
Abstract
Ectopia cordis is an anomaly of embryonic cell growth that causes the heart to form outside the chest cavity during fetal development. Other birth defects are often present with ectopia cordis as well. It is usually a fatal condition, commonly associated with pentalogy of Cantrell, and occurs in 5.5 to 7.9 children per one million live births. Exencephaly is the exposure of a well-developed and differentiated brain outside the skull during the embryonic period. It is the second of three phases in the development of anencephaly and is a lethal neural tube defect with only a small number of cases reported in the literature. Sonographically, it appears as a bilobed-shaped head termed the “Mickey Mouse” head with a relatively large amount of brain tissue present floating in the amniotic fluid.
First described by Stevenson in 1671, ectopia cordis is a rare congenital cardiac anomaly in which the heart is partly or completely located outside the thorax. 1 This anomaly can be separated into five types: cervical, cervicothoracic, thoracic, thoracoabdominal, and abdominal, with an occurrence rate of 5.5 to 7.9 per million live births. 2 Exencephaly (also known as acrania) is the exposure of a well-developed and differentiated brain outside the skull during the embryonic period. 3 It is the second of three phases in the development of anencephaly and is an abnormality of the neural tube. 4 This report presents a rare abnormality in a twin pregnancy, with one twin having no abnormalities and the second twin presenting with ectopia cordis and exencephaly. A detailed literature search fails to reveal prior case reports with these two congenital abnormalities in a fetus.
Case Report
A healthy woman, G2 P0 AB1, in her mid-20s visited her obstetrician because she suspected she was pregnant. Her urine result showed a positive pregnancy test, and she was determined to be nine weeks pregnant. Urinalysis showed a bacterial infection of the genital tract, Gardnerella vaginalis, and the patient was to begin antibiotics after her first trimester. However, blood tests detected elevated levels of maternal serum alpha-fetoprotein (MSAFP). Past medical and genetic history revealed only a miscarriage. She denied any use of alcohol or illicit drugs. She was referred for a sonogram at 16 weeks. She was also referred for genetic counseling and offered an amniocentesis. 5 She declined the amniocentesis.
The initial sonogram was performed using a Philips HDI 5000 (Philips Healthcare, Andover, Massachusetts) at approximately 17 weeks’ gestation. During the examination, the sonographer demonstrated a dichorionic diamniotic twin pregnancy (Figure 1). Imaging of twin A revealed a normal fetus in breech presentation with the placenta located on the posterior uterine wall. Measurements of the head circumference (HC), biparietal diameter (BPD), abdominal circumference (AC), and femur length (FL) placed the fetal gestational age at approximately 17 weeks 1 day. A detailed anatomical survey found normal anatomic structures, with a heart rate of 143 beats per minute.
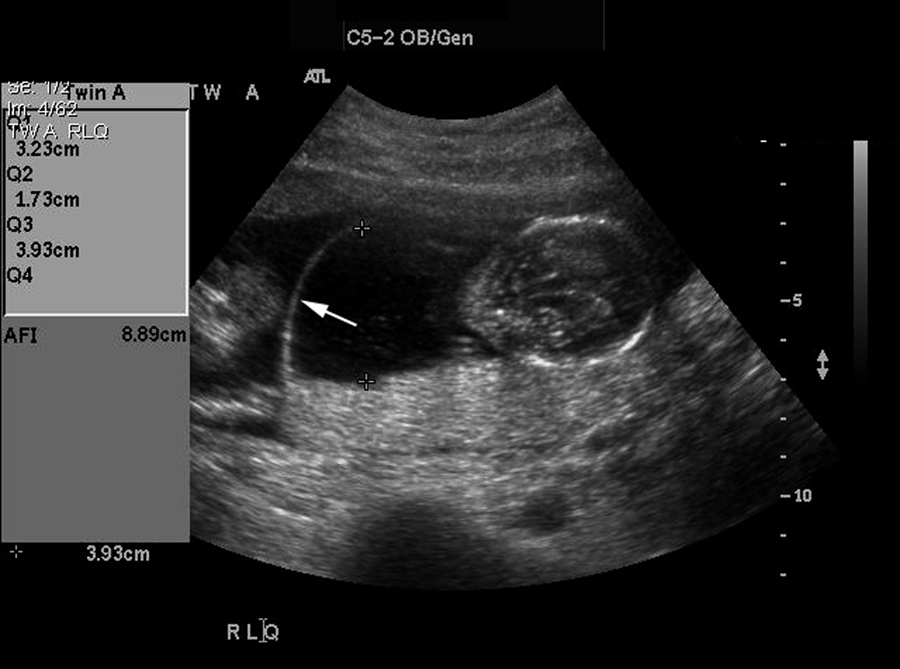
Note the thickness of the amniotic membrane (arrow) separating twin A and twin B, characteristic of a dichorionic diamniotic twin pregnancy.
