
Abstract
Despite representing 30% to 40% of newly diagnosed cases of adult non-Hodgkin lymphoma, diffuse large B-cell lymphoma (DLBCL) rarely presents (1) in the leukemic phase (2) with dysregulation of the TP53 tumor suppressor gene and (3) an elevated serum lactic acid level. In this case report and literature review, we highlight this unfortunate triad of poor prognostic features associated with an aggressive and fatal clinical course in a 53-year-old man with recrudescent DLBCL. A leukemic presentation of de novo or relapsed DLBCL is rare and may be related to differential expressions of adhesion molecules on cell surfaces. In addition, TP53 gene mutations are present in approximately 20% to 25% of DLBCL cases and foreshadow worse clinical outcomes. Finally, an elevated serum lactic acid level in DLBCL that is not clearly associated with sepsis syndrome is a poor prognostic factor for survival and manifests as type B lactic acidosis through the Warburg effect.
Keywords
Introduction
Diffuse large B-cell lymphoma (DLBCL) is the most common type of non-Hodgkin lymphoma (NHL), accounting for up to 40% of all NHL cases worldwide. 1 DLBCL consists of a heterogeneous collection of malignancies with various histological and clinical presentations. In the United States, the incidence and the mortality of DLBCL is approximately 5.4 and 1.8 per 100 000 individuals per year, respectively. 2 Based on a recent Surveillance, Epidemiology, and End Results analysis by the National Cancer Institute, the 5-year survival of all DLBCL cases between 2012 and 2016 was 63.2%. 2
The revised International Prognostic Index (rIPI) has been used to predict overall and progression-free survival in patients with newly diagnosed DLBCL.3,4 In the context of transformed lymphomas, the rIPI does not provide further stratification when DLBCL arises from low-grade NHL. 5 The presence of circulating lymphoma cells in the peripheral blood can occur with mantle cell lymphoma, follicular lymphoma, Burkitt lymphoma, and anaplastic lymphoma. However, a leukemic presentation is much less commonly observed with de novo or relapsed DLBCL, and it is associated with an especially bleak prognosis. 6
Herein, we describe the clinical course of a patient with an antecedent history of DLBCL who demonstrated a partial response to induction chemotherapy, underwent salvage chemotherapy and an autologous stem cell transplant (ASCT), and maintained in remission for nearly 3 years. He subsequently presented to medical attention with a prodrome of fevers, night sweats, and severe pancytopenia. He had a spontaneous clinical remission of 3 months duration, followed by recrudescent fevers, night sweats, upper respiratory symptoms, widespread lymphadenopathy, and an elevated serum lactic acid level. Circulating DLBCL cells were identified on peripheral blood smear and confirmed by flow cytometric analysis. Fluorescence in situ hybridization (FISH) studies were notable for dysregulation of the TP53 tumor suppressor gene.
This case highlights an unfortunate triad of poor prognostic features of lymphoma associated with an aggressive clinical course, including the patient’s leukemic presentation of relapsing and remitting DLBCL, TP53 tumor suppressor gene mutation, and elevated serum lactic acid level. We review the literature on leukemic presentations of DLBCL, discuss the significance of TP53 tumor suppressor gene mutations, and describe the pathogenesis of an elevated serum lactic acid level.
Case Report
A 53-year-old Caucasian man with moderate obesity, hyperlipidemia, and hepatic steatosis was diagnosed with stage IIIA DLBCL in October 2015 after initially presenting to medical attention with cervical and axillary lymphadenopathy. Associated symptoms included a persistent productive cough and mild dyspnea with exertion. The diagnosis was made via excisional biopsy of a right cervical lymph node, with a positron emission tomography and computed tomography (PET-CT) scan demonstrating metabolically active lymphadenopathy above and below the diaphragm.
The patient received 6 cycles of rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) chemo-immunotherapy. He achieved a partial radiographic response, and a subsequent left axillary lymph node biopsy confirmed persistent DLBCL. He received 3 additional cycles of R-ifosfamide, carboplatin, and etoposide (ICE) chemotherapy, followed by an uneventful peripheral blood stem cell collection. In the context of a complete response (CR), he received consolidation chemotherapy consisting of R-carmustine, etoposide, cytosine arabinoside, and melphalan (BEAM), followed by an ASCT in October 2016. He remained disease-free at the time of his last CT imaging study in February 2019.
