
Abstract
Background
Myxomatous mitral valve disease (MMVD) is a degenerative disorder marked by excess tissue fibrosis and matrix remodeling, leading to leaflet prolapse. While recent studies linked transforming growth factor beta (TGF-β) activation to MMVD development and progression, upstream regulators of this and other modulatory/causal pathways remain largely unexplored.
Objectives
In this study, we utilized high-throughput sequencing to conduct unbiased analyses of mRNA and microRNA (miRNA) levels in myxomatous and healthy mitral valve tissues, aiming to uncover novel molecular mechanisms involved in MMVD.
Methods
We defined differentially expressed mRNAs and miRNAs transcripts displaying a fold-change > 1.5 and P < .05. Pathway analysis was performed using Ingenuity Pathway Analysis and DAVID, with key findings validated via qRT-PCR. A total of 2378 transcripts were differentially expressed between myxomatous and normal valves. Established pathways, including TGF-β signaling, were confirmed as major contributors to MMVD. Additionally, Random Forests with Boruta Feature Selection identified transcripts with a 95% likelihood of importance, and subsequent pathway analysis on this subset of genes revealed unique signaling pathways.
Results
Most notably, circadian rhythm disruption emerged as a novel, highly ranked pathway in MMVD. Key miRNAs, such as miR-1, miR-133a, and miR-217 were highlighted as highly relevant, with miRNA–mRNA interactions displaying distinct molecular signatures predictive of MMVD.
Conclusions
Collectively, this study represents the first comprehensive analysis of both miRNA and mRNA expression in MMVD, revealing both established and novel disease-associated pathways. The discovery of circadian rhythm disturbances and new regulatory miRNAs suggests promising directions for further research and potential therapeutic targets for nonsurgical treatment strategies in patients with MMVD.
This is a visual representation of the abstract.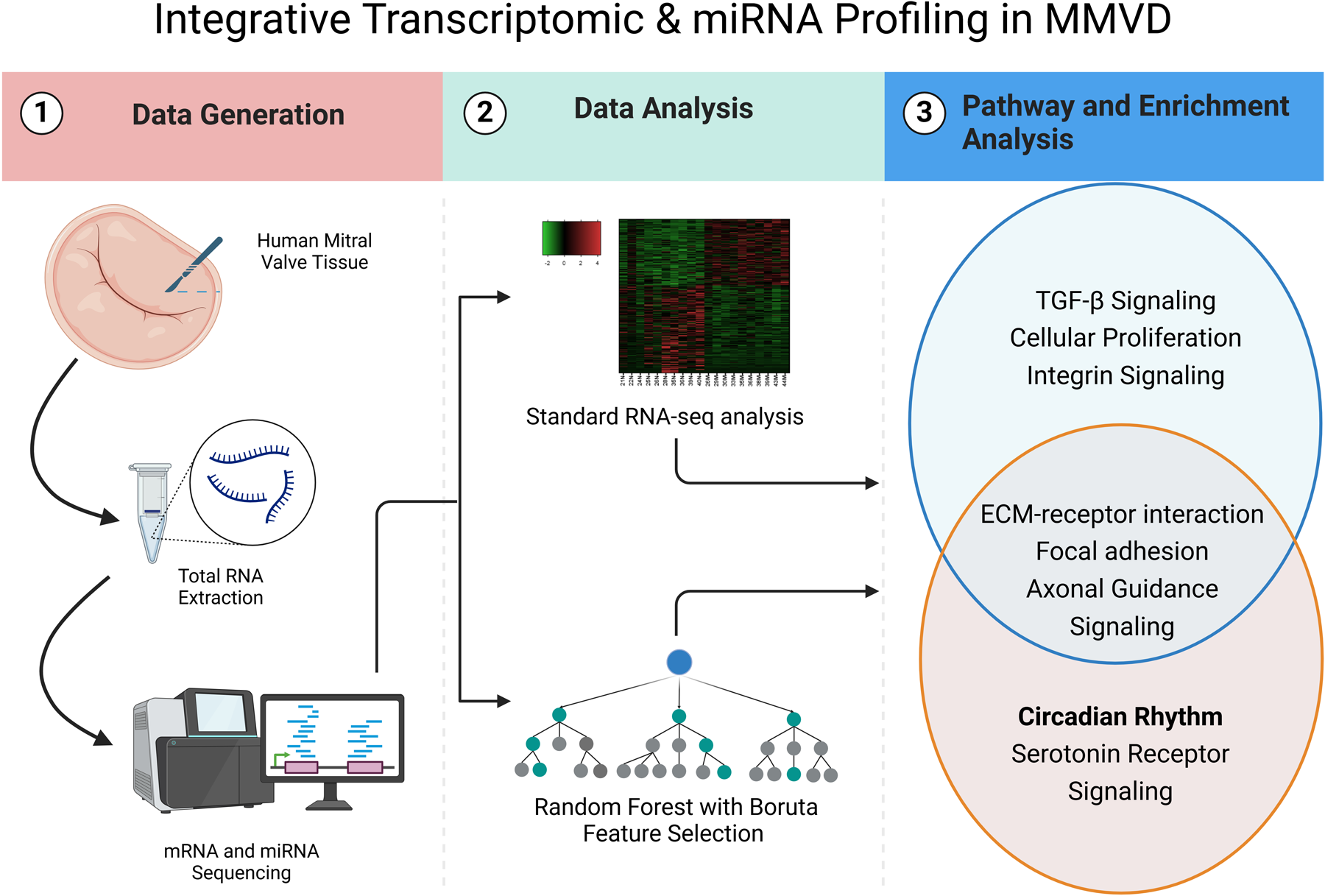
Keywords
Key Points
Myxomatous mitral valve disease (MMVD) is associated with excessive fibrosis and matrix remodeling, with TGF-β signaling playing a key role in disease progression. Previous studies identified molecular pathways contributing to MMVD, but upstream regulators and novel modulators of disease remain largely unexplored. While some circulating miRNAs have been implicated in MMVD, a comprehensive high-throughput analysis of miRNA and mRNA expression within mitral valve tissue has not been performed.
This study provides the first unbiased high-throughput sequencing analysis of both mRNA and miRNA expression in human myxomatous and nonmyxomatous mitral valve tissue. Novel miRNA–mRNA interactions were identified in MMVD tissue, including miR-1, miR-133a, and miR-217. Random Forests with Boruta Feature Selection identified key pathways beyond TGF-β signaling, including GP6 signaling and FOXO hyperactivation. Aberrations in circadian rhythm gene expression emerged as a particularly novel molecular feature of MMVD using this approach, which has mechanistic links to extracellular matrix remodeling and cell proliferation.
The study highlights novel miRNA–mRNA interactions and unique regulatory networks that could serve as future therapeutic targets for MMVD.
Introduction
Myxomatous mitral valve degeneration (MMVD) is characterized by progressive structural changes in the mitral valve leaflets, leading to mitral valve prolapse (MVP), regurgitation, and ventricular dysfunction.1–3 Despite being a common condition with significant clinical implications, the precise molecular mechanisms driving MMVD development and its progression remain elusive, warranting comprehensive investigations to deliver new insights into disease etiology and therapeutic strategies.
Transforming growth factor beta (TGF-β) signaling pathway stands out as a central player in MMVD contributing to tissue fibrosis and matrix remodeling, however, its upstream and downstream effectors in the context of myxomatous degeneration remain incompletely characterized.4–7 Although preclinical studies suggested a role for the use of angiotensin II receptor blockers (ARBs) to mitigate TGF-β-induced fibrogenic signaling,8,9 recent clinical trials produced conflicting evidence regarding their effect on disease progression in syndromic and nonsyndromic valvular and vascular diseases driven by TGF-β.10–15 These data are largely confirmed by clinical veterinary experience, where ACE inhibition showed little to no benefit in delaying heart failure or reduction mortality in dogs with preclinical MMVD and cardiomegaly. 16 This is not to say that subsets of patients may not benefit, as elderly patients with moderate-to-severe MMVD treated with ACE inhibitors (ACE-I) or ARBs showed improved event-free survival, making them a valuable option for patients requiring more conservative treatment. 17 Recent studies highlight other potential therapeutic opportunities for serotonin (5HT) antagonism, supported largely by evidence of increased risk of mitral prolapse in small groups of patients with elevated 5HT signaling (eg, carcinoid disease), hyperactivation of 5HT signaling in valve tissue from patients with end-stage MMVD, mechanistic work linking hyperactivation of 5HT signaling to induction of matrix remodeling genes in canine mitral valve interstitial cells, and findings that 5HTR2B inhibition slows the progression of MVP in experimental animals. 18 Beyond hyperactivation of TGFβ, 5HT signaling, and matrix remodeling genes, however, additional molecular mechanisms underlying the pathogenesis of MMVD remain poorly understood.
