
Abstract
Skeletal dysplasias are a genetically heterogeneous group of disorders that can be diagnosed prenatally by sonography. Differentiating these disorders in the prenatal period can be difficult. However, differentiating known lethal disorders from nonlethal disorders can improve patient care and plan of action. This case study presents multiple pterygia, flexion contractures, and skeletal abnormalities, which are consistent with multiple pterygium syndrome, or Escobar syndrome.
Introduction
Skeletal dysplasias constitute a large, heterogeneous group of disorders comprising the formation and growth of bone. Certain skeletal dysplasias are correlated with abnormalities in other organ systems. Manifestations of these disorders begin in the early stages of fetal development. Causes may include extrinsic factors (maternal autoimmune disorders, teratogen exposure, etc) or intrinsic factors (single gene disorders with autosomal dominant, recessive, or X-linked modes of inheritance). 1 Prenatal diagnosis is centered largely on fetal sonographic findings. Sonography plays an important role in the detection of skeletal dysplasias because a majority of the skeleton begins to ossify early in development. 2
Multiple pterygium syndromes encompass a group of congenital anomalies characterized by multiple pterygia, flexion contractures, and skeletal abnormalities. Skeletal abnormalities include fusion of cervical vertebrae, scoliosis, flexion contractures of fingers, and rocker bottom feet with vertical talus. 3 Inheritance can be autosomal dominant, recessive, or X-linked with autosomal recessive inheritance appearing to be most common. 2 Multiple pterygium syndromes are phenotypically and genetically heterogeneous but are usually separated into prenatally lethal and nonlethal (Escobar) types. 4
Case Report
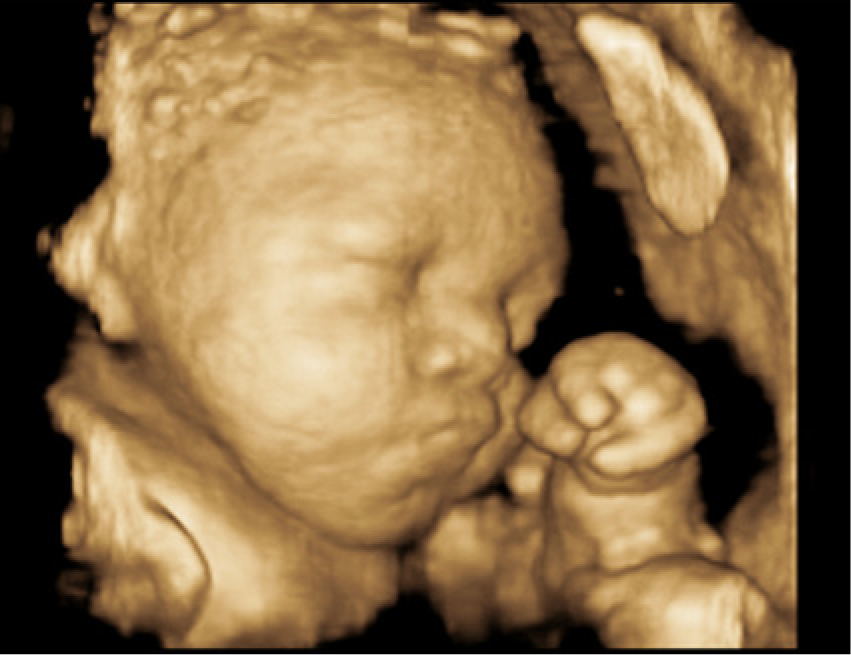
A 19-year-old G1P0000 African American female presented for an initial high-risk maternal fetal medicine consult with a perinatologist due to multiple fetal anomalies detected on an outside sonographic examination. A complete morphology was performed at 19 weeks 2 days according to the estimated due date (EDD). Sonography showed an intrauterine pregnancy (IUP) at 20 weeks 1 day gestation. All the long bones appeared in a locked position (Figures 1 and 2). Fetal gross movement was noted, but no flexion or extension of the arms or legs was visualized. The fetal neck appeared shortened. The hands were noted to be clenched and did not move from that position during the examination, which lasted greater than 30 minutes (Figure 3). The feet were abnormally rotated and remained in that position. The right knee and lower leg appeared abnormally extended at a 45-degree angle laterally; the left leg demonstrated some mild backward hyperextension of the knee, or genu recurvatum (Figures 4 and 5). Edema was noted in the feet, forehead, and scalp (Figure 6). There were no obvious facial, intracranial, or cardiac anomalies (Figure 7).