Twin B was in a cephalic presentation with an anterior placenta. The HC, BPD, AC, and FL measurements dated twin B at 16 weeks 4 days. A detailed sonographic study of the fetus demonstrated multiple anatomic anomalies. Images of the head demonstrated an exencephalic pear-shaped head, with unilateral hydrocephalus (Figures 2 and 3). Detailed imaging of the spine was unremarkable. When the fetal heads were visualized simultaneously, the disparity was readily apparent (Figure 4). The fetal thorax was small, lacking normal intrathoracic cardiac structures. An ectopia cordis (EC) was located anterior to the sternal manubrium without a membrane covering the exposed heart (Figure 5). The fetal heart rate was 130 beats per minute (Figure 6).
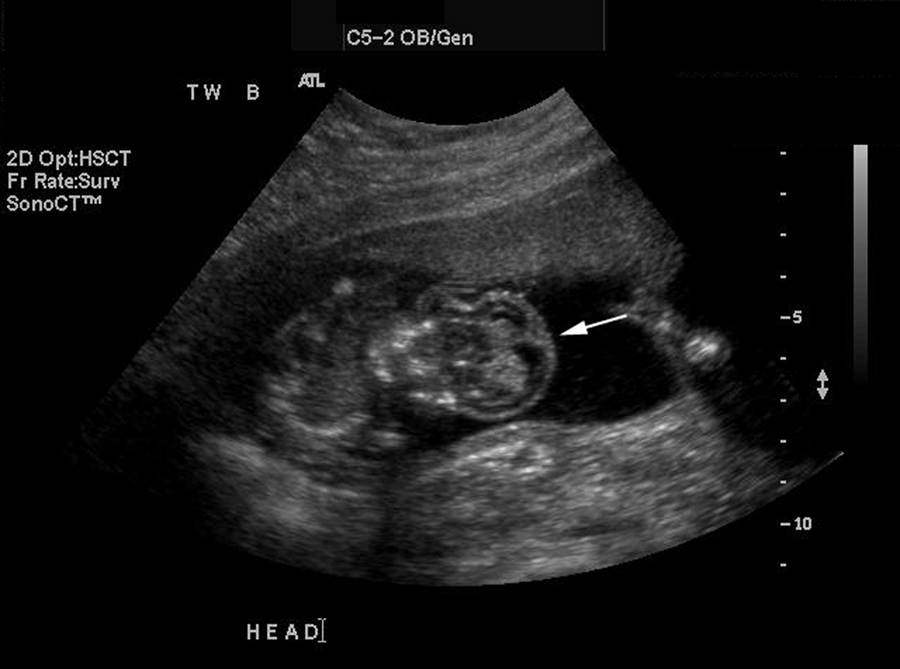
Coronal image of twin B demonstrating cerebellar hemispheres floating in the amniotic fluid (arrow) with absence of normal cranial bone structure.