In our previous efforts to identify novel therapeutic targets in MMVD, 7 we used standard, microarray analyses on valve tissue from patients with MMVD and identified TGF-β, bone morphogenetic protein signaling (a TGF superfamily member), Wnt/β-catenin signaling (which can be transactivated by TGF-β), and immune cell profiles as molecular signatures strongly associated with MMVD. Of particular interest was the substantive overlap of these signaling pathways with those observed in calcific aortic valve stenosis, suggesting that the phenotypic consequences of the sustained hyperactivation of these pathways is highly context dependent. Consistent with this hypothesis, a recent study demonstrated that inhibition of Wnt signaling (a pathway implicated in aortic valve calcification and stenosis) reduced myxomatous valve degeneration and immune cell infiltration in a Marfan mouse model, but that this benefit was only evident when treatment was started in early life (ie, prior to 2 months of age). 19 Intriguingly, this observation compliments the work of Norris and colleagues, who found that genetic mutations impairing function of the primary cilia (which is only active during development) are sufficient to cause subclinical aberrations in the extracellular matrix that cause MMVD later in life.20,21
To build on these findings—and in an attempt to expand the field beyond TGF-β signaling—we comprehensively investigated the regulatory networks governing MMVD pathogenesis, and present herein an integrative approach that combines RNA-sequencing data, microRNA (miRNA) profiling, and advanced analytical techniques to enrich analyses based on statistically defined, highly relevant genes. Our rationale for taking this approach is based on the interconnected nature of miRNA–mRNA interactions, where dysregulated miRNAs may be key orchestrators of the global transcriptomic fingerprints observed in MMVD. To handle the complexity of these extensive datasets and identify highly relevant, differentially expressed mRNAs and miRNAs, we employed Random Forests (RF) with Boruta Feature Selection (RF-B) followed by pathway analysis only on those genes with a likelihood of importance greater than 95%. Ultimately, the goal of using this novel dataset and data processing approach was to deepen our understanding of MMVD at the molecular level, elucidate the intricate molecular networks that underlie this complex valvular disorder, and uncover novel therapeutic targets to mitigate MMVD's clinical burden.
Methods
RNA and miRNA Isolation
Intact human mitral valve tissue was pulverized and transferred to a solution of Lysis/Binding Buffer and 0.1% miRNA Homogenate Additive (Ambion, Life Technologies). Total RNA was extracted from lysates using mirVana™ miRNA Isolation spin columns (Ambion, Life Technologies). A table listing all reagents, vendors, and expanded methods is provided in the online supplemental materials.
Nonbiased High-Throughput RNA and miRNA Sequencing
We utilized whole genome MAPRseq v1.2.1.3 for mRNA sequencing and CAP-miRSeq v1.1 for miRNA sequencing to assess mRNA and miRNA expression in nonmyxomatous (n = 10) and myxomatous (n = 10) human mitral valve tissues. 22 Differential RNA expression was determined using linear modeling, with significance thresholds set at P < .05 and fold-change > 1.5. Canonical pathways were identified through Ingenuity Pathway Analyses 23 and DAVID bioinformatics.24,25 Identification of predicted miRNA targets was accomplished using Ingenuity Pathway Analyses and miRbase, 26 miRDB,27,28 and TargetScanHuman 6.2 bioinformatics tools.11,29
Confirmation of Differentially Regulated mRNAs and miRNAs in MMVD
Differential expression of genes based on highest and lowest fold change between nonmyxomatous (n = 43) and myxomatous human mitral valves (n = 43) was confirmed by quantitative real-time polymerase chain reaction on a StepOne Plus RT-PCR machine (qRT-PCR; see expanded methods and Tables SM1-2 in the Data Supplement for gene expression primers used). Confirmation of miRNAs based on highest and lowest fold change was based on unpaired t tests with an alpha of P < .05.
Random Forest With Boruta Feature Selection
The Boruta 30 approach of variable importance measure has been shown to be one of the most powerful 31 variable selection procedure in RF. 32 The essential idea is to create shadow random variables by permuting the values of each original feature independently, and evaluate whether the original features contains more significant information than these shadow variables. These shadow variables serve as a baseline for comparison since they are completely unrelated to the outcome. After the creation of shadow variables, the dataset is then extended by appending these shadow variables to the original features, and RFs are refit on the augmented data. For each feature, the importance score is calculated using the standard RF mechanism (eg, mean decrease in impurity). The importance of each original feature is compared against the distribution of importance scores for the shadow variables. This allows for the computation of a P-value, which quantifies the probability that a feature's importance could have arisen by chance, which is the case of the shadow variables. This comparison-based approach to variable selection is particularly powerful, as it provides an objective baseline derived from the shadow variables, a property not feasible with standard RF variable importance measures alone. As an application of this method, by systematically eliminating variables with low importance, the Boruta approach significantly reduces the dimensionality of the data without sacrificing the predictive power of the model. Furthermore, this process inherently increases the stability of the model by focusing only on truly relevant features.
The RF-B was executed on a computer system with Intel Core i7-975 CPU with 5GB RAM provided by the National Center for Supercomputing Application at UIUC using custom analytical software consisting of Python and R code.
Statistical Methods
All data are reported as mean ± SE. Significant differences in patient characteristics were detected using unpaired t tests for continuous variables, and with χ2 (when group observations were >5) or Fisher exact test (when group observations were <5) for categorical variables. In our validation datasets using qRT-PCR, significant differences between nonmyxomatous and myxomatous human valve tissue were detected using unpaired t tests.
Results
Differentially Regulated Pathways Based on Fold-Change mRNA-seq Data
Analysis of our mRNA-seq dataset yielded 2378 differentially regulated mRNA transcripts in myxomatous mitral valves (see Supplemental Table SM5). Of all differentially regulated genes, 1002 were upregulated and 1376 were downregulated reflecting a wide range in fold change (see Figure 1A and B). Using qRT-PCR to validate a subset of these genes (5 most upregulated mRNAs and 5 most downregulated mRNAs based on fold-change), we found the upregulated genes with the greatest fold-change in myxomatous versus nonmyxomatous mitral valves were: cellular retinoic acid binding protein 2, phosphoinositide interacting regulator of transient receptor potential channels (PIRT), apelin (APLN), and SH3 and multiple ankyrin repeat domains 2 (SHANK2). ATPase phospholipid transporting 10B (ATP10B), however, was surprisingly decreased in myxomatous valves (P ≤ .05, see Figure 1C-G). The genes exhibiting the most significant downregulation in terms of fold change by RNA sequencing methodologies were: myosin heavy chain 6 (MYH6), matrix metallopeptidase 3 (MMP3), myosin light chain 7 (MYL7), natriuretic peptide A (NPPA), and troponin I3, cardiac type (TNNI3). All were confirmed to have significantly decreased expression in myxomatous mitral valves by qRT-PCR (P ≤ .05, see Figure 1H-L). The activation of fibrosis, matrix remodeling, and cellular proliferation pathways are hallmarks in MMVD which was reaffirmed in our Ingenuity Pathway Analysis (IPA) and DAVID analysis of differentially regulated genes based on fold-change. More specifically, and as shown in Table 1, we found differential regulation of genes related to hepatic fibrosis, extracellular matrix–receptor interaction, focal adhesion and integrin signaling cascades.