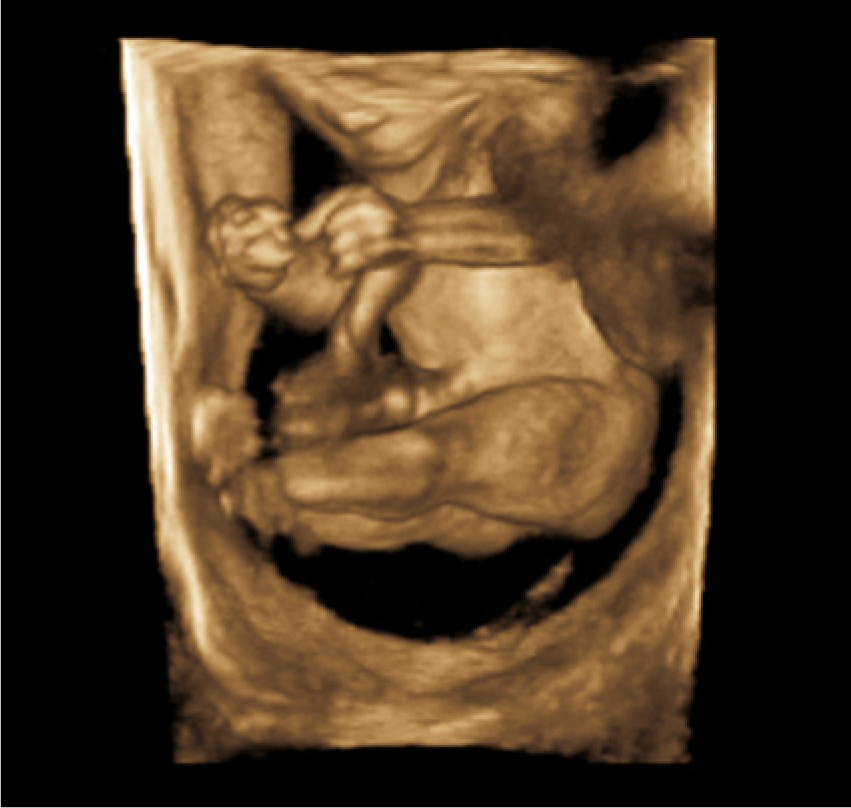
A 3D sonogram showing all long bones in a locked position.
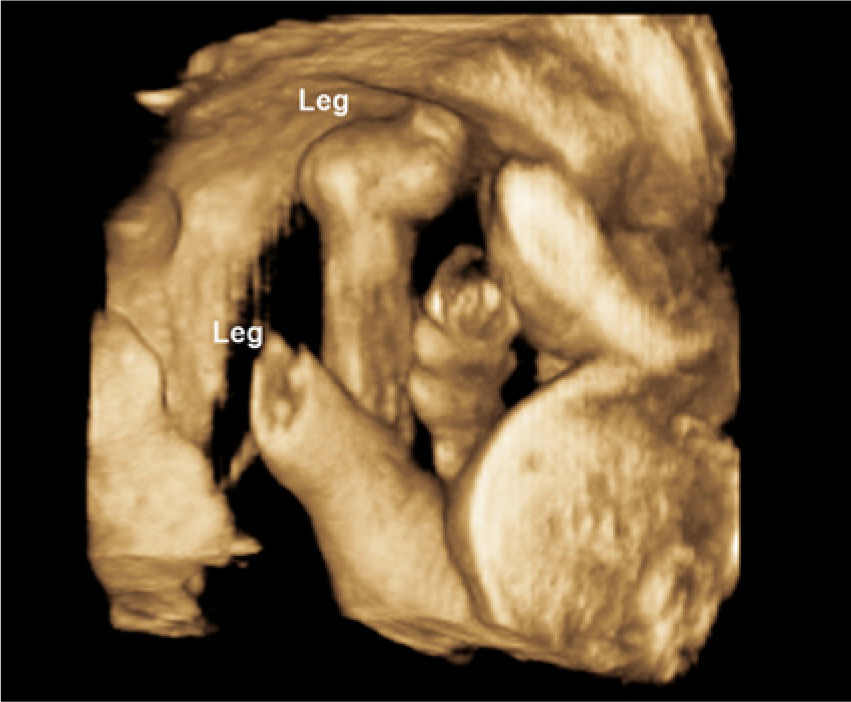
A 3D sonogram showing the lower extremities in a locked position.
A 3D sonogram showing clenched hands.
A sonogram showing the right knee and lower leg abnormally extended at a 45-degree angle laterally.
