
Abstract
Gastrointestinal stromal tumors or GISTs belong to a category of gastrointestinal mesenchymal tumors that can have benign or malignant potential. These tumors were formerly identified as gastrointestinal (GI) leiomyomas, leiomyoblastomas, or leiomyosarcomas. GISTs account for approximately 80% of GI mesenchymal tumors and 3% of all childhood tumors. Pediatric GISTs occur in patients younger than 18 years and are extremely rare. In the United States, pediatric GISTs have been reported in the literature less than 100 times. In addition, much research has failed to distinguish between benign and malignant mesenchymal tumors. This makes it extremely difficult for medical professionals to develop any type of expertise because few see more than one or two of these cases in a career, and of those cases, many never differentiate between the two. This case study involves a young patient who had been diagnosed with pediatric GIST. This report details the initial sonographic findings and the follow-up studies that had initially suggested a pediatric GIST of malignant potential but, upon further outside consultation, suggested a benign pediatric GIST.
Case Report
An 11-year-old boy presented to the emergency room for intractable vomiting. His mother informed the emergency room physician that her son had begun complaining of a stomachache two days earlier. The next day he had a low-grade fever, vague left-sided abdominal pain, and vomiting. The vomiting persisted and she took him to see his primary care physician, who prescribed an antinausea drug. The vomiting progressively worsened and the patient presented to the emergency room for evaluation. His medical and family histories were unremarkable. Physical examination showed a palpable abdominal mass that was initially suspected to be the spleen. Laboratory findings showed an elevated white blood cell count and decreased hemoglobin. Liver function enzymes (LFTs) were unremarkable except for an elevated glucose level. Based on presenting signs and symptoms, a sonogram was ordered to evaluate his pain and assess the spleen.
An abdominal sonogram was performed using an Acuson Sequoia (Siemens Medical Solutions USA, Inc, Malvern, Pennsylvania) and a 4.5 MHz vector transducer. Sonographic imaging demonstrated a 5.6 × 5.6-cm complex, centrally necrotic mass adjacent to the left lobe of the liver (Figure 1). This midline abdominal mass illustrated vascularity around the periphery and appeared to be solitary (Figure 2). The interpreting radiologist initially reported it as a focal nodular hyperplasia (FNH) arising from the caudate lobe or the left lobe of the liver. Differential diagnoses that were reported were a gastrointestinal lymphoma, pancreatic mass, or gastric tumor. The rest of the examination was negative, with the exception of a small amount of abdominal ascites. These abnormal findings prompted an abdominal and pelvic computed tomography (CT) examination, which again demonstrated a large, centrally necrotic mass that was arising from the anterior wall of the gastric body (Figure 3). It compressed the left lobe of the liver and the tail of the pancreas. A moderate amount of free fluid and fat stranding was noted. The CT findings were suggestive of gastrointestinal stromal tumor or gastric lymphoma. However, these tumors are exceedingly rare in the pediatric age group, and further assessment with a biopsy was recommended.