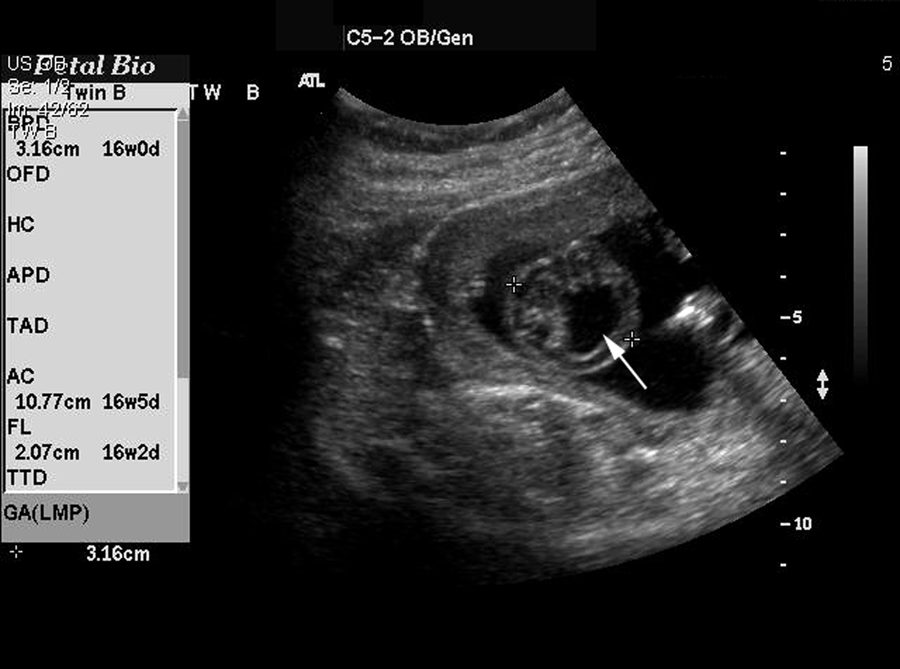
Transverse image of twin B demonstrating severe unilateral hydrocephalus (arrow).
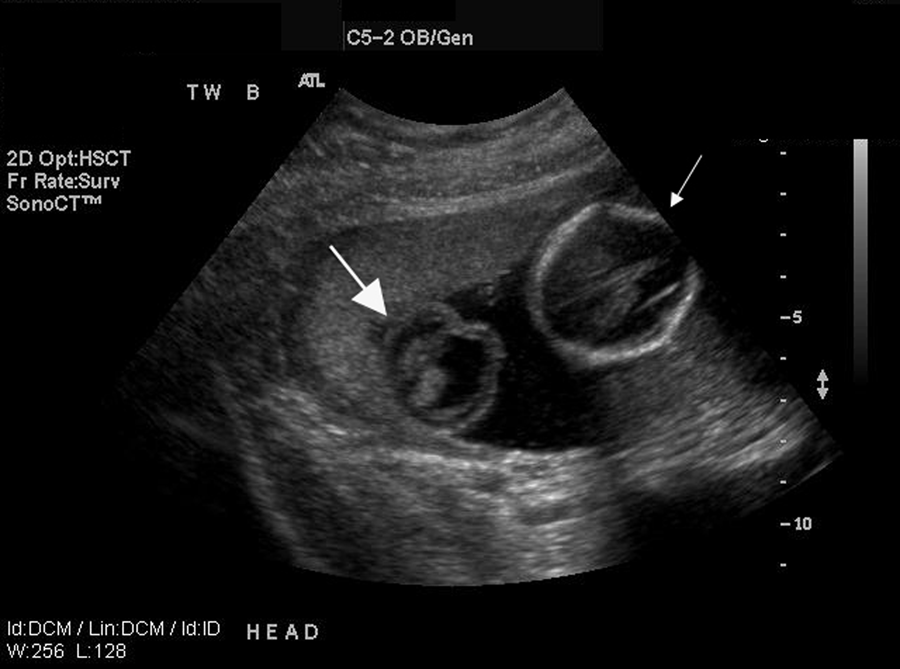
Image of twin A (small arrow) and twin B (large arrow) demonstrating the disparity in the size of the fetal head. Also note the absence of a cranial bone in twin B.