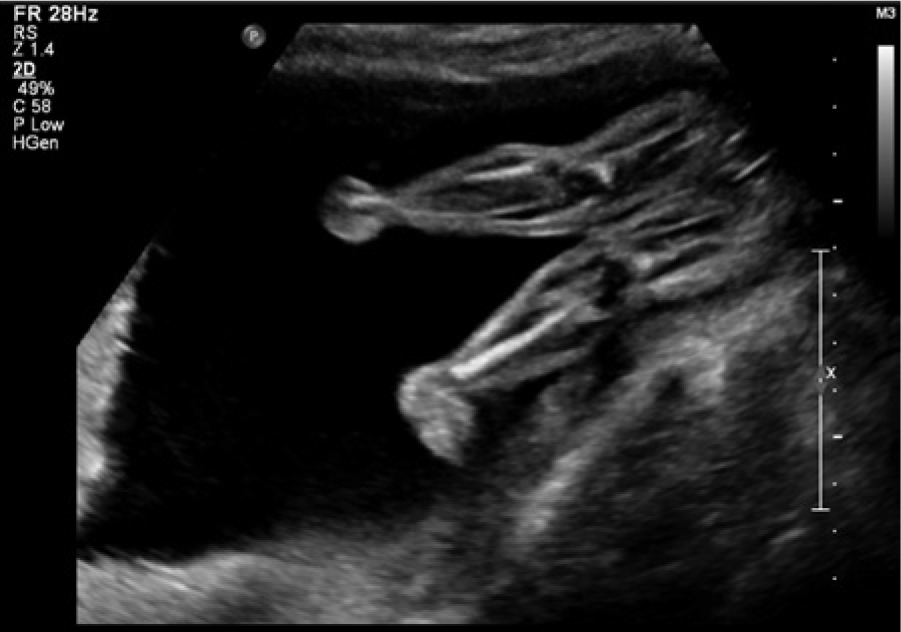
A sonogram of the left leg showing mild genu recurvatum.
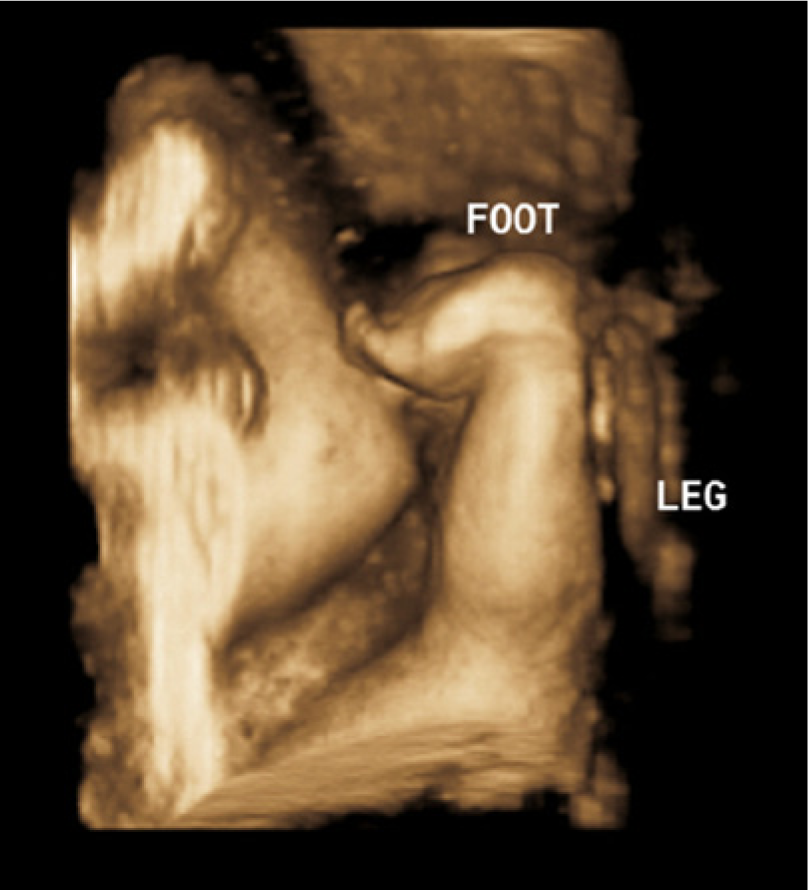
A 3D sonogram showing edema of the foot (rocker bottom).