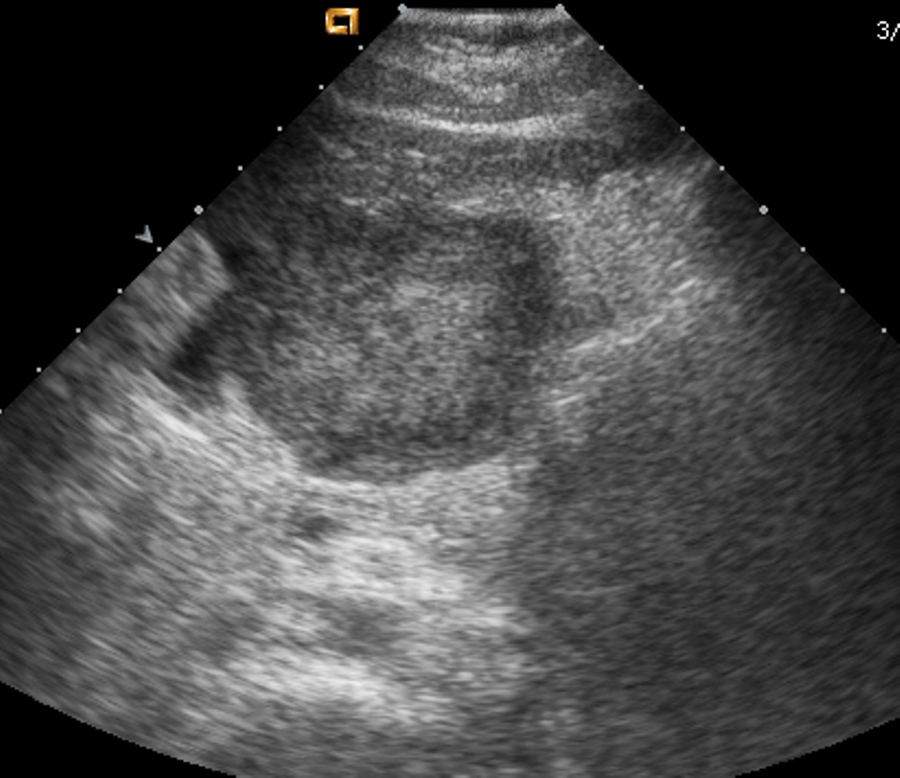
Imaging of the solitary mass in the epigastric region illustrating central necrosis. Longitudinal left lobe of liver.
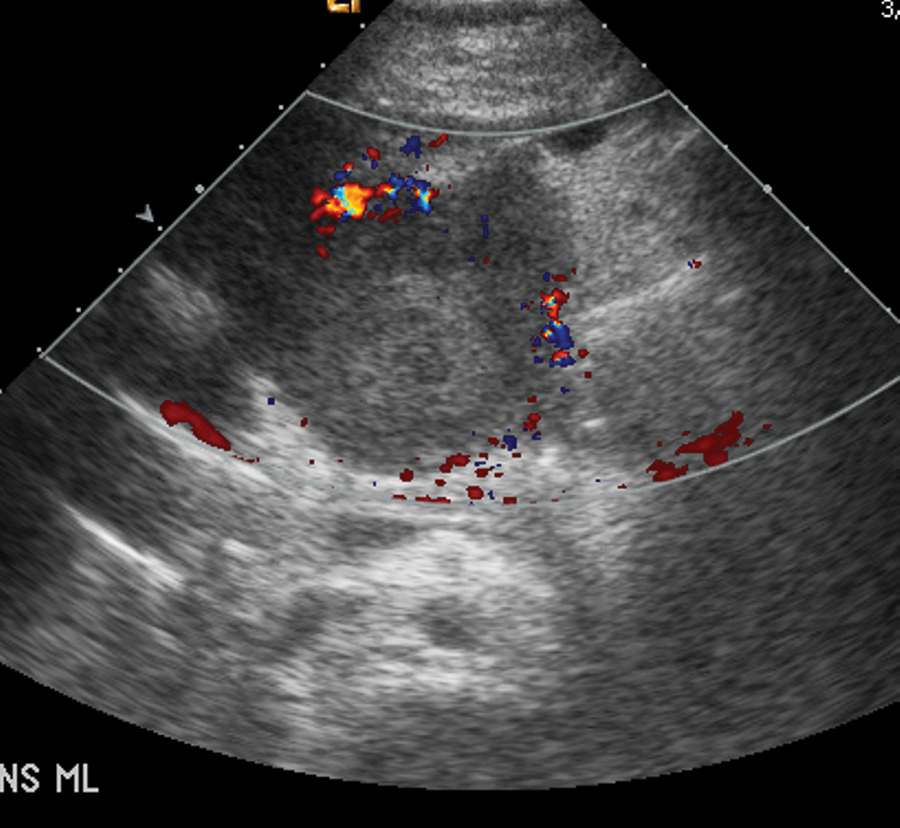
Sonogram of a well-demarcated solitary mass with peripheral vascularity. Transverse midline.