In July 2019, he developed an acute respiratory illness with cough and fever. He was seen at 2 walk-in clinics and received empiric antibiotics, but his condition did not improve. A week later, he presented to a local emergency room with worsening dyspnea, a persistent cough, and fever. Initial laboratory values included a white blood cell (WBC) count of 6.8 × 109 cells/L, hemoglobin of 9.8 g/dL, and platelet count of 49 × 109 cells/L. His peripheral blood smear showed scattered large atypical lymphocytes with round to irregular nuclei, coarse chromatin, and small amounts of basophilic cytoplasm (Figure 1a). He was admitted to the hospital, and a subsequent bone marrow biopsy showed 50% marrow cellularity, adequate megakaryocytes, reduced marrow iron reserves, and no ringed sideroblasts. In contrast to the peripheral blood, his bone marrow aspirate showed a population of small lymphocytes with plasmacytoid features in a background of low cellularity, suggestive of a low-grade lymphoma (Figure 1b). Flow cytometry of the bone marrow aspirate showed an abnormal B-cell population, comprising 10% of WBCs with no light chain expression, negative to dim CD19, bright CD20, dim CD5 on a subset, negative CD10, moderate FMC7, and negative CD200. Cyclin D1 on the bone marrow biopsy was negative by immunohistochemistry. Cytogenetic analysis from culture stimulated with IL2 and DSP30 in lymphoid cells showed an abnormal male karyotype, characterized by 45~47, XY, −1, −2, −4, −5, del(6)(q10), add(7)(p11.2), −9, +7~9mar, inc[cp2]/46, XY[18]. He received empiric broad-spectrum antibiotics and antifungal coverage, but an extensive fever workup, including bronchoalveolar lavage, multiple serological tests, and assorted cultures of various bodily fluids showed no infectious pathogens. CT imaging demonstrated new axillary lymphadenopathy, a 15-cm spleen, and interstitial pulmonary infiltrates. His cytopenias persisted for the duration of his hospitalization, and he received multiple packed red blood cell and platelet transfusions. On hospital day 17, his fevers abated and dyspnea improved. At time of hospital discharge, his WBC count was 4.6 × 109 cells/L, hemoglobin 9.1 g/dL, and platelet count 18 × 109 cells/L. He was advised to follow up with his outpatient oncologist, but was lost to follow-up.
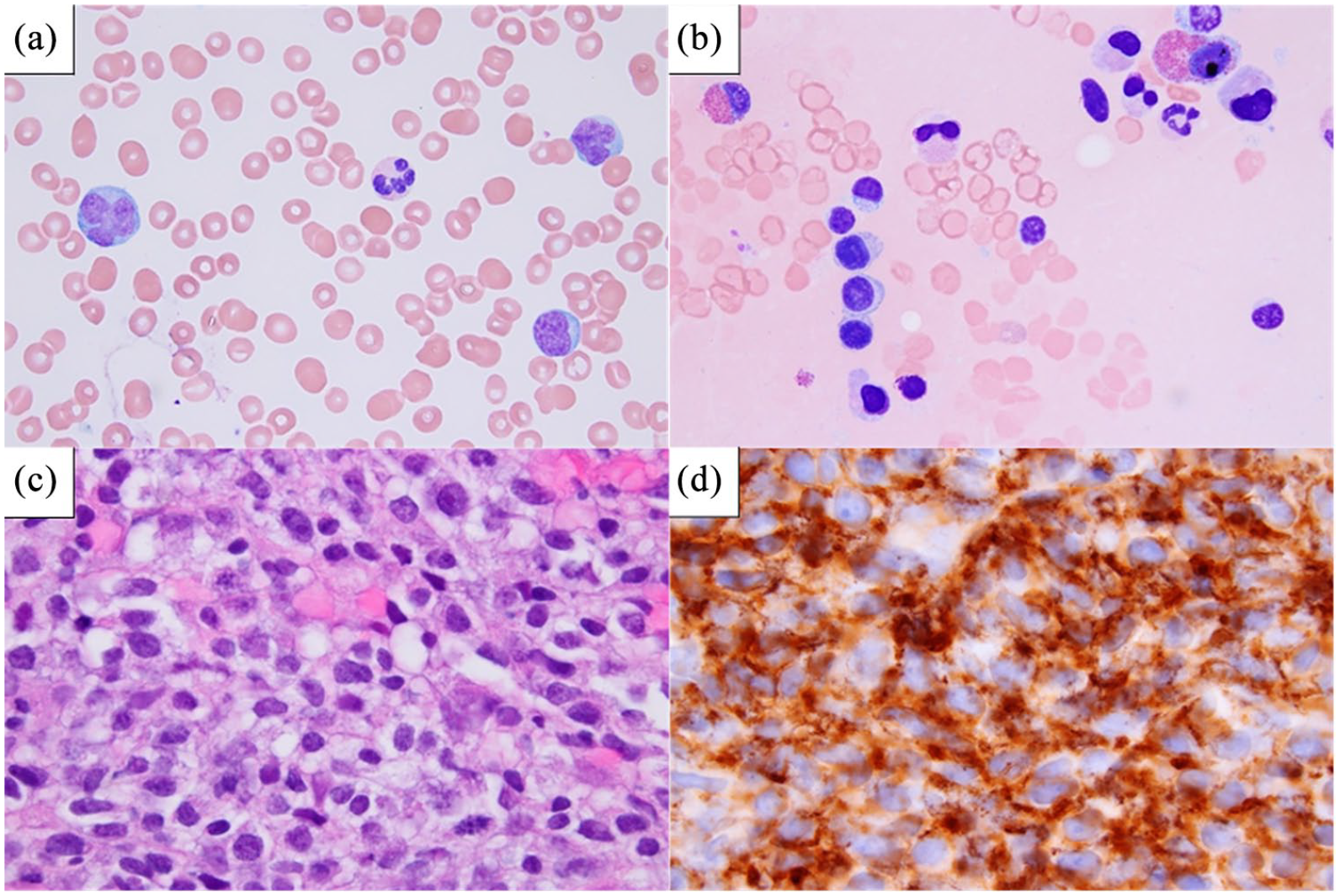
