
Abstract
Congenital pulmonary airway malformation, also known as congenital cystic adenomatoid malformation, is the most common congenital lung malformation of the lower respiratory tract. Congenital pulmonary airway malformation lesions fall into five classifications based on respiratory tract origin and cystic characteristics. Sonographic appearance varies from echolucent cysts to an echogenic, solid-appearing mass within the lungs. Fetal mediastinal shift, pulmonary hypoplasia, and hydrops can occur, depending on the size of the lesion. A case is presented with sonography to characterize the lesion.
Keywords
Congenital pulmonary airway malformation (CPAM) is the most common congenital lung lesion.1–3 There are five types of CPAMs, differentiated by lung anatomy involvement and cystic development. Prenatal sonography can be used to determine the size and type of CPAM, as well as the presence of a mediastinal shift. It is necessary to recognize a CPAM to develop a treatment plan and to predict fetal prognosis.
Case Report
A 34-year-old pregnant woman, gravida 2, para 1, presented to an outside clinic for her initial prenatal care. The patient had no complications at 12 weeks of gestation by last menstrual period. She refused prenatal genetic screening, including sequential and cystic fibrosis screening. She was referred to our clinic at 19 weeks of gestation for a routine sonographic anatomy survey.
Obstetric sonography was performed at 19 weeks of gestation with a Philips iU-22 sonographic system (Philips Ultrasound, Bothell, Washington) with a C5-1 MHz curved array transducer (frequency range, 1.0-5.0 MHz). The images showed an echogenic mass in the right inferior chest, superior to the diaphragm, measuring 1.3 × 1.2 × 0.9 cm (Figure 1). There were no visible cysts within the mass. Color Doppler imaging did not reveal a systemic feeding vessel (Figure 2), and the mass did not show internal vascularity. There was no evidence of mediastinal shift (Figure 3) or hydrops.
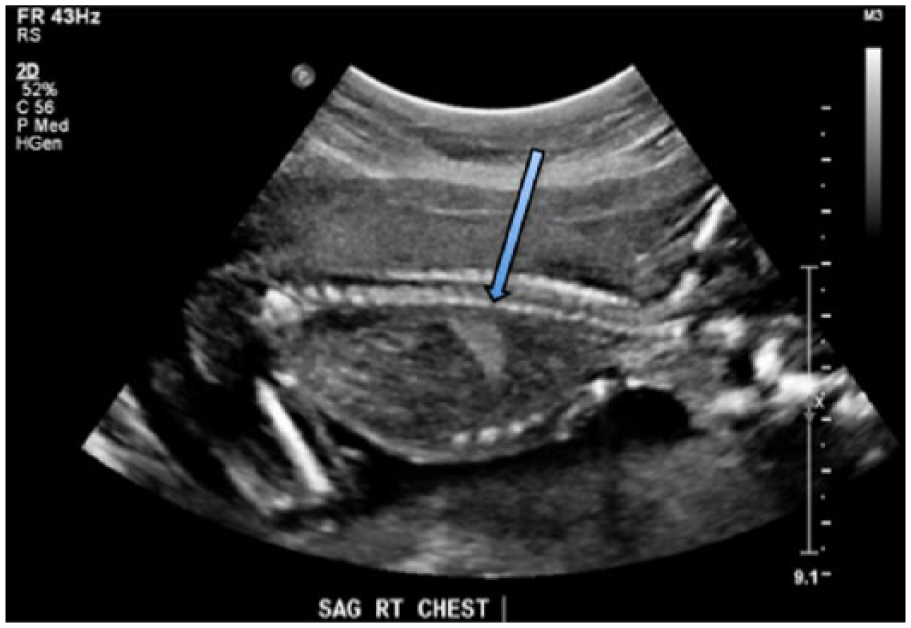
Longitudinal gray-scale image of the right fetal chest showing the echogenic mass diagnosed as a congenital pulmonary airway malformation (arrow).