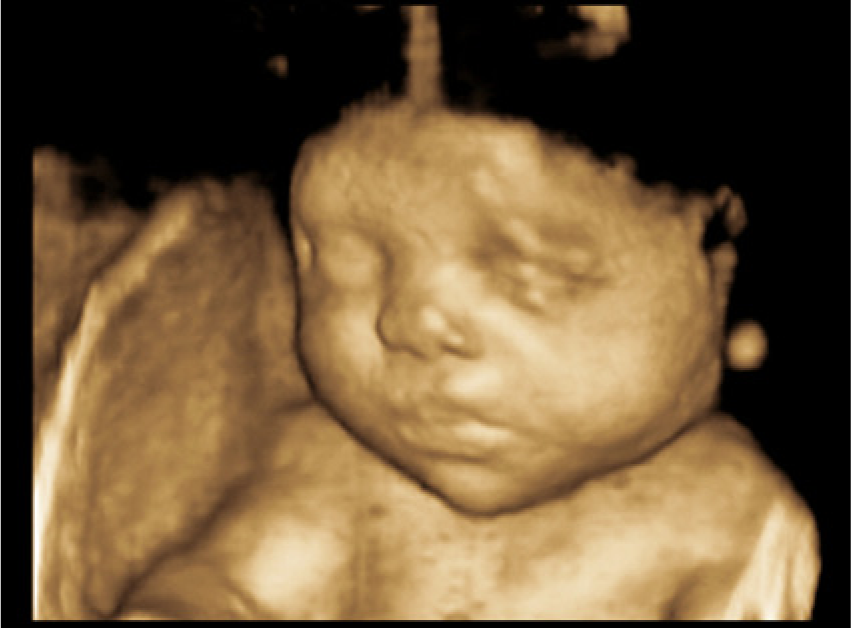
A 3D sonogram showing no obvious facial abnormalities.
A genetics consultation was recommended as well as an amniocentesis for karyotyping. During consultation, no significant teratogenic exposures were reported. The patient denied any prescription or over-the-counter medications (other than prenatal vitamins), alcohol, drugs, infections, illness, fever, exposure to chemicals, or other potential teratogens. The mother’s height was 59 inches (150 cm); the father was a 20-year-old African American with sickle cell trait and a height of 64 inches (162.5 cm). Both family histories were negative for any known birth defects, recurrent pregnancy loss, or other known genetic conditions. The patient had genetic amniocentesis and the results showed normal chromosomal karyotype. Sickle cell trait testing was performed and showed normal hemoglobin electrophoresis. Amniotic fluid alpha-feta protein (AFAFP) was also normal.
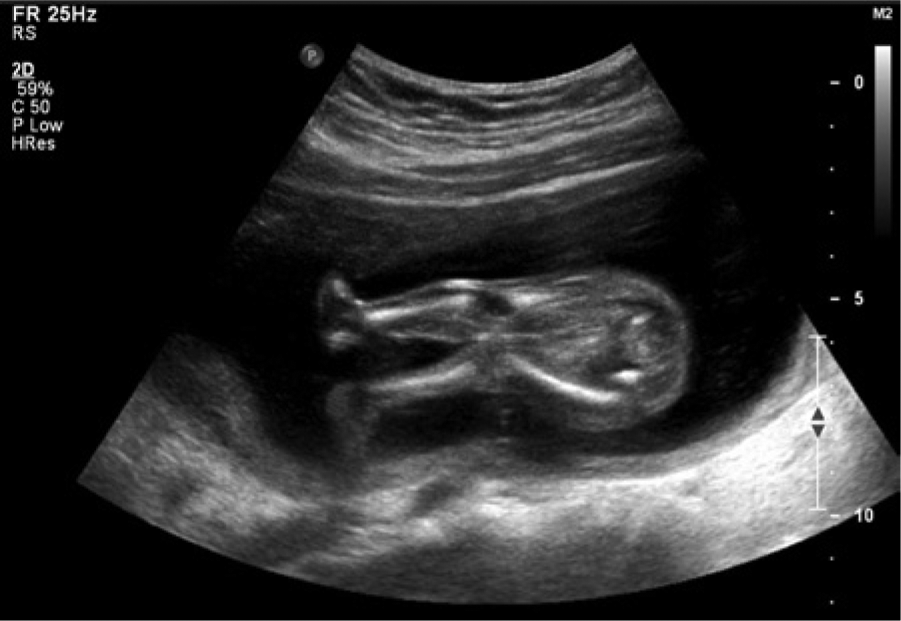
The patient continued with routine prenatal care, which consisted of a normal 1-hour glucose tolerance test, monthly sonographic examinations to assess fetal growth, and weekly biophysical profiles (BPP). At 36 weeks, the estimated fetal weight was at the 6th percentile for gestational age. The interval fetal growth appeared restrictive compared to that of the previous sonographic examination. The long bones measured 4 weeks behind gestational age, restrictive compared to the previous examination (Figure 8). Polyhydramnios was noted to have an Amniotic Fluid Index (AFI) of 35.24 cm with the largest vertical pocket of 13.5 cm.
