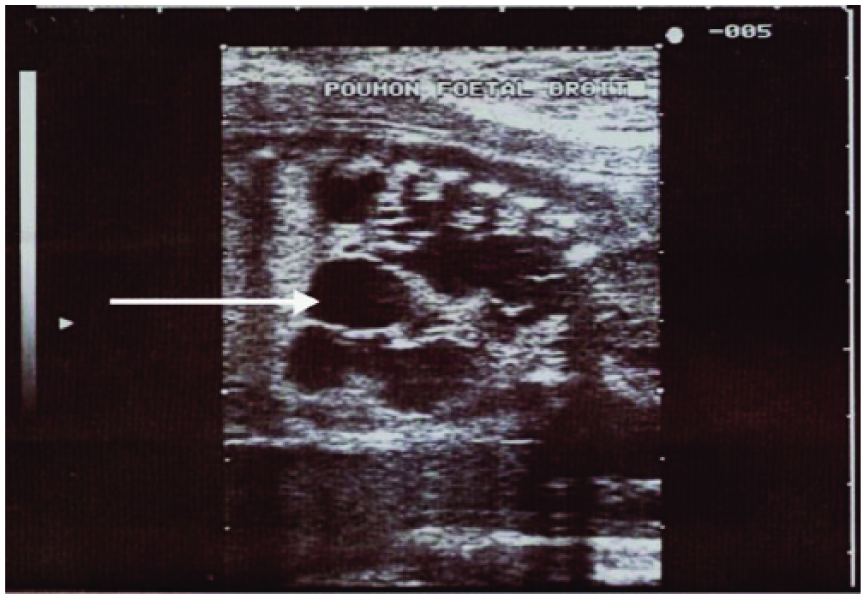
Sagittal gray-scale fetal image showing multiple macrocysts (arrow) scattered throughout the right lung.
Cross-sectional gray-scale image of the right fetal lung showing multiple macrocysts (arrow).
A routine third trimester sonographic check also highlighted a living fetus without any other abnormality. Childbirth occurred vaginally at 41 weeks after a spontaneous labor, with weight, height, and cranial perimeter at birth of 2730 g, 48 cm, and 34 cm, respectively. The Apgar score was 8 at 1 minute and 9 at 5 minutes of life.
Case Report 2
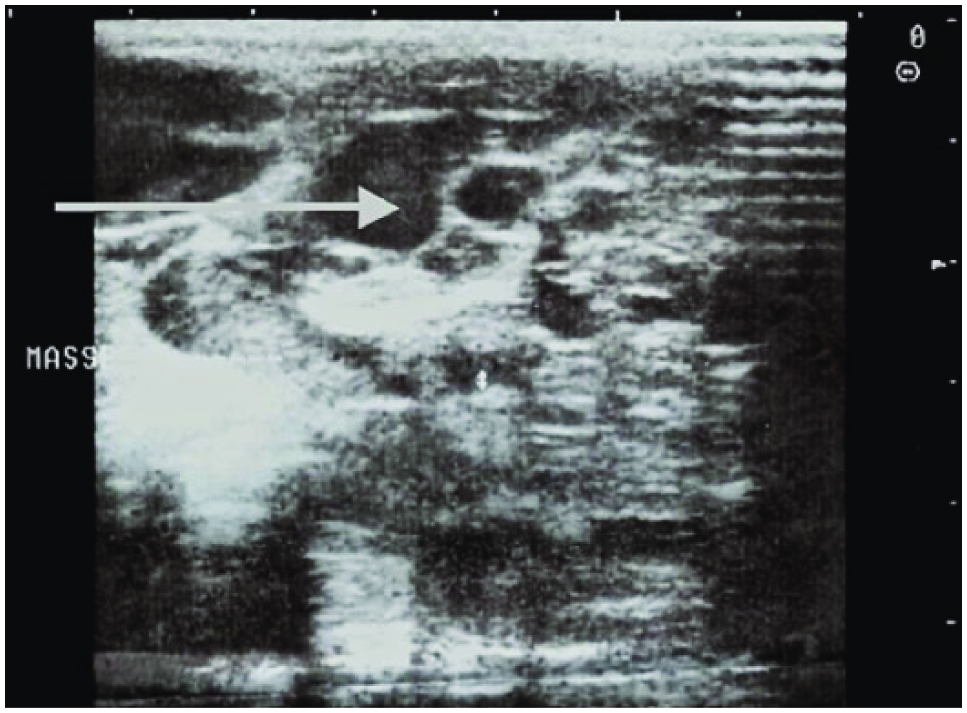
A 28-year-old woman without any known significant medical or surgical history presented for routine sonographic follow-up. A first sonographic evaluation during the first trimester was considered to be normal. The second obstetric sonogram, conducted routinely as part of the second trimester prenatal evaluation, showed a 30 week 3 day fetus with no fetal distress. This examination showed lung cysts bilaterally with diameters between 10 and 15 mm (Figure 3). Other prenatal routine examinations were normal as in the first case above. Color Doppler evaluation highlighted pulmonary vascularization