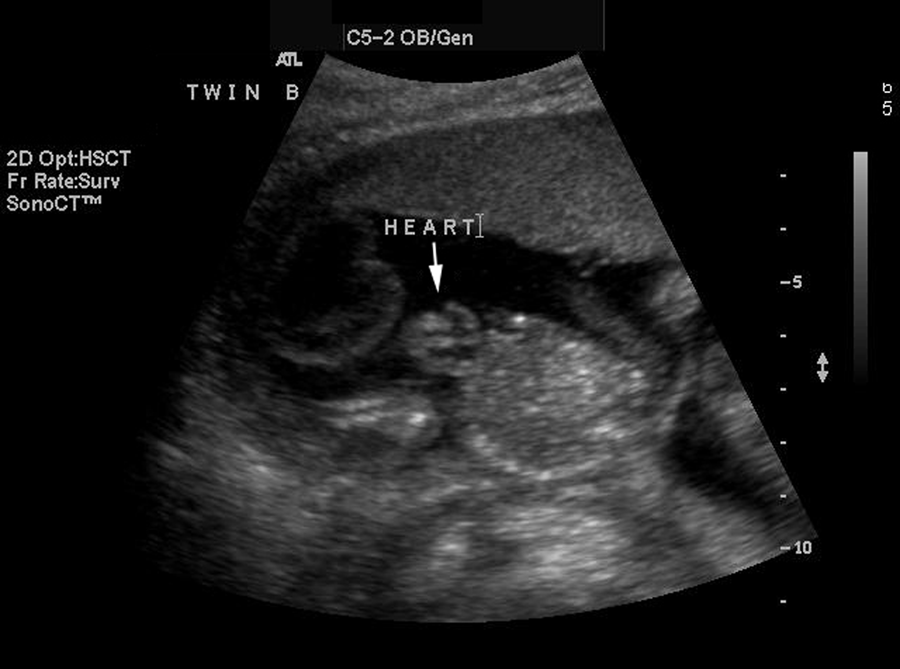
Transverse image of the thoracic cavity of twin B demonstrating the ectopia cordis (arrow).
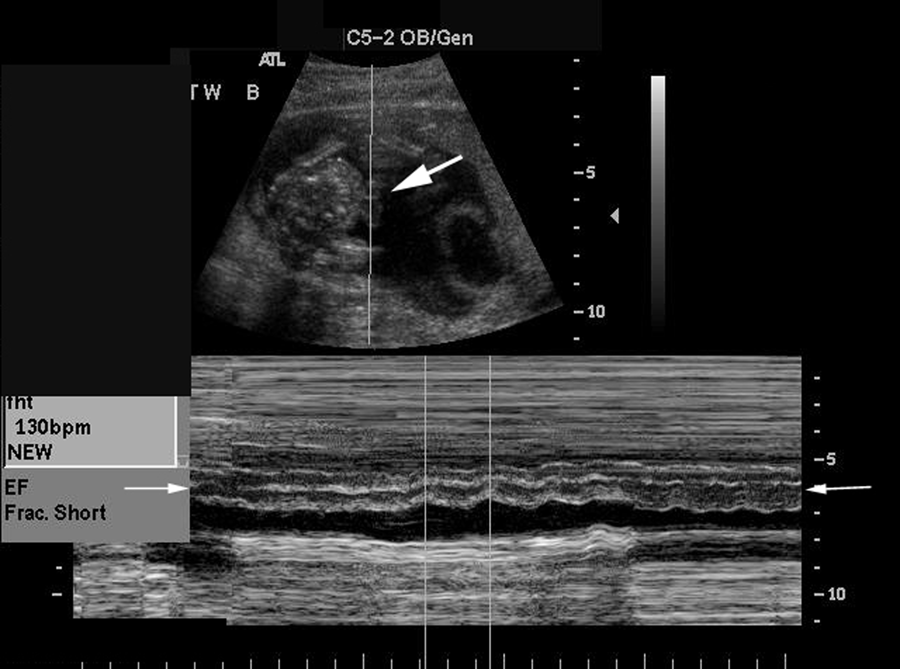
M-mode of the ectopic heart in twin B demonstrating a heart rate of 130 beats per minute (small arrow). Note the placement of the curser outside of the chest cavity (large arrow).