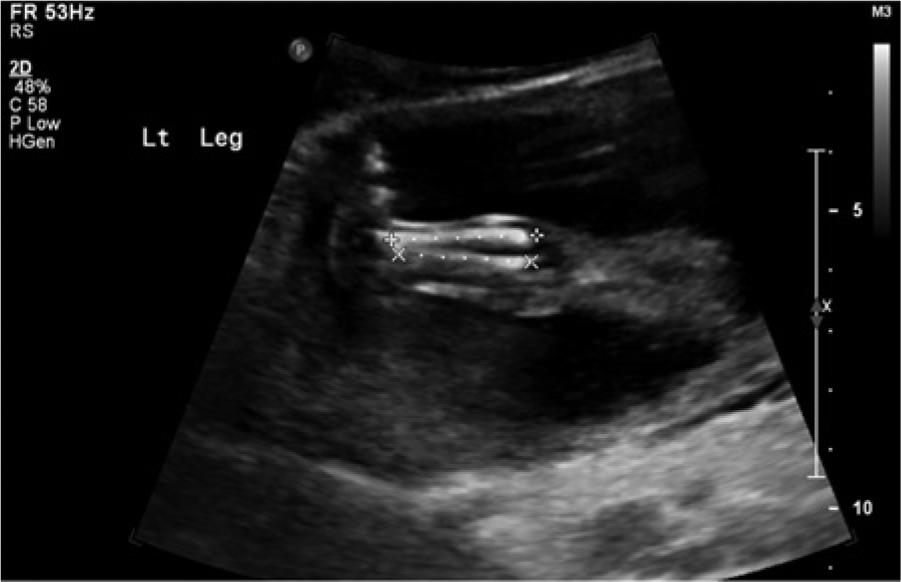
A sonogram showing the long bones measuring 4 weeks behind gestational age.
The patient presented for a scheduled primary cesarean delivery at 39 weeks 4 days for breech presentation. The patient reported feeling well with normal fetal movement, and the surgery was uncomplicated. Estimated blood loss (EBL) was 600 mL, within the expected range. Apgar scores were 5 and 7 at 1 and 5 minutes, respectively. Resuscitation was required in the delivery room for increased respiratory effort, and a meconium stained cord was noted. The infant was subsequently transferred to the neonatal intensive care unit.
Orthopedics and genetics consultations were recommended. Prior to the initiation of treatment, bilateral sonograms and radiographs of the hips were obtained to assess for dislocation of the hip joint and anatomy of upper and lower extremities, as well as the spine. Findings were consistent with severe developmental dysplasia of the hips, greater on the left, with complete dislocation of the left femur and severe subluxation of the right femur (Figure 9). The upper and lower limbs were unable to be passively mobilized in positions other than those currently maintained, with rigid extension of the elbows, flexion of the wrist, and the fingers in the fist position. Orthopedics concluded that due to her many musculoskeletal deformities, and the inability to mobilize the joint, she would likely require long-term orthopedic treatment to get her in a functional position. Initial management entailed short-leg casting to correct her lower extremity deformities prior to any institution of proximal knee, hip, and upper extremity treatment. The patient will require prolonged stretching with frequent casting into positions of function as she continues to age.
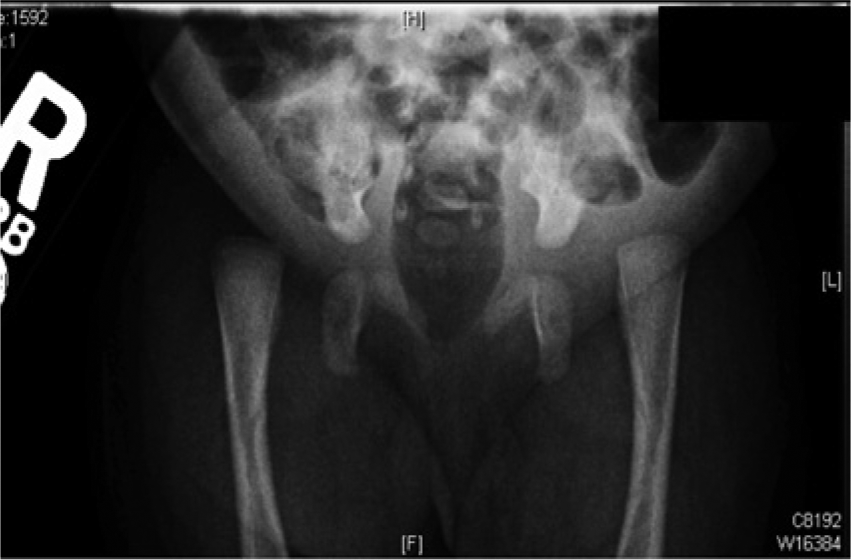
Radiograph demonstrating bilateral proximal femoral dislocation and hip joints, left greater than right, and dysplastic acetabula compatible with congenital hip dysplasia.