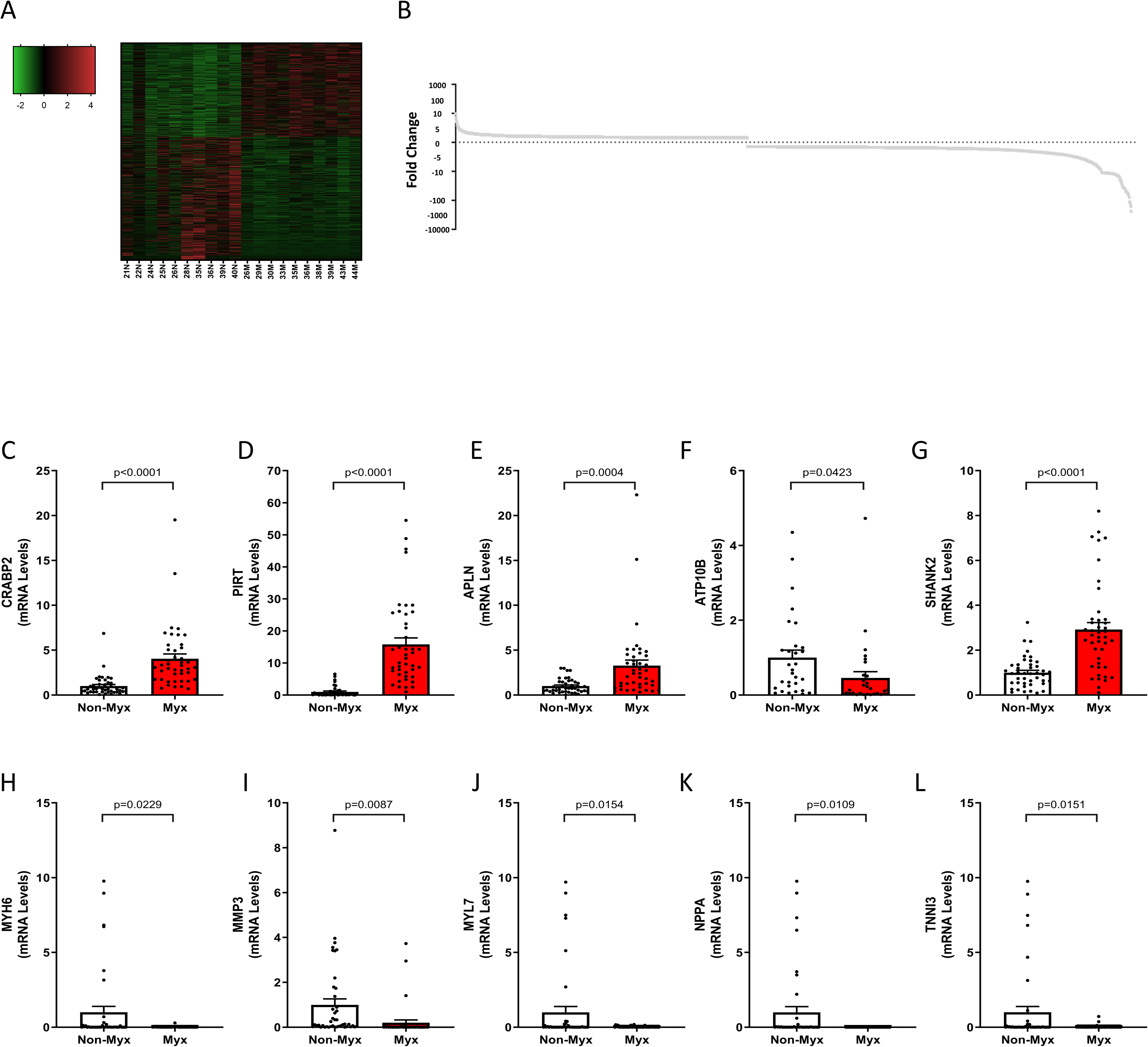
Standard mRNA-seq analysis identified 2378 differentially regulated transcripts in myxomatous mitral valves, with 1002 upregulated and 1376 downregulated genes. We performed validation experiments using independent tissue samples from 43 normal and 43 myxomatous mitral valves and were able to validate 9 of the 10 most differentially regulated genes. Notably, key upregulated genes in the RNA-seq dataset that were successfully validated included CRABP2, PIRT, APLN, and SHANK2, while ATP10B was unexpectedly decreased. Reciprocally, key downregulated genes in the RNA-seq dataset that were successfully validated included MYH6, MMP3, MYL7, NPPA, and TNNI3. All changes were significant (P < .05), comparing myxomatous to non-myxomatous valves (n = 43, 43).
Differentially Regulated Pathways Based on a Subset of Highly Relevant mRNA Species From RF-B
After RF-B of 2378 differentially regulated mRNA transcripts, 588 mRNAs (394 upregulated, 194 downregulated) were identified by the Boruta Feature Selection procedure as highly relevant. Among these, 459 mRNAs (comprising 325 upregulated and 134 downregulated) had a likelihood of importance of at least 95% (P ≤ .05). Examining the differentially expressed genes using RF-B, we noticed a significant predominance of upregulated genes in myxomatous valves (Figure 2A). However, when sorted by likelihood of importance, we observed a more balanced distribution of upregulated and downregulated genes, suggesting that gene significance is not solely determined by fold-change magnitude (Figure 2B and C). We validated the highly relevant mRNAs identified through our RF-B analysis using RT-qPCR. In myxomatous valves, we observed significantly elevated gene expression levels of the most highly relevant upregulated mRNAs: synaptotagmin 17 (SYT17), plexin domain containing 1 (PLXDC1), WW domain containing E3 ubiquitin protein ligase 2 (WWP2), TGFB3, and collagen type V alpha 1 chain (COL5A1) (P ≤ .05, see Figure 2D-H). Conversely, we noted marked reductions in mRNA levels of FOS-like 2, AP-1 transcription factor subunit (FOSL2), Rho GTPase activating protein 10 (ARHGAP10), interleukin 10, salt inducible kinase 1 (SIK1), and homeodomain interacting protein kinase 2 (P ≤ .05, Figure 2I-M). Ingenuity Pathway Analysis revealed differentially regulated genes involved in signaling cascades similar to the analysis based solely on fold-change such as hepatic fibrosis and ECM–receptor interaction. However, there was robust evidence of highly relevant differentially regulated genes associated with circadian rhythm and axonal guidance signaling pathways (Table 2).
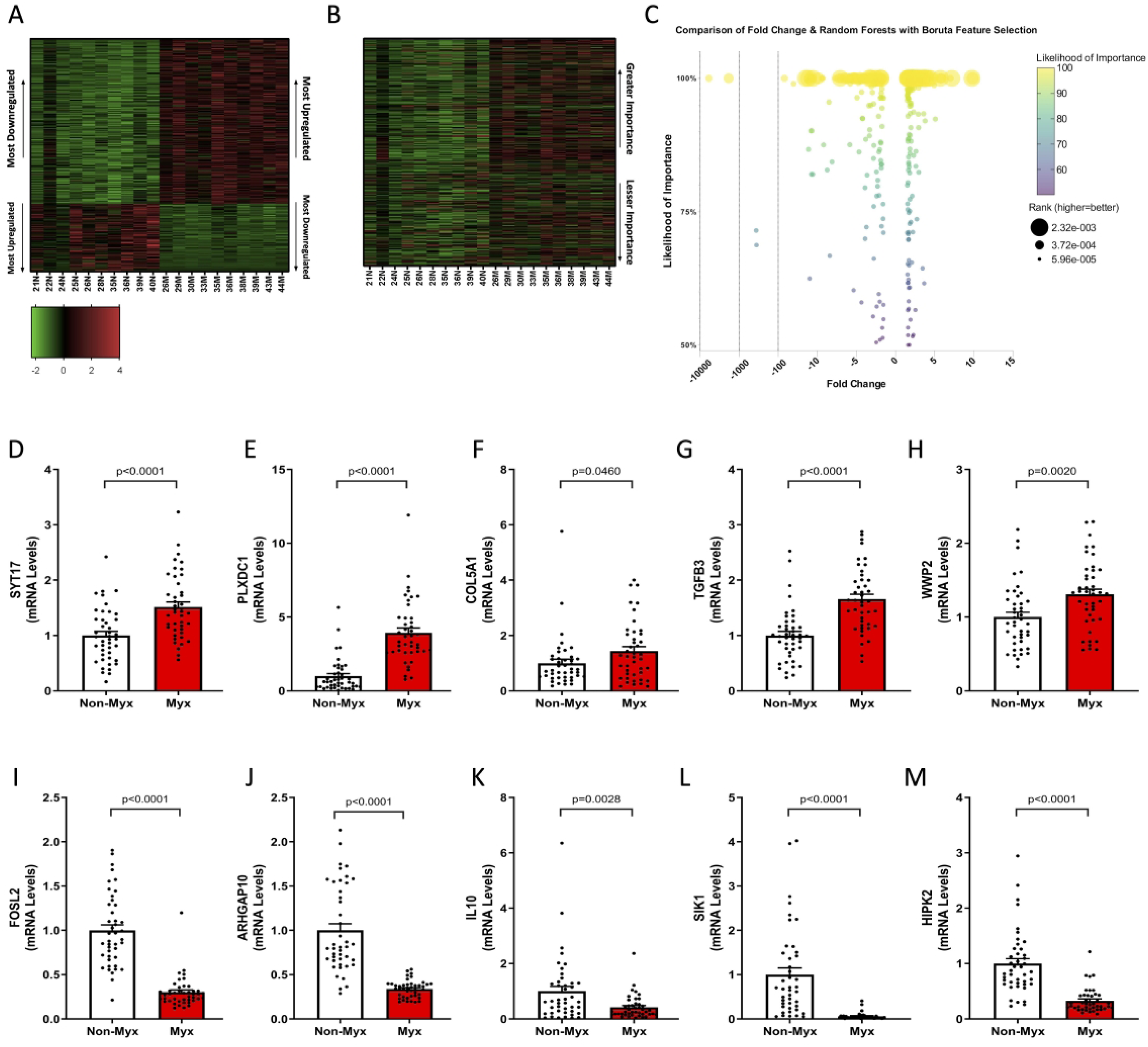
Random forests with Boruta feature selection of the 2378 differentially regulated mRNA transcripts revealed 588 highly relevant mRNAs (394 upregulated, 194 downregulated). Of these 588 highly relevant mRNAs, 459 mRNAs (325 upregulated, 134 downregulated) showed a likelihood of importance of at least 95% (P ≤ .05). While upregulated genes were predominant in myxomatous valves (A), sorting by likelihood of importance (B) suggested a nuanced distribution, with upregulated genes outweighing downregulated genes when plotted by importance/ranking. Further evidence suggesting consideration of biological significance beyond fold-change can be inferred from Panel C, where there is clear unlinking of the association between fold change, likelihood of importance, and rank. We performed validation experiments using independent tissue samples from 43 normal and 43 myxomatous mitral valves and were able to validate 10 most important differentially regulated genes. Notably, key upregulated genes in the RNA-seq dataset that were successfully validated included SYT17, PLXDC1, WWP2, TGFB3, and COL5A1. Reciprocally, key downregulated genes in the RNA-seq dataset that were successfully validated included FOSL2, ARHGAP10, IL10, SIK1, and HIPK2. All changes were significant (P < .05), comparing myxomatous to non-myxomatous valves (n = 43, 43).