A follow-up sonogram performed at 19 weeks demonstrated normal interval growth for twin A. Twin B, however, demonstrated a spontaneous demise. Amniocentesis was again suggested to evaluate the health of the surviving twin. The patient declined.
Bimonthly sonography examinations were performed in the third trimester to closely monitor the remaining viable fetus. Routine growth measurements were obtained as well as amniotic fluid volume assessments. The surviving fetus continued to develop normally through the remainder of the pregnancy.
At 40 weeks’ gestation, the patient was schedule for induction. Five days prior to her scheduled induction, she went into labor. Both fetuses were delivered vaginally without complications. The mother and surviving child were discharged two days later. This patient has since gone on to have a subsequent pregnancy that resulted in a normal fetus without evidence of any neural tube defects (NTDs) or thoracic defect.
Discussion
An MSAFP blood test is used to assess a fetus at risk for aneuploidy or structural defect. Normal values vary depending on gestational age. When elevated, sonographic imaging is often performed to confirm the gestational age and evaluate the fetus for structural defects. 6 One such abnormality that occurs during fetal development is an NTD. This defect allows open communication of the brain or spinal cord with the amniotic fluid, causing the MSAFP to rise above the normal range. One of the more common NTDs is anencephaly, with a reported incidence of 1 in 1000.
Ectopia Cordis
Embryologic Development of the Thorax
In the sixth week of human embryonic development, the “anlage” of the sternum becomes visible. It appears in the form of two mesenchymal bands that arise on either side in the ventral body wall far from the midline. The sternum originates from these two separate mesenchymal bands. 7 A pair of blastemas are located between the ventral ends of the primordia of the left and right clavicles and are described as “suprasternal structures.”8–10 These structures are localized in line with the cranial ends of the two sternal bands, which will later fuse. An unpaired mesenchymal condensation located between the caudal ends of the suprasternal structures is known as the “precostal process.”8,10 The development of the manubrium involves the suprasternal structures, the precostal process, and the cranial parts of the sternal bands situated at the level of the first rib. 11
Fusion of the cranial end of the thorax starts in the seventh week. This fusion results from the paired sterna bands fusing with their adjoining suprasternal structures (sterna primordia). As the fusion of both sternal primordia proceeds caudally, the ventrally localized unpaired precostal process is enclosed. The fusion of the more caudal parts of the sternal bands is completed during the ninth or tenth week.12,13
During the fourth week of fetal development, folding of the embryonic disk occurs. The lateral body walls converge and fuse in the ventral aspect of the embryo. If incomplete fusion occurs during this stage of development, complete or partial evisceration of the heart may occur. 2
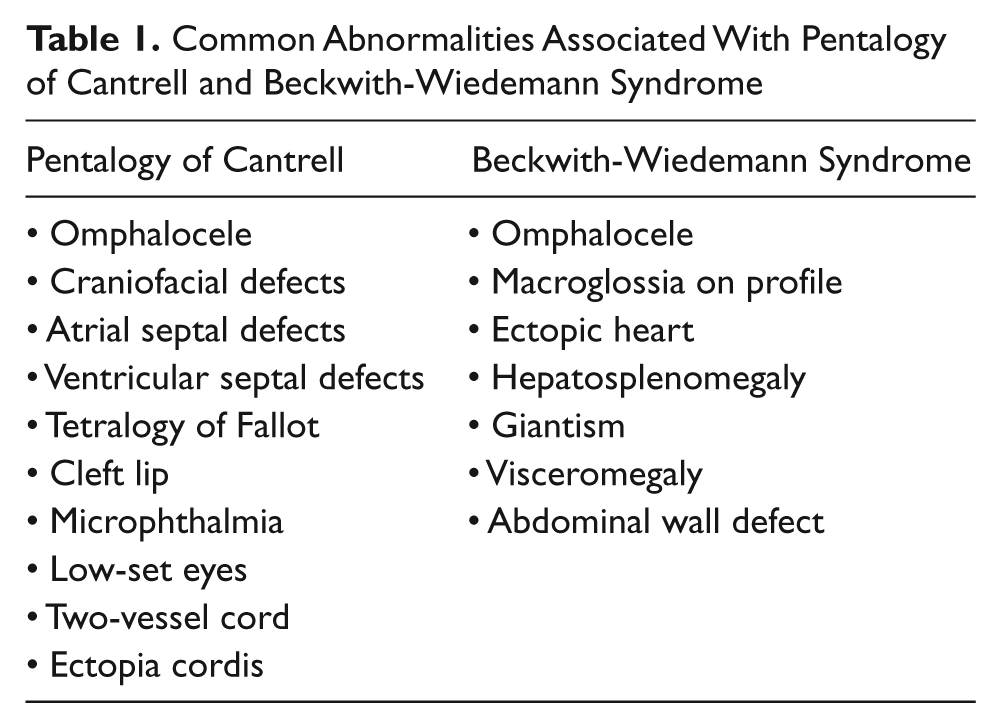