Discussion
Skeletal dysplasia is rare, with the prevalence estimated at 2.4 per 10,000 births.5,6 The prevalence appears to be slightly higher at 7.5 per 10,000 pregnancies screened by sonography. 7 The patient above presented at term delivery with a complex constellation of medical findings, including an appearance of a short stature and multiple contractures of the proximal and distal extremities. The extremities were remarkable for pterygium of the inguinal region with a web across the superior portion of the pubic region, joining the two medial segments of the upper thigh. Anomalies of the distal lower extremities included syndactyly of digits 2 to 3, markedly broad feet with medially displaced first digits and increased space between the first and second digits, as well as talipes deformities bilaterally. The lower extremities were hyperextended in a fixed position with extension past 180 degrees by approximately 10 degrees. Upper extremities were remarkable for relative fixed extension at the elbows and no evidence of antecubital webbing. The wrist was essentially fixed in a flexed position. There was also fisting with extended digits and overlapping of the second and fifth digits with digits 3 and 4 (see Figures 10–13).
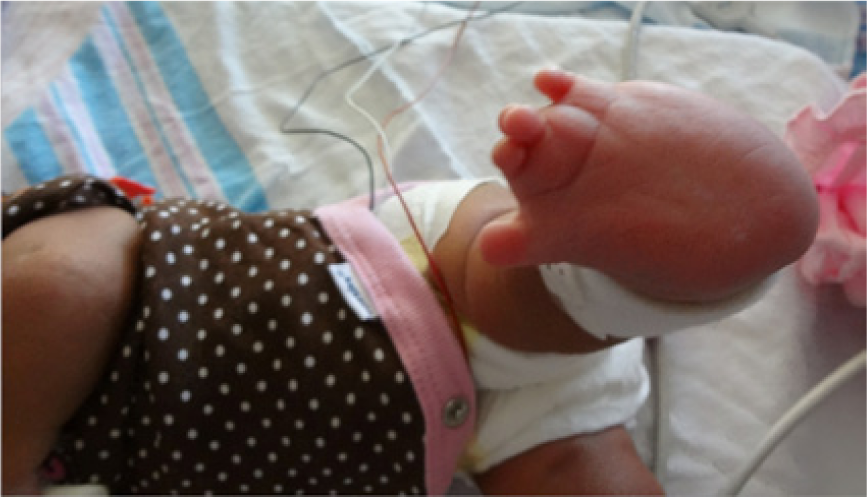
Postnatal photograph showing syndactyly of digits 2 to 3 and markedly broad feet with medially displaced first digits and increased space between the first and second digits.
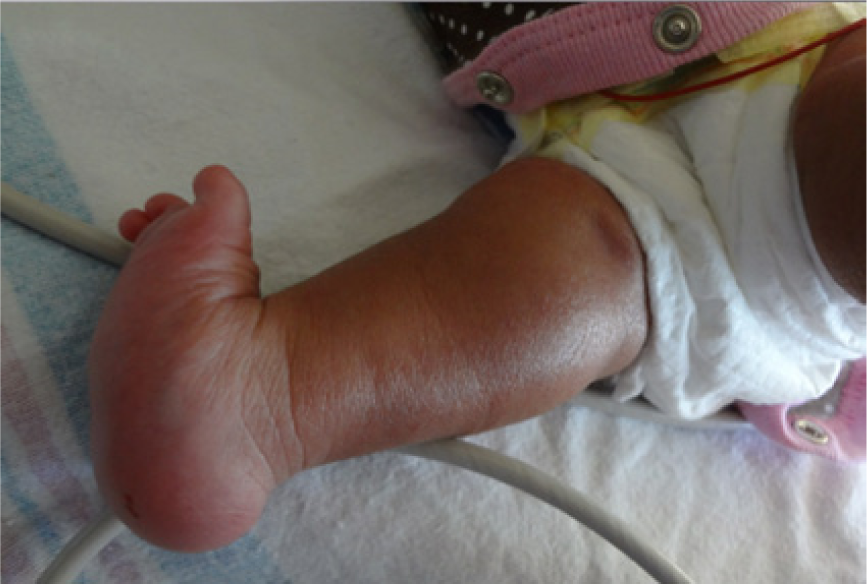
Postnatal photograph showing edema of the foot (rocker bottom).