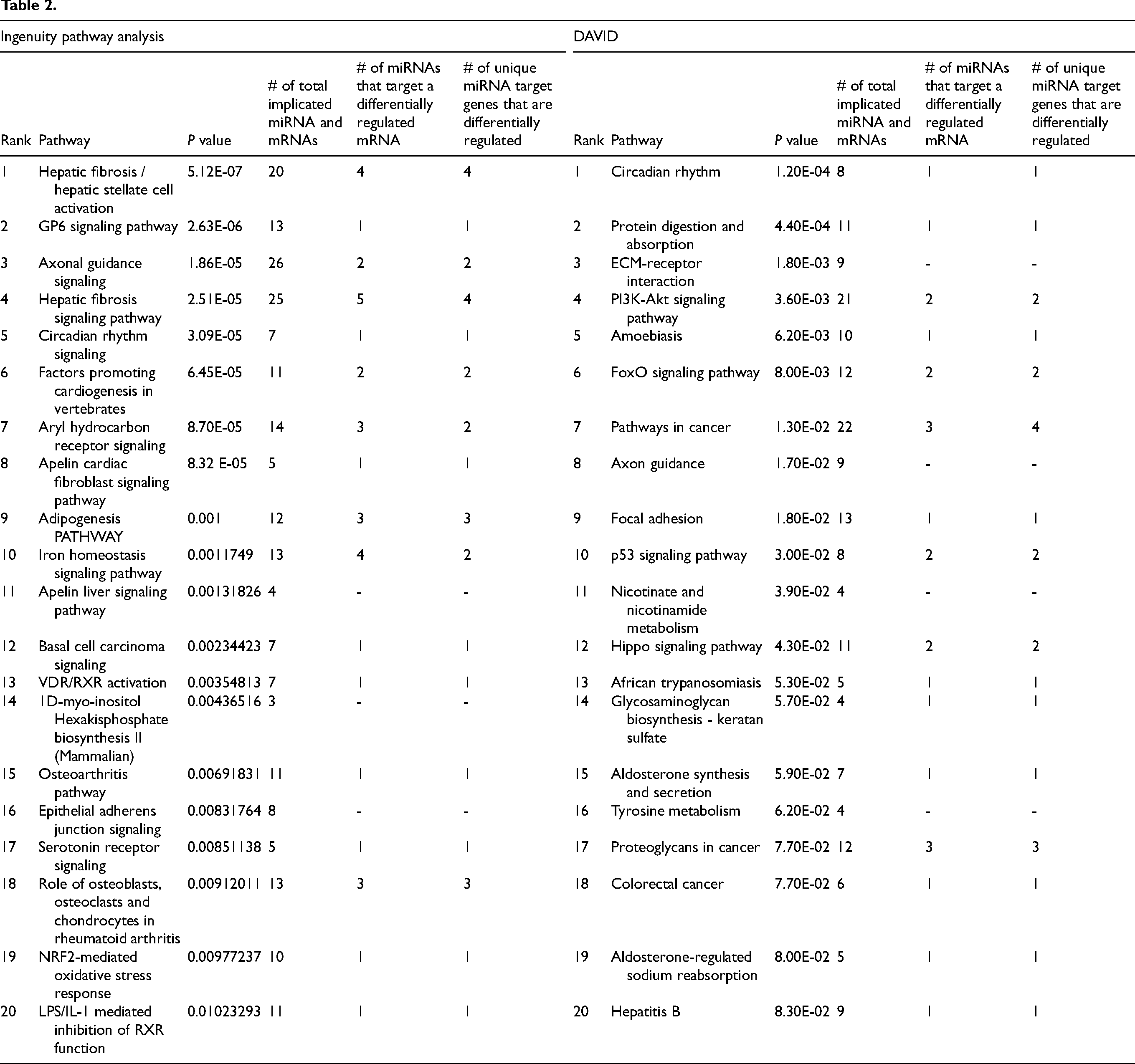
Analysis of miRNA-seq Data Based on Fold-Change
From the miRNA-seq dataset we observed 67 miRNAs differentially regulated between nonmyxomatous and myxomatous mitral valves with 24 miRNAs upregulated and 43 downregulated (see heat map in Figure 3A). For data validation, we examined the miRNA molecules with the greatest fold change using quantitative real-time PCR (5 most upregulated mRNAs and 5 most downregulated mRNAs based on fold-change). The upregulated miRNAs with the greatest fold change were hsa-miR-210 and hsa-miR-34c-5p, and both were confirmed to be significantly increased in myxomatous mitral valves (n = 43) compared to nonmyxomatous (n = 43, P ≤ .05, Figure 3F and G). Conversely from our validation data, we found no difference or slight increases in hsa-miR-216a-5p, hsa-miR-216a-3p, and hsa-miR-217 (Figure 3C-E). The most downregulated miRNAs with the greatest fold-change included hsa-miR-1, hsa-miR-133a, hsa-miR-499a-5p, and hsa-miR-143-5p, and all were significantly decreased in myxomatous mitral valves compared to nonmyxomatous mitral valves when validated with qRT-PCR (Figure 3H-J). miR490-3p failed to meet the threshold for statistical significance when validated by qRT-PCR (Figure 3L). MicroRNA-seq analysis of differentially regulated miRNA molecules using IPA identified 60 significant diseases and biological functions including cancer, organismal injury, and abnormalities, as well as cellular proliferation, fibrosis, and cellular senescence.
Standard miRNA-seq analysis revealed 67 differentially regulated miRNAs in myxomatous mitral valves, with 24 upregulated and 43 downregulated. Validation experiments on independent tissue samples (n = 43 Each) confirmed 6 of the top 10 differentially regulated miRNAs. Among validated upregulated miRNAs were HsamiR-210 and hsa-miR-34c-5p, While hsa-miR-217, hsa-miR216a-5p and hsa-miR216a-3p tended to increase but lacked significance. Conversely, validated downregulated miRNAs included hsa-miR-1, hsa-miR-133a, hsa-miR-499a-5p, and hsa-miR-143-5p, while hsa-miR-490-3p showed a decreasing trend but did not reach statistical significance. All changes significant (p < 0.05), comparing myxomatous to non-myxomatous valves (n = 43, 43).