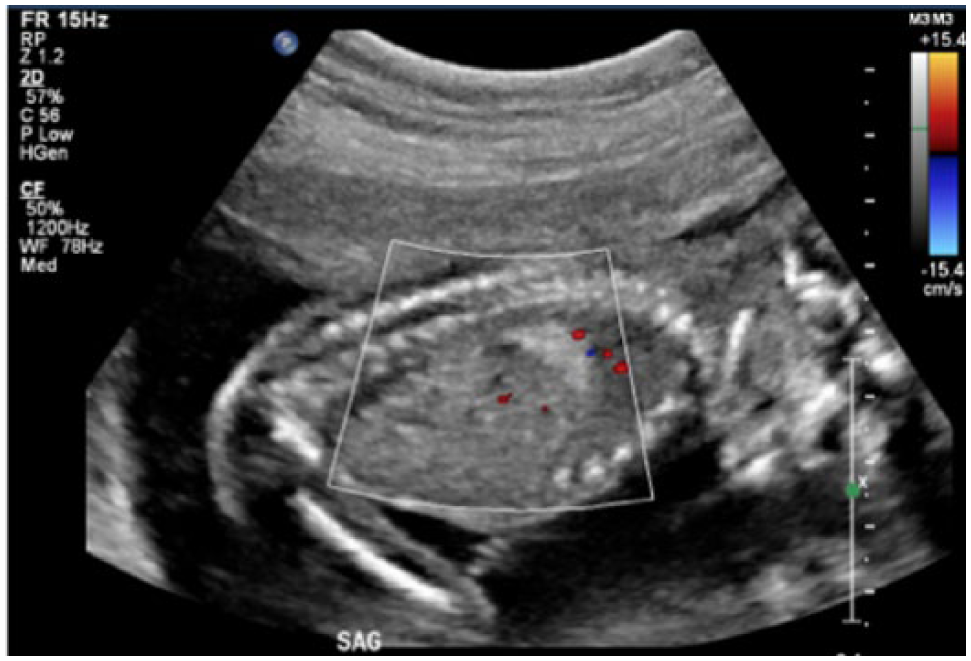
Longitudinal color Doppler image of the congenital pulmonary airway malformation showing a lack of systemic and internal blood flow.
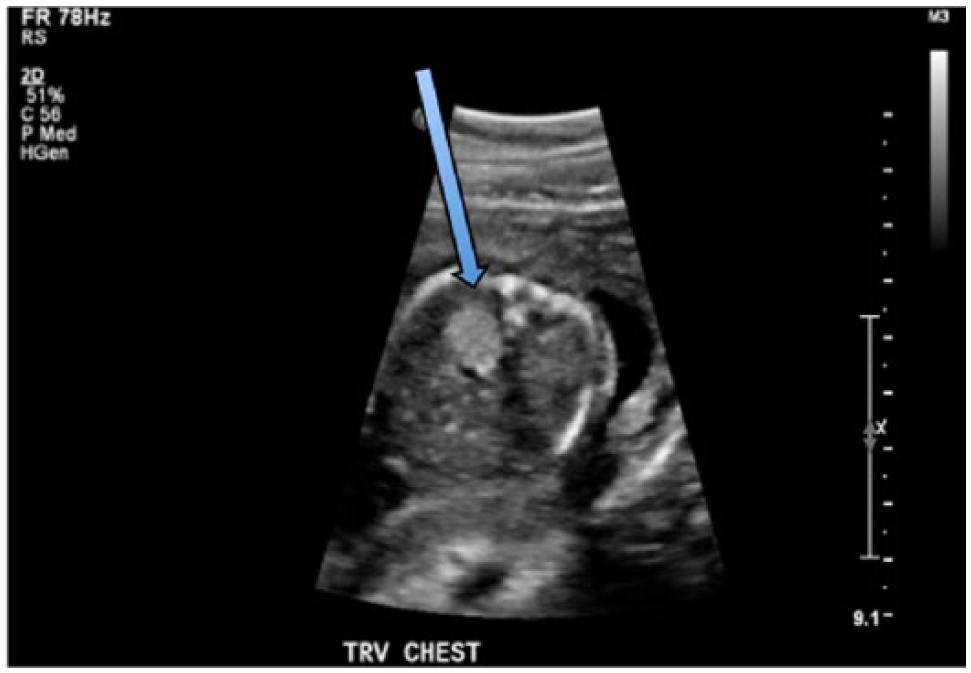
Transverse gray-scale image of the fetal chest showing the congenital pulmonary airway malformation (arrow) within the right side of the thoracic cavity without a mediastinal shift.
The fetus was diagnosed with CPAM. Differential diagnoses included bronchopulmonary sequestration, congenital diaphragmatic hernia, and congenital lobar emphysema. The CPAM volume was estimated with the formula for a prolate ellipse: CPAM volume = length × height × width × 0.52. The CPAM volume ratio (CVR) is then obtained by dividing the CPAM volume by the head circumference (measured in centimeters). 4 The CVR was calculated for this patient to be 0.04. The patient was referred to an outside clinic for further evaluation.
She was seen at the outside facility 5 days later where an additional sonographic examination was performed. The CPAM measured 1.6 × 1.2 × 1 cm, and the CVR was 0.06. At that time, the fetus was further diagnosed with type 3 CPAM. The patient was advised to have a fetal magnetic resonance imaging (MRI) examination for additional evaluation of the lesion. The MRI showed the CPAM originating in the anterior right lower lobe of the lung with mild medial displacement of the esophagus. The patient was also advised to return for weekly sonograms to evaluate for hydrops and to monitor the CPAM growth until 26 weeks.