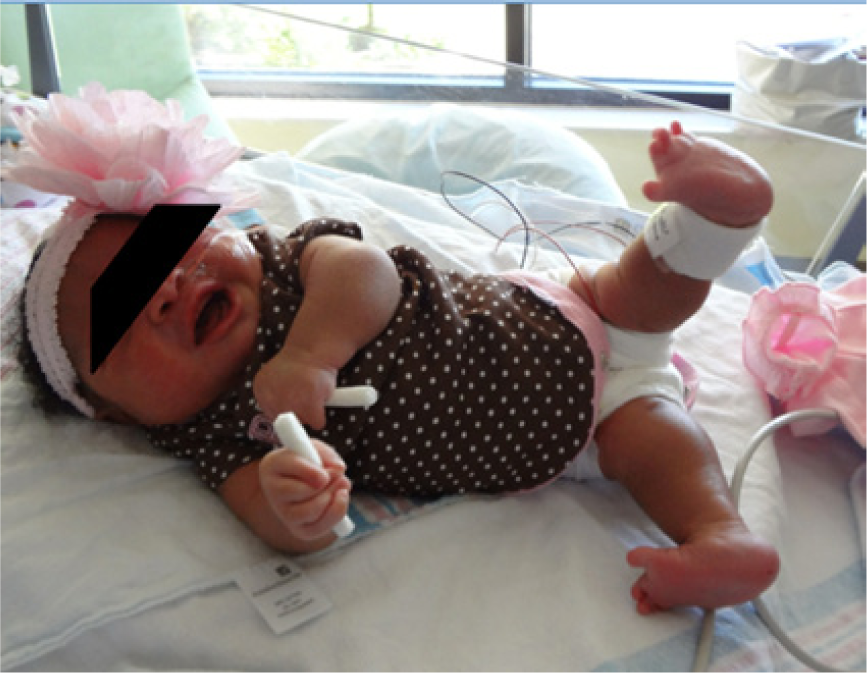
Postnatal photograph showing hyperextension of the lower extremities.
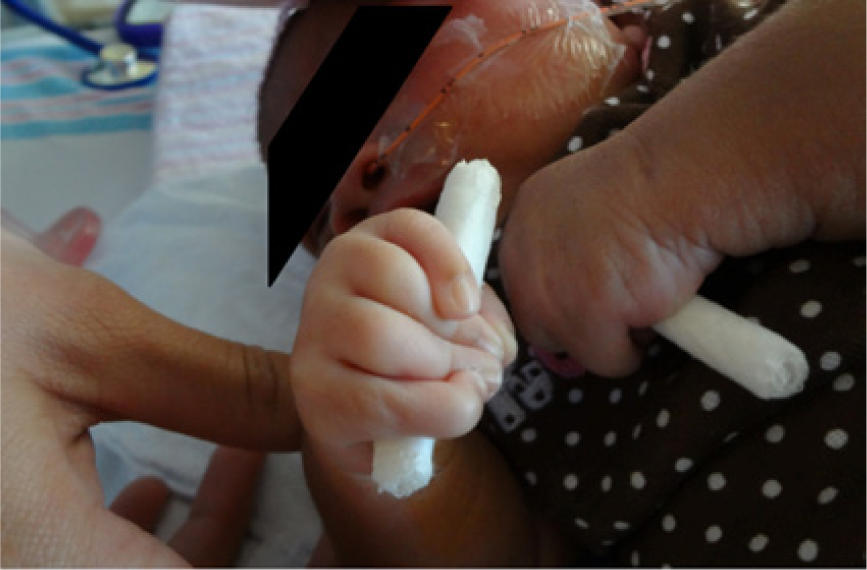
Postnatal photograph showing clenched hands and overlapping of the second and fifth digits with digits 3 and 4.