feeding those cystic malformations with a pulsed Doppler spectrum typical of pulmonary arterial flow. There was no aberrant vascularization deriving directly from the aorta, and as above, there appeared to be normal lung segmentation. The results of all other routine prenatal checks, including rubella serology, toxoplasmosis, syphilis, HIV, blood count, hemoglobin, serum glucose, urea, and creatinine, were normal. The results of the routine third trimester sonogram were as those of the second evaluation with no evidence of any other fetal malformation. Childbirth occurred vaginally at 40 weeks 2 days, with an Apgar score of 9 at 1 minute and 10 at 5 minutes of life. The weight, height, and cranial perimeter at birth were 3030 g, 49 cm, and 34 cm, respectively. In the immediate postnatal period, thoracic sonography showed a heterogeneous hypoechoic mass with numerous cystic structures of various size in the right and left lungs. The thoracic sonogram further showed intrapulmonary opacities with bilateral lower lobe rounded water tone.
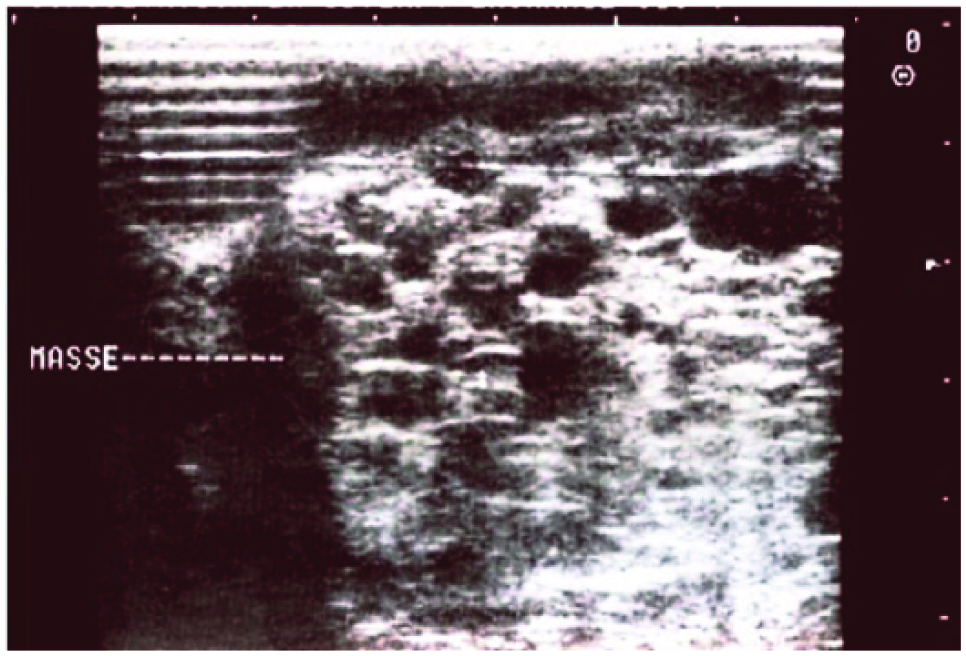
Cross-sectional gray-scale image of the fetal lung showing an echogenic mass (dashed line) with multiple macrocysts bilaterally.