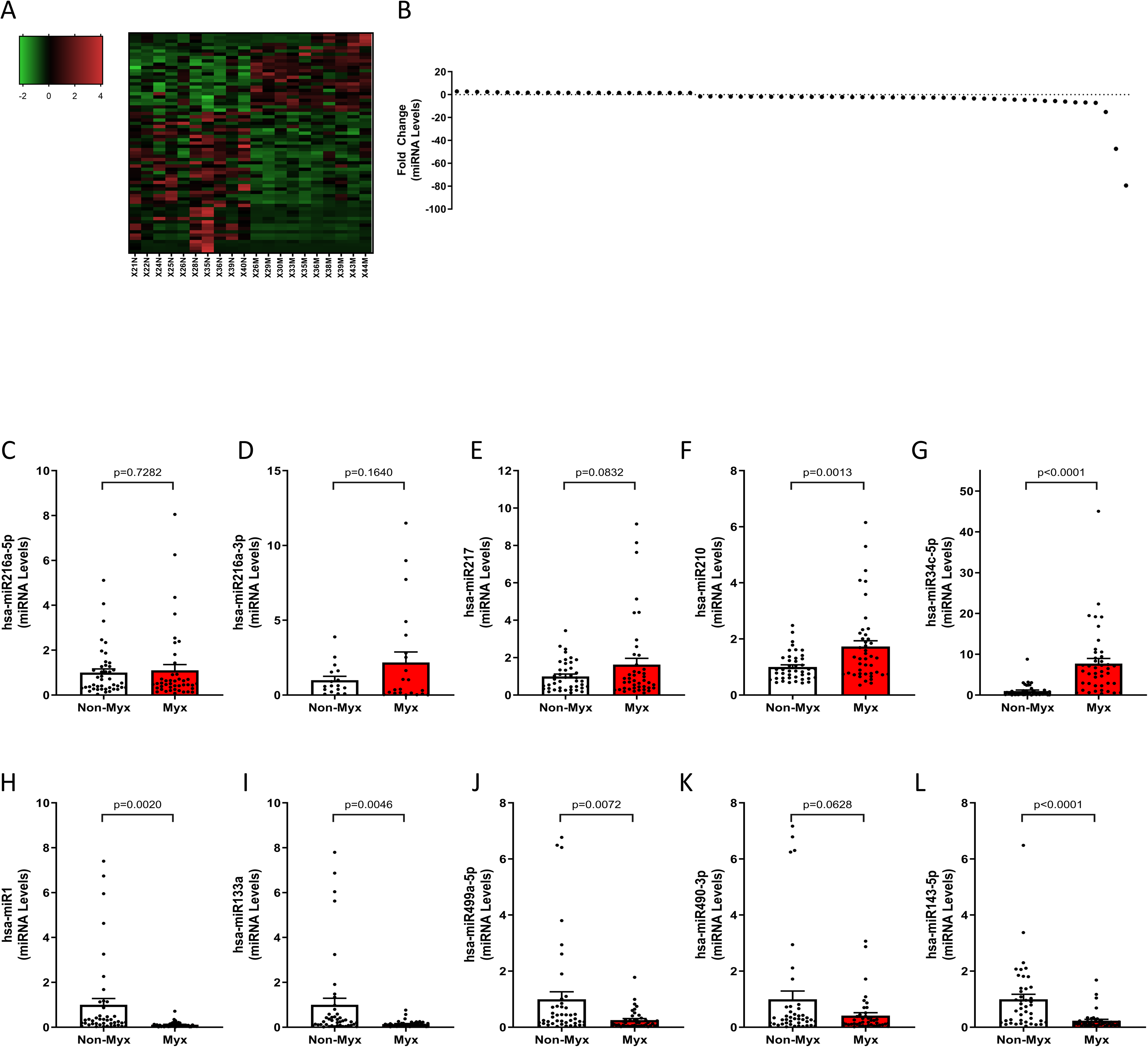
Analysis of miRNA-seq Data Based on RFs With Boruta Features Selection
Random Forests with Boruta Feature Selection identified 43 miRNAs (17 upregulated, 26 downregulated) as highly relevant, of which 34 miRNAs (15 upregulated, 19 downregulated) had a likelihood of importance of at least 95% (P ≤ .05, Figure 4A-C). Using qRT-PCR we confirmed that levels of the miRNAs hsa-miR-363-3p, hsa-miR-223-5p, hsa-miR-337-3p, hsa-miR-133a, hsa-miR-1, hsa-miR-654-3p, and hsa-miR-411-5p were significantly different between myxomatous and nonmyxomatous tissues, whereas hsa-miR-758-3p, hsa-miR-7-5p, and hsa-miR-296-3p did not reach statistical significance in this validation step (P ≤ .05, see Figure 4D-M). Two miRNAs, hsa-miR-133a and hsa-miR-1, were featured as highly significant in both analytical approaches (ie, pure fold-change and RF-B). Of the 55 disease and biological functions found, IPA largely categorized these miRNAs into psychological disorders, cancer, organismal injury and abnormalities, cellular proliferation, and cardiovascular diseases.
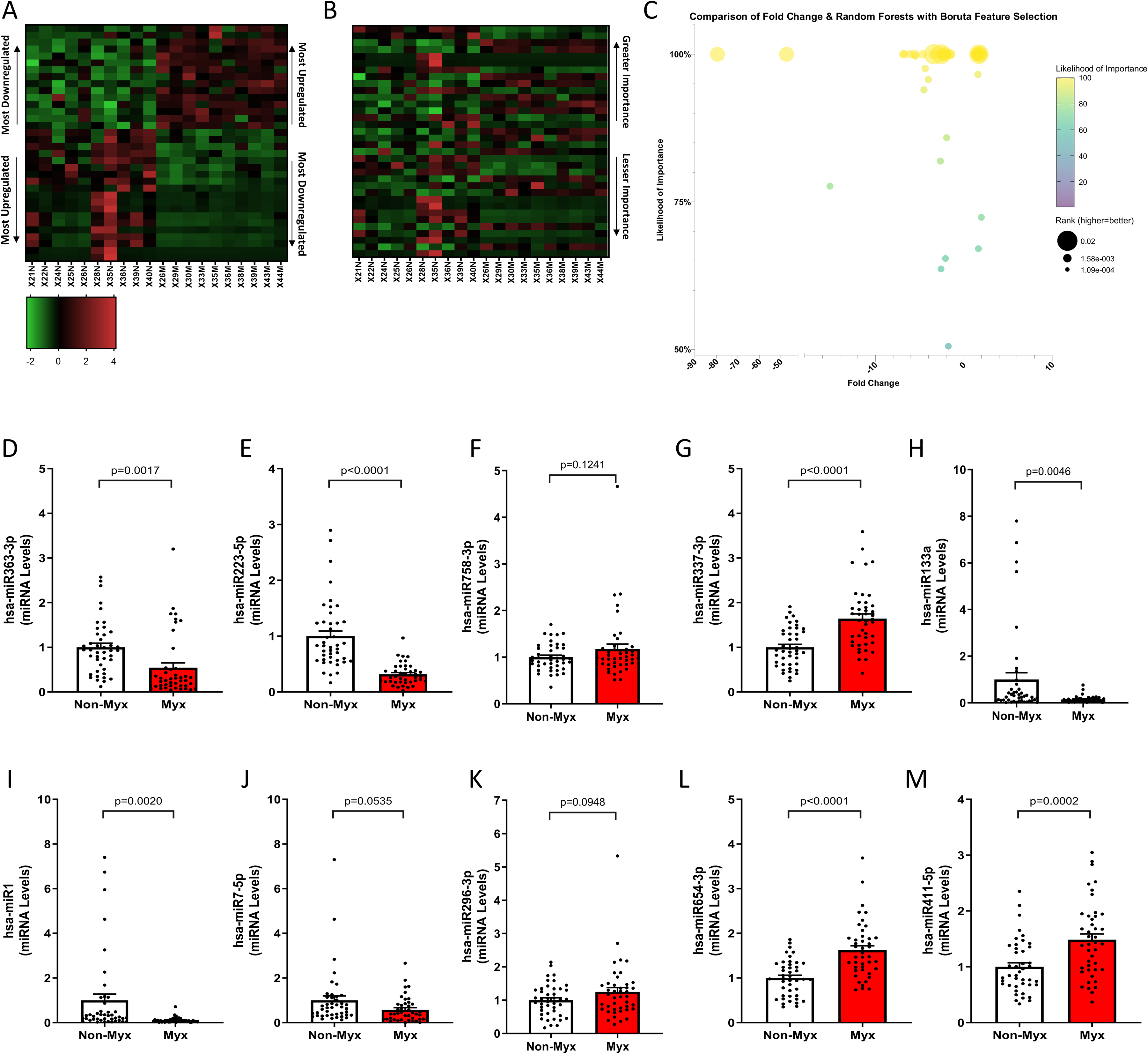
Random forests with Boruta feature selection of the 67 differentially regulated miRNA transcripts identified 43 highly relevant miRNAs (17 upregulated, 26 downregulated), with 34 miRNAs (15 upregulated, 19 downregulated) showing at least 95% likelihood of importance (P ≤ 0.05). While downregulated miRNAs outweighed in myxomatous valves (A), sorting by importance (B) revealed a nuanced distribution with greater importance. Further evidence suggesting consideration of biological significance beyond fold-change can be inferred from Panel C, where there is clear unlinking of the association between fold change, likelihood of importance, and rank. Validation experiments on independent tissue samples (n = 43 each) confirmed 7 of the 10 most important differentially regulated miRNAs. Validated miRNAs included hsa-miR- 363-3p, hsa-miR-223-5p, hsa-miR-337-3p, hsa-miR-133a, hsa-miR-1, hsa-miR-654-3p, and hsa-miR-411- 5p, while hsa-miR-758-3p, hsa-miR-7-5p, hsa-miR-296-3p did not reach significance (P < .05), comparing myxomatous to non-myxomatous valves (n = 43, 43).