(a) Peripheral blood smear from July 2019 showed scattered atypical large lymphocytes with irregular nuclei. Peripheral blood smear from December 2019 showed similar abnormal cells, (b) Bone marrow aspirate from July 2019 showed increased numbers of small lymphocytes with round nuclei and variable plasmacytoid morphology, suggestive of a low-grade lymphoma. The large lymphocytes with irregular nuclei present on peripheral blood were not seen in the bone marrow aspirate. Flow cytometry of the bone marrow aspirate showed 10% monoclonal B-cells with negative to dim CD19, bright CD20, dim CD5 on a subset, negative CD10, moderate FMC7, and negative CD200, (c) Left axillary lymph node biopsy from December 2019 showed large atypical B-cells, consistent with involvement by diffuse large B-cell lymphoma, and (d) Immunohistochemistry for CD20 of the left axillary lymph node biopsy showed diffuse involvement by sheets of abnormal B-cells.
By September 2019, he had relocated to the Pacific Northwest for a business opportunity, and he established care at our clinic. He was feeling well, had re-gained 15 pounds, and his only medications were over-the-counter vitamins and supplements. He had quit smoking in 2013 after a 40 pack-year smoking history, and he drank alcohol in moderation. His vital signs were normal, including a temperature of 36.5 Celsius and an oxygen saturation of 98% on room air. His physical examination was normal with no palpable lymphadenopathy or hepatosplenomegaly. Laboratory studies included the following: WBC count 5.9 × 109 cells/L with 21% lymphocytes; hemoglobin 13.4 g/dL; platelet count 250 × 109 cells/L; serum iron 88 mcg/dL (normal: 60-165 mcg/dL); TIBC 293 mcg/dL (normal: 300-480 mcg/dL); iron saturation 30% (normal: 20%-50%), and ferritin 1136 ng/mL (normal: 31-356 ng/mL). His elevated ferritin was attributed to the multiple blood transfusions he had received during his recent hospitalization and during his antecedent ASCT. His lactate dehydrogenase (LDH) was 177 U/L (normal: 125-243 U/L) and serum glucose was 130 mg/dL (normal: 75-139 mg/dL). His remaining liver transaminases and renal function tests were also within normal range. Serum protein electrophoresis showed no monoclonal bands, and flow cytometric analysis of his peripheral blood showed no evidence of lymphoma.