Sonographic and radiologic monitoring aided in the management of this infant and helped determine the appropriate time for referral to pediatric surgery, where the anatomic and histopathologic examinations confirmed the diagnosis of type II adenomatoid dysplasia according to the Stocker classification. 3 Postoperative follow-up has shown the baby to be in good health.
Case Report 3
A 24-year-old woman with no known significant medical and surgical history presented at 29 weeks of pregnancy for routine second trimester sonography. The obstetric sonogram showed a fetus of 30 weeks with bilateral pulmonary microcysts with diameters less than 5 mm in both lungs, complicated by fetal hydrops and polyhydramnios (Figures 4a and 4b). Other prenatal routine examinations were normal. Color Doppler evaluation showed pulmonary vascularization feeding the malformations with a Doppler spectrum typical of pulmonary arterial flow. There was no aberrant vascularization deriving directly from the aorta, and there appeared to be normal lung segmentation. During follow-up, the fetus died in utero at the 37th week of pregnancy. (The sonograms for all cases reported here were done using a Logiq 500 [GE Ultrasound, Waukesha, WI] with a 5-MHz curved linear or broadband 5- to 9-MHz linear array probe approved for obstetric work. 4 )
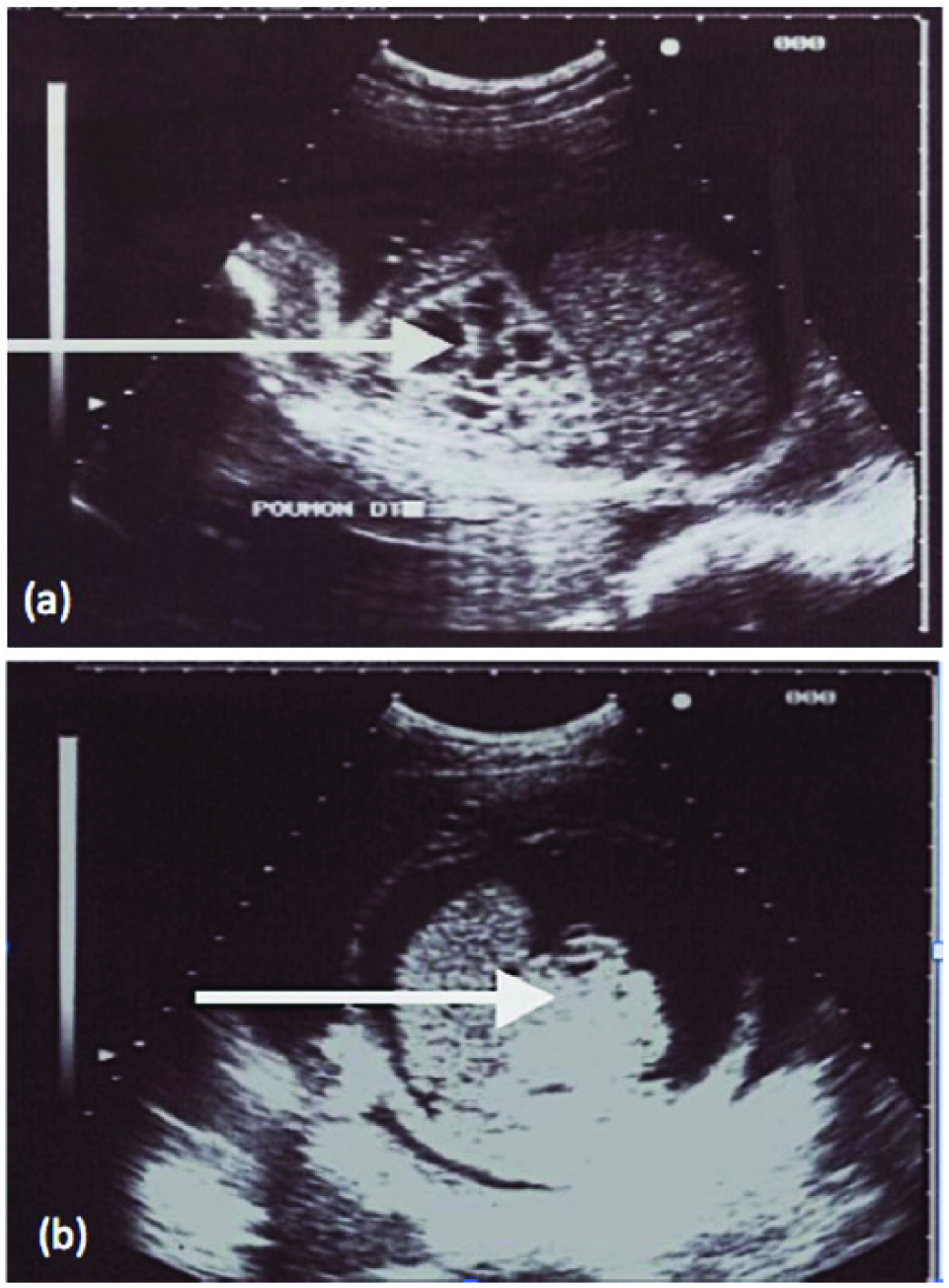
Cross-sectional gray-scale images of the fetal lung showing (a) multiple microcysts (arrow) associated with hydrops (b).