First described by Stevenson in 1671, ectopia cordis (EC) is a rare congenital cardiac anomaly in which the heart is partly or completely outside the thorax. 1 In 1958, Cantrell described a pentalogy characterized by a midline supraumbilical abdominal wall defect, a defect of the lower part of the sternum, a defect of the anterior diaphragm, a defect of the diaphragmatic pericardium, and a congenital heart malformation (Table 1). The hallmark of this syndrome is an omphalocele with associated ectopia cordis. 14 Although the spectrum of this syndrome consists of these five anomalies, it is often not complete. Toyama 15 suggested three classifications to the pentalogy of Cantrell (Table 2).
Common Abnormalities Associated With Pentalogy of Cantrell and Beckwith-Wiedemann Syndrome
List of Classifications to the Pentalogy of Cantrell Suggested by Toyama 15

Beckwith-Wiedemann syndrome was first described as a syndrome by J. B. Beckwith and then again by Wiedemann in 1964.16,17 A wide range of anomalies may be observed when a fetus presents with this condition (Table 1). Omphalocele and polyhydramnios are common in fetuses with this condition; however, macroglossia and visceromegaly are nearly always present. Cardiac defects and microcephaly also sometimes occur. 18 Sonographic findings are often subtle and nonspecific. Although most cases occur sporadically, some familial cases have been observed.18,19 Chromosomal analysis suggests that the gene producing insulin-like growth factor II (IGF II) on the short arm of the 11th chromosome may be abnormally expressed in Beckwith-Wiedemann syndrome. The patient presented with this case was offered an amniocentesis for chromosomal evaluation on two separate occasions; however, she declined the procedure on both occasions. 20
When a diagnosis of EC is considered, care should be taken to evaluate the fetus for other developmental abnormalities. EC is found in both the pentalogy of Cantrell and Beckwith-Wiedemann syndrome (Table 1). All additional sonographic findings should be compared with these conditions.
In complete thoracic EC, the naked heart is displaced outside the thoracic cavity without pericardial coverage. 21 This anomaly can be separated into five types: cervical, cervicothoracic, thoracic, thoracoabdominal, and abdominal, with an occurrence rate of 5.5 to 7.9 per million live births. 2 The most common are thoracic (59%) and thoracoabdominal (38%). 2 The thoracoabdominal type is associated with the pentalogy of Cantrell. Reported cases suggest that this developmental abnormality has its origin at about 14 to 18 days of embryonic life when the primary mesoderm splits into its splanchnic and somatic layers.14,22
Once fetal abnormalities are detected, a team is assembled to assist in the diagnosis of the condition and management of the pregnancy (Table 3). This team is put together to help the patient make an informed decision on whether to continue or terminate the pregnancy. 23
Specialists That May Comprise the Team to Manage Care Throughout the Pregnancy

After surgical correction of an EC, the survival for infants past the perioperative period is rare. 24 After delivery, an echocardiogram is performed to evaluate the neonate for pulmonary or ventricular abnormalities that may have been overlooked on the prenatal sonograms. The decision will then be made if surgical correction is a viable option. If the surgical placement of the heart in the chest is a success, the child may need to undergo many follow-up reconstructive surgeries, including the use of skin covers, grafts, and/or prosthetics.