MicroRNA–mRNA Interactions Within Each Analytic Subset (Fold Change/RFs)
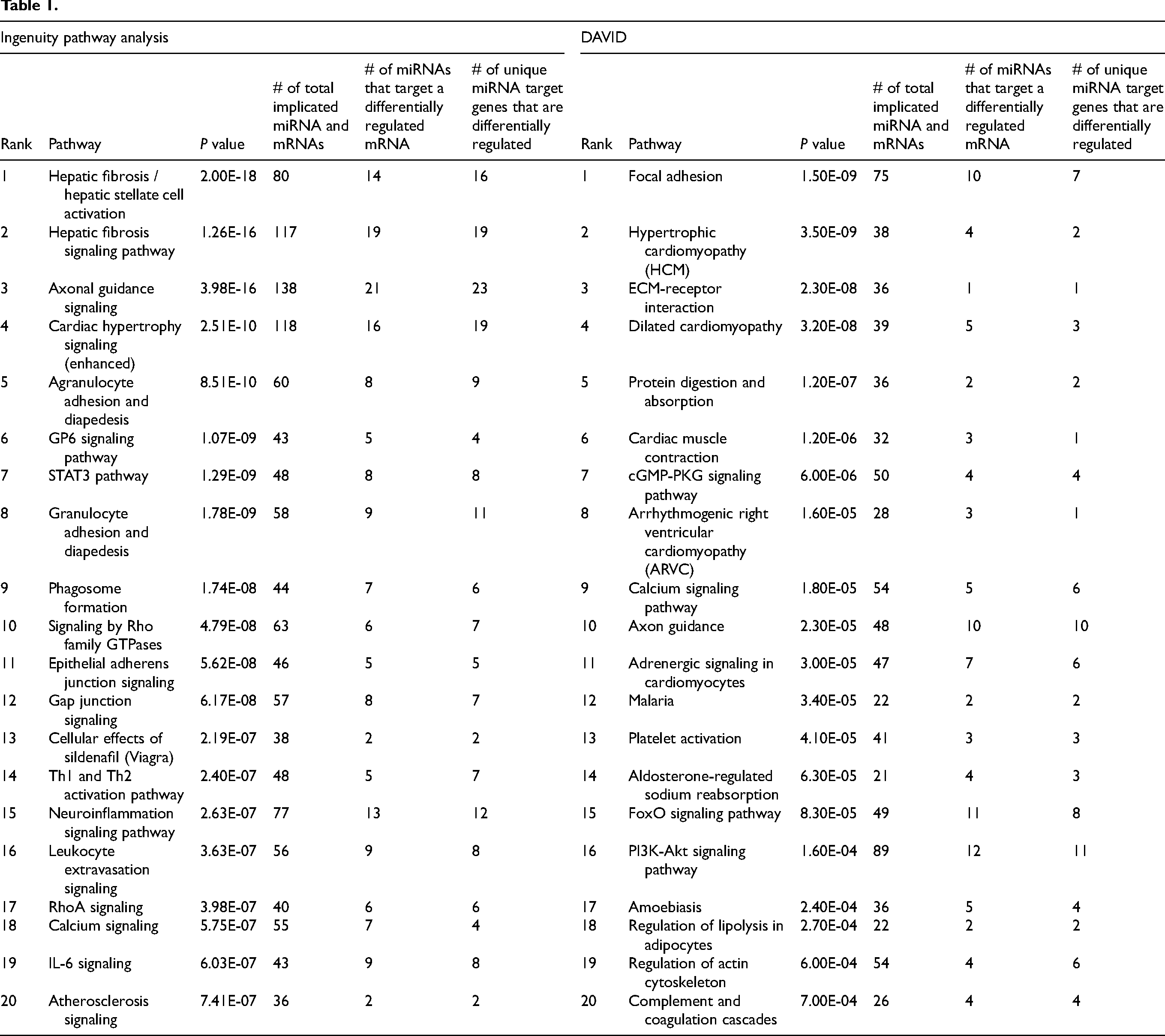
To integrate these two datasets, we first took highly differentially regulated miRNA species and determined whether they were strongly associated with genes clustered in highly differentially regulated signaling pathways from our pathway analyses (IPA, DAVID). As one miRNA can have multiple mRNA targets and likewise one mRNA can be targeted by various miRNAs we examined the total number of implicated miRNA and mRNAs species and also noted the number of unique miRNAs and mRNA in each differentially regulated pathway. In our highest ranked differentially regulated pathway, Hepatic Fibrosis / Hepatic Stellate Cell Activation, we identified a total of 80 miRNA–mRNA interactions between 14 distinct miRNAs and 16 mRNAs, and four such miRNA–mRNA interactions included miRNA and mRNA species that were classified as highly relevant based on RF-B modeling. The noteworthy pathways and interactions, prominently ranked through each analytical method, are showcased in Tables 1 and 2.
Validation of Changes in Circadian Rhythm and Integration of miRNA–mRNA Interactions in MMVD
A particularly unique finding was dysregulated expression of genes implicated in the circadian rhythm pathway in myxomatous mitral valve tissues. Upon further investigation of this pathway, we discovered a total of 13 differentially regulated mRNAs (6 upregulated, 7 downregulated, Figure 5A). mRNA levels of a subset of these differentially regulated genes were confirmed using RT-qPCR with the downregulated mRNAs, basic helix-loop-helix ARNT like 1 (BMAL1), neuronal PAS domain protein 2 (NPAS2), nuclear factor, interleukin 3 regulated (NFIL3 [E4BP4]), and RAR-related orphan receptor B (RORB) were significantly decreased in myxomatous valves compared to nonmyxomatous (n = 43, 43) (P < .05, see Figure 5B-E). Upregulated genes included period circadian regulator 2 (PER2), period circadian regulator 3 (PER3), and basic helix-loop-helix family member e41 (BHLHE41 [DEC2]), which were confirmed to be significantly increased in MMVD with a modest increase in cryptochrome circadian regulator 2 (CRY2) and nuclear receptor subfamily 1 group D member 2 (NR1D2 expression) (see Figure 5F-J).