Discussion
The three cases reported above showed the benefit of sonographic imaging in the prenatal diagnosis and monitoring of cystic pulmonary airway malformations. This series confirms that prenatal ultrasound is efficacious for the diagnosis of this condition for pulmonary cysts of various presentation and size, from microcysts (less than 5 mm diameter) to macrocysts (greater than 5 mm diameter). Monitoring is important in this disease as its evolution is unpredictable and can lead to the death of the fetus in utero, as in case report 3 above. Alternatively, there may be spontaneous involution of cysts ex utero with healing over a variable but unpredictable timeline, as shown in the first two cases reported above. However, it must be kept in mind that the efficacy for sonographic studies can be operator and machine dependent, and therefore, the possibility of a bias related to the operator and/or to the performance and quality of the devices used cannot be excluded. To illustrate this point, it was noted in the series of three cases that two were diagnosed during the third trimester of the pregnancy with microcysts, whereas second trimester sonograms were said to be normal, based on readings by a junior radiologist. To control for this possible bias in our own institution, all the cases above were reviewed and confirmed by a senior radiologist with extended experience in prenatal sonography.
Cystic pulmonary airway malformation is a relatively rare condition. It represents 25% of congenital lesions of lung pathologies diagnosed in utero. 1 The cases reported above are the first such observations reported in the prenatal period in Côte d’Ivoire (Ivory Coast). In Sub-Sahara Africa specifically, the prevalence of this condition is difficult to specify because none of the cases are identified or published. The three cases in this work do not belong to the same family, and no familial origin of the disease was recognized. No ethnic or genetic predisposition was noted, although family-related cases have been described.5,6 We are unable to completely discount a genetic origin in the cases above but it appears to be unlikely because the parents of all those children are young and there is no consanguinity between them.
Prenatal diagnosis of cystic pulmonary airway malformation is based on medical imaging, which in almost all cases is sonography, as there are no specific signs or symptoms during pregnancy. 7 There are reports of an association between cases of trisomy 18 and 21 and cystic pulmonary airway malformation,8,9 but most reported cases are the result of an incidental discovery during routine prenatal surveillance.
The cases reported show that pulmonary cysts are of variable presentation and size, ranging throughout the spectrum from micro- to macrocysts as seen by sonography. Adzick et al. 10 suggested a sonographic classification based on the size of the cysts. They proposed that when the diameter of the cyst is larger than 5 mm, the cyst should be considered macroscopic, whereas when it is less than 5 mm, the cyst should be considered a hyperechoic microscopic cyst, as was the case in this report.
The site of pulmonary cysts is typically unilateral but may be bilateral, with no significant bias toward left or right lung involvement or the lung segment(s) affected.9,11 In our series, the site was bilateral in two cases, atypical according to the literature, and unilateral in only one case. In the literature, segment involvement is often unique, rarely bilateral, and relates in many cases to the lower left lobe segment.