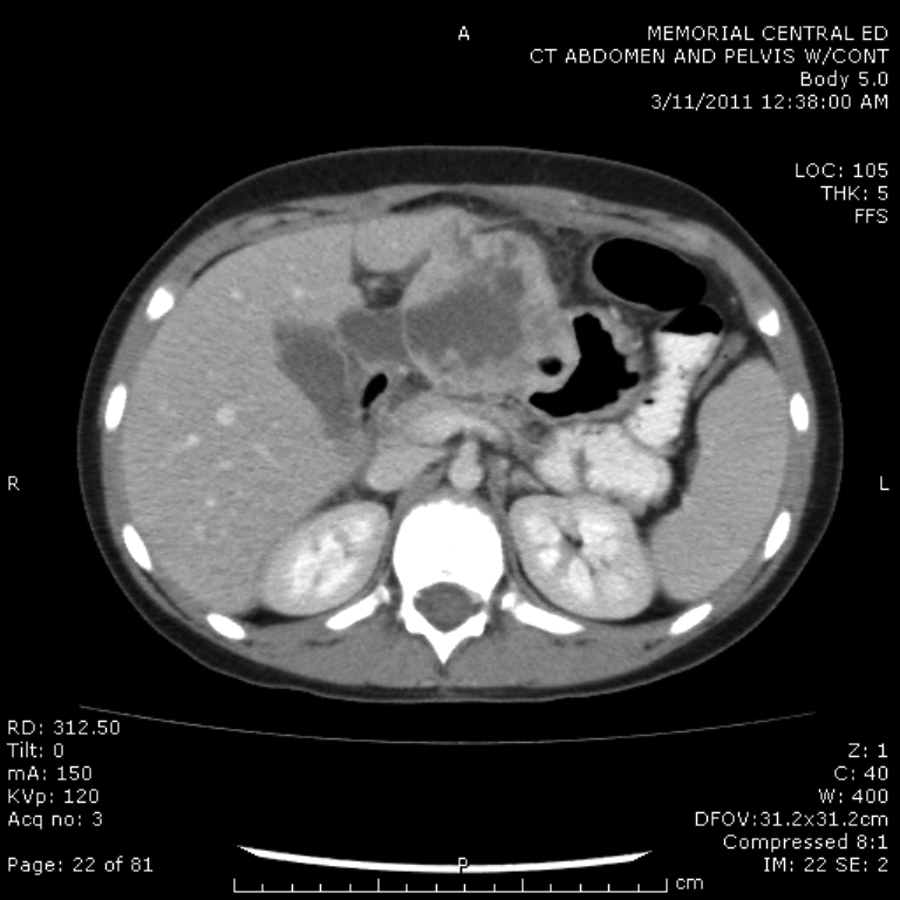
Computed tomography imaging showing a solitary, well-circumscribed lesion that appears to arise from the wall of the stomach. The mass has the appearance of a gastrointestinal stromal tumor.
The patient was admitted to the hospital for an endoscopic biopsy. The biopsy specimens were inconclusive but suggestive of a GIST or spindle cell tumor. Surgery was then scheduled and performed for gastric tumor resection and partial gastrectomy. The tumor arose from the lesser curvature of the stomach and was heavily wrapped in omentum, from which it derived a huge portion of its blood supply. There did not appear to be any metastases to the liver or in the retroperitoneum on palpation. The patient handled the surgery well with blood loss estimated at 500 mL.
The histomorphology of the excised gastric tumor was similar to that of a GIST, but the negative CD117 and CD34 staining and positive smooth muscle actin (SMA) staining favored a diagnosis of a smooth muscle tumor. Given the necrosis and large size of the tumor, it was considered a tumor of uncertain malignant potential. The microscopic description stated that the gastric mass showed a submucosal spindled cell neoplasm with a central area of hemorrhage with necrosis present. The spindled cells were bland with a rate of mitotic activity of <1/high-power field (HPF).
Because of the inconclusive surgical pathology report, an outside expert consultation was performed. The outside impression of the slides and the two blocks from the excised gastric tumor was that the tumor was a cellular smooth muscle neoplasm. There were no obvious or convincing features of malignancy, and the likelihood of recurrence was low.