The postnatal plan is to monitor the CPAM growth by computed tomography (CT) when the infant is >50 postconceptual weeks of age. Surgical resection of the CPAM has been recommended by a minimally invasive approach to confirm the diagnosis and to eliminate risk of infection and malignant transformation.
Discussion
Congenital cystic adenoid malformation is the original terminology for CPAM. Congenital cystic adenoid malformation was initially organized into three categories based on cystic appearance and size. The name was recently changed to CPAM to include the different types of pulmonary involvement and to recognize the variable cystic appearances. 1 The incidence of CPAM is relatively low, at 1 in 25,000 to 35,000 pregnancies. CPAM is the most common congenital lung lesion.1–3 It is a result of abnormal development of bronchial tree tissue at the level of the trachea, bronchioles, and/or alveoli. CPAM may be present in both lungs; however, it is found unilaterally in the majority of cases. 5 CPAM has a sporadic incidence and is associated with congenital anomalies about 1% of the time. 3 The lesion is benign in the majority of cases and does not have a connection to the fetal airway. CPAMs are supported by pulmonary circulation, and internal blood flow is rarely seen. CPAM is organized into five categories; each reflects the type of pulmonary involvement and the sonographic appearance3,6:
Type 0 CPAM is a rare form and is incompatible with life. There is acinar atresia throughout the lungs and small echolucent cysts.
Type 1 CPAM is the most common form and arises from the distal bronchus to the proximal bronchiole. The lower lungs may have multiple echolucent cysts, ranging from 3 to 10 cm. A single cyst of up to 10 cm may also be seen.
Type 2 CPAMs contain multiple echolucent cysts in the lower lungs, measuring 0.5 to 2 cm and appearing sponge-like. The cysts develop from the distal bronchioles. Type 2 CPAMs have the highest association with chromosomal anomalies.
Type 3 CPAMs appear as an echogenic mass in the lower lungs. There are microscopic cysts within these lesions that are not detected by sonography. These lesions may appear similar to bronchopulmonary sequestration; however, bronchopulmonary sequestration lesions have systemic vascularization directly from the aorta, which can be seen with color Doppler imaging.
Type 4 CPAMs contain echolucent cysts measuring up to 10 cm and originate from alveolar tissues. They may be associated with a malignant pleuropulmonary blastoma, also known as primary pulmonary rhabdomyosarcoma.
With the increased use of prenatal sonography, CPAMs are usually detected before birth. CVR measured by sonography uses the ratio of the lesion size to the head circumference. 6 This ratio is used to determine the risk of fetal demise due to hydrops. With a CVR >1.6, the risk for fetal hydrops increases significantly. Fetal MRI is often used in conjunction with sonographic examinations to confirm the diagnosis, determine the lesion morphology, and measure fetal lung volume. 6
These lesions can progress during pregnancy, with maximal growth between 20 and 26 weeks of gestation. Often, the patient will have weekly sonographic examinations up to 26 weeks to evaluate for hydrops and to monitor lesion size. After 26 weeks, CPAM lesions can plateau, decrease in size, or appear to regress completely. Larger CPAMs may cause mediastinal shift, hydrops, or pulmonary hypoplasia. Polyhydramnios without hydrops may also result from esophageal compression by the lesion.6,7 Fetal prognosis depends largely on the presence of polyhydramnios, hydrops, and pulmonary hypoplasia. 8 The CPAM may seem to disappear completely by sonographic evaluation by 32 weeks, as the lesion usually becomes isoechoic to the normal lung tissue. Postnatal CT scans of infants with a history of complete CPAM regression have revealed that the lesions are still present in the thoracic cavity. 6 Nearly 95% of patients with CPAM survive to delivery. 3
Treatments of CPAMs are controversial and vary depending on the type and size of the lesion. Treatment options include postnatal minimally invasive surgical resection or an observational approach.3,6,9 If the CPAM is symptomatic at birth with mediastinal shift or pulmonary hypoplasia, prompt surgical resection is the most common treatment.
There are several opinions regarding when surgery should be performed on asymptomatic patients. One management option is long-term monitoring of the patient by serial CT scans until a complication develops. Some cases of asymptomatic CPAMs will become symptomatic within a year after birth, and malignant transformation of these lesions has also been reported. 2 There is no way to detect which lesions will be at risk for malignant transformation.
Conclusion
CPAM is the most common congenital lung lesion and is usually easily seen during fetal sonographic examinations. With the increased use of prenatal sonography, the fetal chest should be routinely examined for abnormal lesions and cystic areas. Accurate differential diagnosis is important for patient care and management. A CVR may be used to help determine fetal prognosis. With appropriate prenatal imaging, the classification of CPAMs can be determined, and the necessary monitoring or treatment planning may be initiated.
Footnotes
Declaration of Conflicting Interests
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author received no financial support for the research, authorship, and/or publication of this article.