Of the five types of EC, the thoracoabdominal group has had limited success in being surgically corrected. This is related to the severity of the associated cardiac abnormalities for this group when compared with the others. 14 The abdominal group may have a slightly better prognosis as a number of patients with this type of anomaly have lived into adult life. 25 Other authors have reported successful elective surgical repair of partial thoracic EC. 26
Complete thoracic EC, if left untreated, is universally fatal, 27 and there are few reported survivors following surgical correction. Of four reported survivors beyond one month, only one had an intracardiac lesion corrected successfully. Three patients who survived the initial neonatal surgical correction later died from unrelated causes. 28
Exencephaly
Exencephaly (also known as acrania) is the exposure of a well-developed and differentiated brain exposed to amniotic fluid during the embryonic period. 3 It is a developmental sequence of events that leads to anencephaly consisting of three phases: (1) dysraphia, or a failure of the neural groove to close in the rostral region; (2) exencephaly, or well-developed brain outside the skull during the embryonic period; and (3) disintegration of the exposed brain during the fetal period, resulting in anencephaly. It is in the second phase, when imaged sonographically, that exencephaly can be visualized (Figure 6).29,30
Exencephaly is a lethal neural tube defect abnormality that manifests as absence of the cranial bones. Only a small number of cases have been reported. Although it may have a similar appearance to acalvaria, it can be distinguished by the large amounts of disorganized brain tissue projecting from the base of the skull. 4
This condition starts in week four of gestation when the mesenchymal tissue fails to migrate, preventing bone formation over the cerebral tissue. 31 The overlying ectoderm of the future cerebral hemisphere, which normally becomes the epidermis of the scalp, becomes an amnion-like transparent sac. 32 If imaging occurs while the brain is still intact, it would be demonstrated sonographically as a bilobed-shaped head, termed the “Mickey Mouse” head, with a relatively large amount of brain tissue present.
Exencephaly is the second of three phases in the development of anencephaly. 5 “Prolonged exposure of the externalized cerebral tissue to amniotic fluid and perhaps repeated trauma is thought to cause compression and/or destruction of the tissue, which then is replaced by the area cerebrovasculosa, as seen in the anencephalic fetus.” 33 Anencephaly is one of the most common NTDs, with a reported incidence of 1 in 1000. There is an increased risk with a family history of NTDs, and if one parent has an NTD, the risk is as high as 5%. 4
Amniocentesis
When a fetus has multiple developmental anomalies, an amniocentesis is often suggested. Although the patient in this case twice declined the procedure, it is often a recommended part of patient management, and for completeness, the amniocentesis procedure is discussed as it relates to this case.
Amniocentesis is the aspiration of a small amount of amniotic fluid through a needle inserted through the maternal abdomen into the amniotic sac for diagnostic analysis of fetal genetics, maturity, or disease. 6 This procedure is usually performed between 15 and 18 weeks’ gestation for genetic reasons. 31 The risk of a miscarriage when an amniocentesis is performed during this period has not been clearly defined. 34 When performed in the middle of the second trimester, fetal loss has been reported to be 8.0% in the United States. 31 Although it is possible to perform an amniocentesis after 20 weeks, there is a risk of poor cell growth in the sample.
Using sonography to guide the needle into the amniotic sac is a technique that reduces the risk of complications to mother and fetus. Care is taken to avoid the fetus, central portion of the placenta, and umbilical cord with the needle. The maternal abdomen is prepared with a chlorhexidine solution, the sonographic probe is covered with a sterile sleeve, and the guide is attached (Figure 7a). Using an aseptic technique, the needle is then inserted into the amniotic sac under continuous sonographic monitoring. This monitoring allows for repositioning of the needle if required (Figure 7b).