A genetics consultation recommended sending a chromosomal microarray since chromosomal rearrangements including large deletions or duplications could mimic any of the Mendelian disorders. DNA diagnostic studies were also recommended to rule out multiple pterygium syndrome. This would include sequencing of the CHRNG gene, the gene for the fetal acetylcholinesterase receptor, which is the gene mutated in Escobar syndrome. It is difficult to place all of these findings under a singly unifying diagnosis. However, they are most consistent with a diagnosis of multiple pterygium syndrome, or Escobar syndrome. These features could also be seen in distal arthrogryposis type 8, or in a variety of congenital skeletal dysplasias including spondyloepiphyseal dysplasia with congenital joint dislocations.
The pathogenesis of multiple pterygium syndrome has been uncertain, yet early onset fetal akinesia, fragile collagen, and edema have all been associated with it. 8 In an analysis of lethal cases, a limited number were found to have another associated diagnosis (eg, specific primary myopathy or metabolic or neurodevelopmental disorder), and there was associated clinical and pathological heterogeneity. 9 A substantial minority of cases of lethal and nonlethal multiple pterygium syndrome had germline CHRNG mutations that represented advances diagnostically as well as direction into the etiology of multiple pterygium syndrome in these patients.
CHRNG gene mutations produce most cases of multiple pterygium syndrome, Escobar type, and a smaller percentage of cases of lethal multiple pterygium syndrome. The CHRNG gene instructs the production of the gamma (γ) protein component (subunit) of the acetylcholine receptor (AChR) protein. 10 CHRNG gene mutations result in an impaired or missing γ subunit. The severity of the CHRNG gene mutation affects the severity of the condition. Mutations that prevent the production of any γ subunit will result in the lethal type, whereas the Escobar syndrome is a result of the production of some γ subunits. These are essential for signaling to take place between nerve and muscle cells. This lack of signaling leads to akinesia and pterygium before birth and can result in other signs and symptoms of multiple pterygium syndrome. 10
There are four forms of pterygium syndrome: two lethal and two nonlethal. This syndrome is characteristic of webbing in the joints, called pterygia. Arthrogryposis is the permanent fixation of a joint in a contracted position and is characteristic of all forms of pterygium syndrome. The four forms are classified into lethal and nonlethal (Escobar) multiple pterygium syndrome and lethal and nonlethal popliteal pterygium syndrome. Popliteal pterygium syndrome of the nonlethal type has an incidence of 1 in 300,000. It is autosomal dominant with sonographic findings of a cleft lip/palate, popliteal pterygium, missing teeth, syndactyly, malformed genitalia, spina bifida occulta, and talipes. 4 This form of pterygium syndrome has normal intelligence, delayed language development, and a potential for learning disabilities. The other nonlethal syndrome is multiple pterygium syndrome, or Escobar syndrome/nonlethal arthrogryposis multiplex congenital. This is a Mendelian disorder with sonographic findings of extremity contractures, pterygia, micrognathia, camptodactyly, syndactyly, rocker bottom feet, vertical clubfoot, abnormal genitalia, microcephaly, low set ears, cleft palate/lip, spinal fusion, ocular hypertelorism, and diaphragmatic hernia. This form is associated with normal intelligence. 4
Prenatal diagnosis is based primarily on fetal sonography findings. In most cases, initial sonograms are performed due to family history of skeletal dysplasias. Fetal skeletal dysplasias are also questioned because of qualitative bony abnormalities observed during morphology examinations, most often performed during the second trimester. An increase in the nuchal translucency has also been linked to skeletal dysplasias. When this association is present, roughly 85% of these cases are lethal.11,12 An extensive assessment of the fetal skeleton is essential to establish the bones affected and their severity. The existence of accompanying anomalies in other systems is also necessary to composite the findings that may or may not define a specific skeletal dysplasia. Prenatal sonography in the diagnosis of skeletal dysplasias has been enhanced drastically, with a 68% rate of correct prenatal diagnoses, a 31% rate of partially correct diagnoses, and only a 0.07% false positive rate.12-19 When a skeletal dysplasia is identified, the main goal is to establish whether the dysplasia is lethal or nonlethal. The accuracy of diagnostic sonography is vital for diagnosis in order to educate the parents, as some will elect to terminate the pregnancy.
Conclusion
Prenatal diagnosis is centered largely on fetal sonographic findings. 2D and 3D ultrasound can recognize abnormal skeletal abnormalities, and by analyzing the constellation of findings, a diagnosis can be achieved. This case study supports sonography as an indispensable diagnostic imaging modality in obstetric studies.
Footnotes
Acknowledgements
The author thanks Margaret Harry, MHS, RDMS, and Dr. R. Floyd, MD, for their assistance, leadership, and guidance.
Declaration of Conflicting Interests
The author declares no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author received no financial support for the research, authorship, and/or publication of this article.