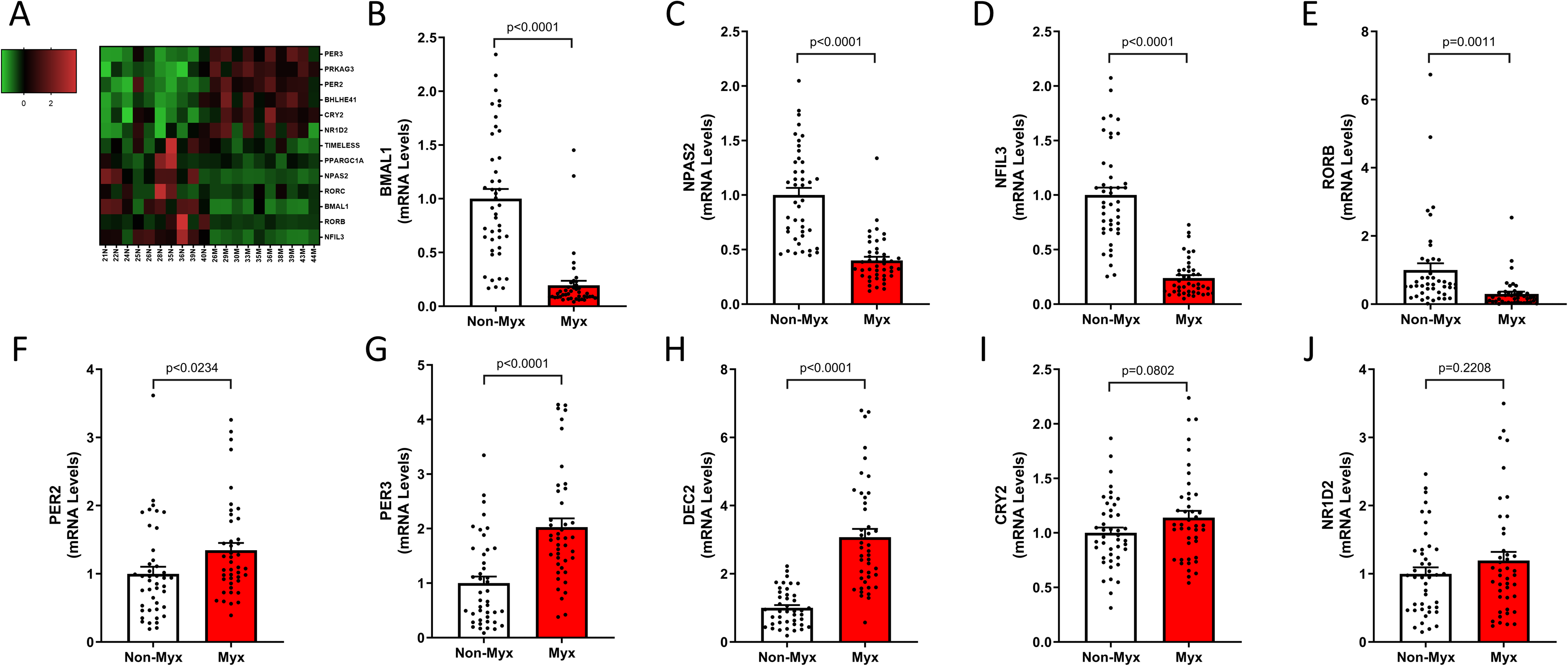
In the circadian rhythm pathway, we found 13 differentially regulated mRNAs (6 upregulated, 7 downregulated). RT-qPCR confirmed downregulation of BMAL1, NPAS2, NFIL3, and RORB, and upregulation of PER2, PER3, and BHLHE41 (DEC2) in myxomatous valves compared to non-myxomatous (n = 43, 43, p < 0.05).
From our integration of the highly differentially regulated circadian rhythm pathway and miRNAs via IPA, we have identified the highly relevant miRNA–mRNA interaction of hsa-miR-133a and PER2. As stated previously, hsa-miR-133a was identified as both a highly ranked based on fold-change and highly relevant miRNA based on our RF-B modeling. We further confirmed by qRT-PCR that miR-133a was significantly decreased and mRNA levels of PER2 were significantly increased in myxomatous mitral valve tissue (P < .05, see Figures 3I and 5F). Figure 6 delineates a model representation of these highly relevant transcriptional changes in relation to circadian rhythm.
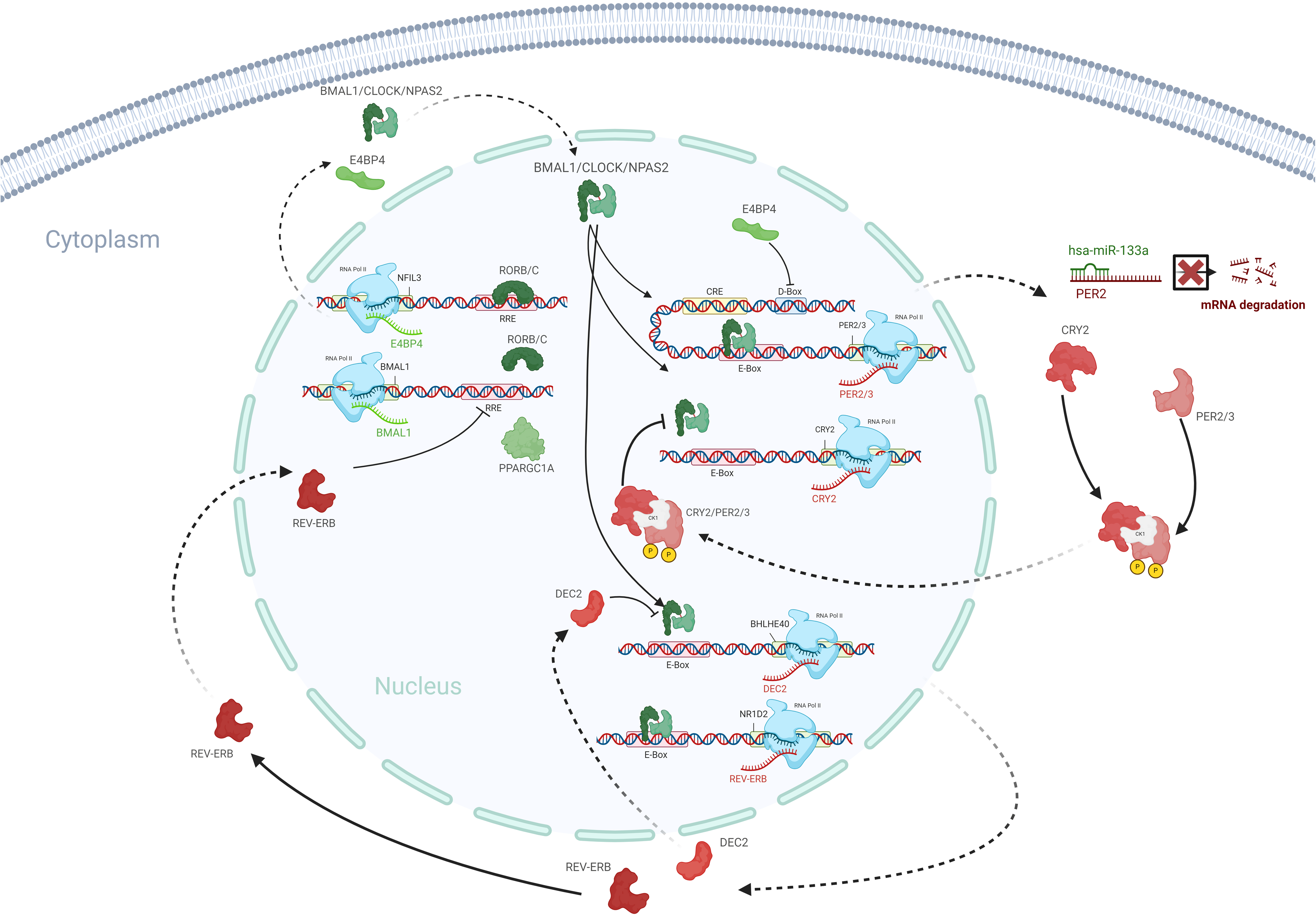
This model outlines the significant transcriptional changes in the circadian rhythm pathway occurring in MMVD, with red denoting upregulated and green indicating downregulated molecules. Integration of mRNA and miRNA data revealed hsa-miR-133a's interaction with PER2, validated by qRT-PCR, showing decreased hsa-miR-133a and increased PER2 expression in myxomatous valves (n = 43, 43, P < .05). Created in BioRender. Hagler (2025) https://BioRender.com/g01f275.