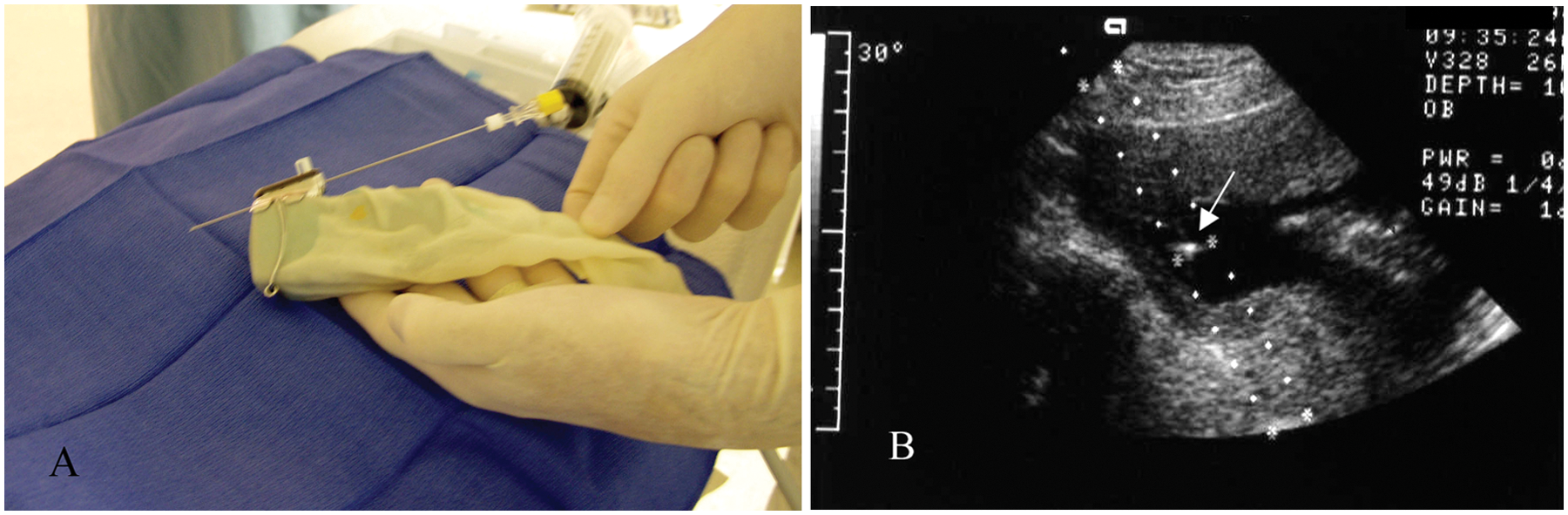
(a) Sonographic transducer in a sterile sleeve demonstrating the needle inserted through the guide attachment. (b) Amniocentesis using a needle guide attached to the transducer while inserting the needle; note the echogenic tip (arrow) of the needle in the amniotic fluid.
The technique used for amniocentesis of multiple gestations is similar to that of a singleton fetus. However, care must be taken to ensure sampling is performed from each fetal sac. To ensure that this is done, indigo carmine dye is injected into the first sac after the sample has been obtained. 31 When accessing the second fetal sac, the lack of blue dye confirms entry into a different sac.
Conclusion
An MSAFP blood test is used to assess a fetus at risk for aneuploidy or structural defect. An NTD with an opening in the spinal cord or brain will result in elevated levels. One of the more common NTDs is anencephaly, with a reported incidence of 1 in 1000. 4 Exencephaly (also known as acrania) is the exposure of a well-developed and differentiated brain exposed to amniotic fluid during the embryonic period. 3 It is a lethal NTD that manifests as absence of the cranial bones. Only a small number of cases have been reported.
First described by Stevenson in 1671, ectopia cordis is a rare congenital cardiac anomaly in which the heart is partly or completely located outside the thorax. 1 The occurrence rate for EC is from 5.5 to 7.9 per million live births. 4 Because of the rarity, there appears to be no specific technique for surgeons to handle a neonate with ectopia cordis, and every case appears to have unique features. 35 To our knowledge, this is the first reported case in which EC and exencephaly have been documented in a single fetus in a twin pregnancy.
The presence of an NTD significantly increases the risk for recurrence, warranting close monitoring for all subsequent pregnancies. 36 This patient’s subsequent pregnancy resulted in a normal fetus without evidence of NTD or thoracic defect.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.