Discussion
Gastrointestinal stromal tumors are rare tumors of the gastrointestinal tract. These tumors are found in the cells located in the wall of the gastrointestinal (GI) tract commonly referred to as the “pacemaker cells” or the interstitial cells of Cajal. Not all GISTs are cancerous, and more than half start in the stomach, although they can occur anywhere in the GI tract. 1
Gastrointestinal stromal tumors are the most common mesenchymal tumors arising from the gastrointestinal tract in adults.1,2 In children younger than 18 years, they are exceedingly exceptional. It is estimated that there are fewer than 100 cases of pediatric GIST. Not even a third of these are reported in the literature. The most current literature involves the differentiation between GISTs, leiomyomas, and leiomyosarcomas. 3
Pediatric GISTs usually present with one or more tumors in the stomach. The most common symptom is anemia with associated clinical signs such as paleness, fatigue, and vertigo. 4 In some cases, there may be a palpable tumor, abdominal distention, abdominal pain, and/or vomiting. GISTs in children and young adults have a definite predilection toward females and are more likely to be high-grade tumors. In boys, spindle cell tumors are reported more frequently, whereas epithelioid cells are seen in girls.2,5
Pediatric GIST is divided into subtypes. The most common subgroup, referred to as pediatric GIST, occurs between the ages of 6 and 18 years. It is described as an abdominal tumor that almost always originates in the stomach. The second type of pediatric GIST, first described in 1977, is referred to as Carney’s triad. This diagnosis includes having any two of the following: GIST, pulmonary chondroma, and/or functioning extra-adrenal paraganglioma. In 2002, another form of Carney’s triad was introduced. It is similar to the first described Carney’s triad but appears to be inheritable. Termed familial paraganglioma and gastric stromal sarcoma, it occurs between the ages of 9 and 46 years. The last subgroup is called neonatal or congenital GIST because the patient is born with the tumor and requires surgery soon after birth. It affects males and females similarly and seems to originate from the intestines instead of the stomach.6–8
GISTs are well-demarcated spherical masses arising from the muscularis propria layer of the GI tract. GISTs can range in size from a few millimeters to more than 25 centimeters. Larger GISTs almost always exhaust their vascular supply, leading to extensive areas of central necrosis and hemorrhage. Larger tumors tend to be malignant, although the conventional criteria for determining risk of malignancy such as tumor size, mitotic activity, and anatomic location are not reliable in pediatric GIST. 9
The goal of radiologic examinations is to locate the tumors, evaluate local invasion, and detect distant metastases. The single most documented characteristic of GIST is well-circumscribed intramural masses, possibly with overlying ulceration. Larger GISTs appear as complex masses with areas of necrosis, which occur as a result of the tumor outgrowing its blood supply. Most pediatric GISTs tend to arise from the stomach and present with multifocal tumors.
Unfortunately, radiologic findings are nonspecific and may have multiple differential diagnoses. Also, the distinction between benign or malignant GISTs cannot be made unless metastases or tumor invasion into adjacent structures are identified.
Pediatric GIST can be a life-threatening disease. These young patients tend to follow a slow course of disease and therefore have a favorable prognosis. Like any cancer, GIST can metastasize. It will most commonly metastasize to the liver, lymph nodes, and peritoneum. 8 Pediatric GIST does not always metastasize but because recurrence and metastasis are common, follow-up care is critical. 6
The primary treatment for pediatric GIST is surgery. The goal of surgery is to remove the tumor and achieve clear margins. The types of surgery vary from excision of the tumor to a total gastrectomy. In some cases, this provides a good long-term prognosis. GISTs are virtually unresponsive to chemotherapy and radiotherapy. 10 There has been some research with drug therapy; however, the initial data show that drugs are not significantly effective in the treatment of pediatric GIST. 11
In conclusion, although pediatric gastrointestinal tumors are rare, they are becoming increasingly recognized. Until recently, research has been limited. Pediatric gastrointestinal tumors make up a distinct clinical and molecular subset that is independent from adult tumors. Health care professionals who have exposure to these unique tumors need to document their findings. Sonography is an invaluable diagnostic tool for the pediatric population and is an indispensable component of the pediatric workup. It is vital that atypical and remarkable findings be chronicled and reported so that diagnostic guidelines for the management of these challenging tumors can be developed. This case report is offered to provide sonographers the tools and information necessary to be successful in that quest.
Footnotes
The author declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
The author received no financial support for the research and/or authorship of this article.