In addition to cystic pulmonary airway malformation, diagnoses to be considered in the prenatal period include bronchogenic cyst, diaphragmatic hernia, esophageal duplication, and pulmonary sequestration.7,10,11 A bronchogenic cyst results in a rounded or oval anechoic image with a well-circumscribed thin wall. Usually unilocular and posterior, the cysts may be multifocal. A left diaphragmatic hernia results in a rightward deviation of the heart, a disappearance of the intra-abdominal visualization of the stomach, and intrathoracic visualization of one or more abdominal organs and eventually a visualization of the diaphragmatic defect. Right-sided diaphragmatic hernia causes the ascension of the liver and an upward deviation of the portal vein and hepatic veins. Esophageal duplication is characterized by a posterior mediastinal fluid image, while pulmonary sequestration is visualized as a left hyperechoic, posterolateral, homogeneous triangular track. Color Doppler imaging confirms the diagnosis by showing an arterial pedicle deriving from the aorta, which may at times be subdiaphragmatic. The differential diagnosis between cystic pulmonary airway malformation and other pathologies can be based on the pulmonary appearance and the normality of the diaphragm and intra-abdominal digestive structures. It can be further based on the absence of hepatic and/or porto-systemic abnormalities. 1
A thorough sonographic imaging study, coupled with color and spectral Doppler evaluation, should also look for other related malformations. Color Doppler imaging may highlight the pulmonary or aortic vascularization of such malformations, with resulting characteristic Doppler spectra. 12 The cases reported above did not show any related malformations as reported by Dommergues et al., 13 and other reports relate associated malformations such as intraventricular communication, anal imperforation, renal agenesis, and pulmonary sequestration. 14 In addition to sonography, magnetic resonance imaging may be used for prenatal diagnosis of the pulmonary sequestration of Craig’s disease. However, this examination usually provides little complementary information beyond what has already been seen by sonography. Macrocystic forms appear as lobulated, heterogeneous hyperintense masses without feeder vessels, and microcystic forms appear as simple homogeneous lobulated masses; a mediastinal shift may also be visible. 15 The positive predictive value and negative predictive value of prenatal ultrasound for detecting lung malformations in late gestation have been reported as 96% and 43%, respectively, by Kunisaki et al. 16
Because the evolution of adenomatoid malformations is unpredictable, sonographic monitoring after initial diagnosis should be done. Spontaneous resolution or regression is possible, although it has an unpredictable frequency, varying in literature reports from 3% to 60%. 13 However, even in cases of resolution by prenatal sonography, it may be prudent to continue routine monitoring as very minimal lesions may persist and serve as sites for later infections. At the other extreme is the risk of death in utero, as occurred in case report 3. The fetus, with associated hydrops and polyhydramnios, could not be delivered and died at the 37th week of pregnancy as the parents refused a caesarean operation.
Fetal hydrops represents the major progressive complication of cystic pulmonary airway malformation in utero. Its frequency varies according to published series as about 7% to 8% and can be correlated with the volume of the cystic mass.17–19 Adzick et al. 10 reported on a series of 134 cases and found that 109 fetuses without ascites survived while all of the other 25 with ascites died in utero or right after their birth. De Santis et al. 20 and Bruner et al. 21 in their reports came to a similar conclusion. The ascites appears likely to be secondary to obstruction of venous return caused by cardiac compression by the mass(es).22,23 In addition to these related or progressive abnormalities, the size of the cysts, the position of the mediastinum, and the presentation of the lesions may affect the fetal prognosis. Bunduki et al. 24 found the prognosis to be related to the presence of microcystic lesions, bilaterality of the pulmonary lesions, fetal ascites, polyhydramnios, and mediastinal deviation. Of note, case report 3 included all of these factors of poor prognosis except mediastinal deviation. This study is an additional argument to support the regular use of sonography in prenatal care. This examination is essential for the prenatal diagnosis of cystic pulmonary airway malformation, to monitor for the development of any complications, and to plan for perinatal treatment.
Conclusion
Sonography done in the prenatal period allows for the early diagnosis of cystic pulmonary airway malformation and can determine the presence or absence of any related abnormalities. The monitoring of this malformation is best done by duplex sonography using B-mode with color and spectral Doppler evaluation, with possible postnatal thoracic radiography and computed tomography. It is important to ensure high-quality sonography by trained personnel for both scanning and interpretation for the diagnosis as well as planning for prenatal and postnatal management.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.