Discussion
To our knowledge, this is the first report of high-throughput sequencing of both miRNA and mRNA species in human myxomatous and nonmyxomatous mitral valve tissue. Our study yielded five key findings: (1) several of the highly differentially regulated mRNAs map on to highly differentially regulated pathways previously implicated in MMVD but also identify several other novel molecular mechanisms not characteristically associated in MMVD, (2) numerous miRNA species are differentially regulated in MMVD, (3) many highly differentially regulated transcripts—for both miRNA and mRNA species—do not predict presence of MMVD using RF-B, (4) miRNA–mRNA interactions and pathway analyses from “highly relevant” transcripts result in significantly different molecular fingerprints predicting presence of MMVD, and (5) our data specifically implicate aberrations of circadian clock gene expression as a novel molecular change strongly associated with presence of MMVD. Collectively, using this exploratory method our findings lay a novel foundation for subsequent research efforts targeting these highlighted molecular pathways and highly relevant regulatory miRNA mechanisms that have previously gone unrecognized in MMVD.
Established and Novel Molecular Contributors to MMVD
Using standard analyses for RNA-seq data, our dataset strongly implicated pathways associated with hyperactivation of TGF-beta signaling and cellular proliferation, in MMVD. This finding is not particularly novel given the extensive body of previous reports implicating TGF-beta in the extensive matrix production and remodeling that are hallmarks of MMVD, including previous reports from our research group.5,7 There were, however, several novel genes identified from our analyses that significantly modulate TGF-beta signaling, including SIK1, WWP2, and LOXL2. Given the challenges associated with the direct, systemic targeting of TGF-beta signaling, exploring therapies that would alternatively target modulators of this pathway may prove to be a more fruitful approach toward attenuating this major biological pathway that is so centrally implicated in the evolution of MMVD.
While standard IPA analyses of a subset of highly relevant genes (identified through RFs) yielded similar findings of TGF-beta hyperactivation, cellular proliferation, and cellular hypertrophy, several other novel pathways associated with MMVD also emerged, including GP6 signaling, molecular signals associated with chemotaxis, hyperactivation of adipogenic signaling, FOXO hyperactivation, and suppression of circadian rhythm. Importantly, these findings extend upon previous reports from our laboratory implicating TGF-beta signaling, BMP signaling, Wnt/beta-catenin signaling, and immune cell infiltration in the presence and progression of MMVD, but also draw interesting parallels to a growing body of literature implicating these pathways in extracellular matrix remodeling and cellular proliferation in various cancers.33–35 It is also interesting to note that the upregulation of adipogenic signaling may be a key modulator of classically “osteogenic” signaling pathways activated in MMVD, thereby shifting the phenotypic consequence of these pathways toward cellular proliferation and matrix remodeling in the myxomatous valve. Ultimately, we feel the continued molecular characterization of diseased valvular tissue using such nonbiased, high-throughput methods in a variety of patient cohorts will provide key insights into molecular drivers and modulators of disease progression.
Novel Characterization of miRNAs in MMVD
To our knowledge, this is the first report to characterize differentially regulated miRNAs in myxomatous mitral valve tissue using any type of high-throughput, nonbiased analytical techniques. Namely, we were successful in identifying 67 differentially regulated miRNAs in our cohorts. Several of these miRNAs—such as miR1 and miR133a—have been implicated and exhaustively studied in a broad range of cardiovascular phenotypes and other pathologies (eg, tissue fibrosis, 36 matrix remodeling, valvular calcification, 37 cellular proliferation, 38 cellular hypertrophy 39 ). Interestingly, the majority of validated miRNA transcripts identified in our study did not have substantive overlap with recent reports of circulating miRNAs that predict presence of MMVD, suggesting that circulating miRNAs may be more reflective of systemic stressors and adaptations than the valve microenvironment itself. 40
In an effort to more precisely identify miRNAs that are highly likely to influence disease progression in MMVD, we again performed RF-B to identify a subset of transcripts of greatest relevance. Using this approach, only 34 of the 67 transcripts reached our threshold of 95% likelihood of importance in MMVD, and only miR1 and miR133a remained in the top 10 of both analytical approaches. Importantly, our analyses—combined with proof-of-concept validation in other clinical conditions41–45—suggest that the outputs of this work are likely to have significant clinical and mechanistic relevance. Future work focused on in vitro and in vivo mechanistic validation of the relevance of these putative targets will nonetheless be critical in confirming their therapeutic utility and advancing our understanding of MMVD.
Aberrations in Circadian Clock Gene Expression are a Novel Molecular Characteristic of MMVD
While standard IPA analyses of a subset of highly relevant genes (identified through RF-B analysis) yielded similar findings of TGF-beta hyperactivation, we were surprised to find aberrations in circadian rhythm as the lead candidate pathway following DAVID analysis of RF-B-enriched genes. There are numerous reports that disturbance of the circadian rhythm—either from clinical or epidemiological factors—is strongly associated with an increased prevalence of adverse cardiovascular outcomes.46–50 Given the strong emergence of aberrations in circadian rhythm in our analyses, we were surprised by the paucity of epidemiological evidence suggesting a link between disturbances in the circadian rhythm and prevalence of MMVD. Systemic alterations in circadian rhythm (eg, losses in variability of HR and blood pressure 51 ) can frequently be a consequence of sympathetic activation and/or parasympathetic withdrawal and increased blood pressure, which has direct implications for the mechanical stresses placed on the mitral valve. 52 Interestingly—and perhaps most directly related to molecular mechanisms influencing progression of MVP, loss of circadian gene patterns in local tissues has been shown to directly regulate extracellular matrix deposition and turnover through a broad range of mechanisms.53,54 Thus, future work focused on the mechanistic interactions between both systemic and local changes in circadian rhythm will be critical to advancing our understanding of MMVD.
Limitations
A primary limitation of this study was our inability to control for differences in age, left ventricular function, or medications between our nonmyxomatous and myxomatous cohorts. Given that the nonmyxomatous tissue typically originated from explanted hearts not suitable for transplant, ejection fraction was significantly lower, which can place strain on the mitral valve apparatus and/or induce secondary regurgitation. While a number of patients in the nonmyxomatous group did have echocardiographic evidence of mild or moderate regurgitation secondary to left ventricular dilatation, exploratory assessments of our data suggest that these points were evenly distributed throughout the data range for patients without mitral regurgitation (see Supplemental Figures SM11). Furthermore, patient medical records included reports from anatomic pathology stating an absence of mitral valve thickening and/or myxomatous degeneration. Similar qualitative subset analyses suggested negligible effects of age or pharmacological management on the directionality or distribution of data within a gene set. Notably, the proportion of patients receiving angiotensin II modulating pharmacotherapies (angiotensin converting enzyme inhibitors or angiotensin receptor blockers)—which have been implicated in the modulation of TGF-beta signaling14–17—did not differ significantly between groups.
We also note that a small subset of transcripts detected in our sequencing data were not confirmed in our larger validation cohort. This is not uncommon when performed appropriately in developmental and/or molecular screening studies and can be due to increased variability in a separate cohort, population heterogeneity, or a reduced effect size.55–59 Regardless of the factor driving the failure to validate a gene, this regression toward a “true mean”/”true difference” suggests reduced relevance at a general population scale. Collectively, however, we feel our approach is robust and minimizes the risk of a Type I Error for the most novel molecular signals identified and emphasized within this report (ie, circadian rhythm).
Conclusion
This study is the first to report nonbiased screening of mRNA and miRNA signatures in MMVD and implicates several pathways that are novel but importantly harbor mechanistic links to known molecular drivers of mitral valve disease. Future efforts aimed at investigating the putative targets that we highlight herein will be pivotal to advancing our understanding of MMVD and guiding the development of novel nonsurgical/noninterventional treatment options for patients with this disease.
Supplemental Material
sj-docx-1-hvs-10.1177_30494826251362814 - Supplemental material for Unraveling the Molecular Complexity of Myxomatous Mitral Valve Degeneration: Integrating Transcriptomic and miRNA Profiling Using Random Forests with Boruta Feature Selection
Supplemental material, sj-docx-1-hvs-10.1177_30494826251362814 for Unraveling the Molecular Complexity of Myxomatous Mitral Valve Degeneration: Integrating Transcriptomic and miRNA Profiling Using Random Forests with Boruta Feature Selection by Michael A Hagler, Nassir Thalji, Nate T Russell, Michael E Welge, Ruoqing Zhu, Colleen Bushell, Matthew Berry, Bryan A White, Aleksey V Matveyenko, Rakesh M Suri and Jordan D Miller in Journal of the Heart Valve Society
Footnotes
Abbreviations
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Heart, Lung, and Blood Institute, (grant number R01HL141819).
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental Material
Supplemental Material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.