We requested outside medical records and plans were made for follow-up, but he was not seen again until December 2019 when he presented to our emergency room with worsening dyspnea despite a 1-week course of empiric antibiotics, hectic fevers, obvious neck and axillary lymphadenopathy, and a non-blanching petechial rash. His temperature was 38.0 Celsius and oxygen saturation was 100% on room air. Initial laboratory studies included the following: WBC count 17.8 × 109 cells/L with 47% lymphocytes; hemoglobin 8.1 g/dL; platelet count 28 × 109 cells/L; serum iron 76 mcg/dL; TIBC 220 mcg/dL; iron saturation 35%; and ferritin 3012 ng/mL. His glucose was 97 mg/dL, albumin was 3.1 g/dL (normal: 3.5-5.0 g/dL) and alkaline phosphatase was 197 U/L (normal: 40-150 U/L). His serum chemistries and total protein were otherwise within normal limits. Additional laboratory testing included an LDH of 1047 U/L and a serum lactic acid of 3.24 mmol/L (normal: ⩽1.6 mmol/L).
His peripheral blood smear during his December 2019 hospitalization demonstrated large atypical lymphocytes with irregular nuclei, concerning for involvement by a large cell lymphoma. Flow cytometry of his peripheral blood showed an abnormal B-cell population, positive for CD20 (dim) and CD22, and negative for both kappa and lambda light chains, CD5, CD10, CD19, CD200, and CD23 (Figure 2). This phenotype was similar to the flow cytometry of the patient’s bone marrow core biopsy from July 2019, consistent with involvement of B-cell lymphoma. FISH studies from his peripheral blood showed 17p, 6q, and 14q deletions, among which his 17p deletion suggested a TP53 mutation. A CT angiogram was negative for pulmonary embolism, but demonstrated a right lateral mid-chest opacity that was initially suspicious for pneumonia. Whole body CT imaging showed numerous enlarged lymph nodes above and below the diaphragm, the largest of which measured approximately 5 cm in diameter. A left axillary lymph node biopsy showed large atypical B-cells (Figure 1c). Immunohistochemistry studies were positive for CD20 (Figure 1d), BCL6, and MUM1, and negative for CD10, with a Ki-67 proliferation rate of 80%. C-MYC was positive in 30% of tumor cells by immunohistochemistry. There was insufficient tissue for FISH studies. The overall findings were most consistent with DLBCL, not otherwise specified, of non-germinal center phenotype.

Peripheral blood flow cytometry from December 2019 showed an abnormal B-cell population, positive for CD20 (dim) and CD22, and negative for both kappa and lambda light chains, CD5, CD10, CD19, CD200, and CD23. This phenotype was similar to the flow cytometry of the patient’s bone marrow core biopsy from July 2019, consistent with involvement of B-cell lymphoma.
The patient’s initial blood and sputum cultures were unremarkable. Bronchoalveolar lavage, extensive serologic and molecular tests, and additional blood and sputum cultures did not point to an infectious source. In the backdrop of increasing oxygen needs and worsening pulmonary infiltrates, he received corticosteroids in conjunction with broad spectrum antibiotics and empiric antifungal therapy. For his cytopenias, he received multiple packed red blood cell and platelet transfusions. Despite best supportive care, his condition worsened. When adequate oxygenation could not be maintained with a venti-mask and with dramatic progression of his lymphadenopathy, he was transitioned to comfort care on day 14 of his hospitalization. He died the following morning due to respiratory arrest secondary to recurrent DLBCL. The patient’s WBC count increased dramatically during his final days, with a value of 50.8 × 109 cells/L with 82% lymphocytes on the morning prior to his death.
Discussion
This patient’s case highlights an unfortunate triad of poor prognostic features of lymphoma associated with a fulminant clinical course, including the patient’s leukemic presentation of relapsing and remitting DLBCL, TP53 tumor suppressor gene dysregulation, and elevated serum lactic acid level.
Despite representing 30% to 40% of newly diagnosed cases of adult NHL, DLBCL rarely manifests in the leukemic phase, which is characterized by bone marrow infiltration and circulating DLBCL tumor cells. Using a Medline search with the key words “diffuse large B-cell lymphoma AND leukemic/leukemic phase” and “diffuse large B-cell lymphoma AND leukemic/leukemic presentation”, we identified a total of 16 case reports and 2 case series of de novo or relapsed DLBCL presenting in the leukemic phase that were published in English (Table 1).7-24 In one case series describing the clinical course of 40 individuals with leukemic presentations of de novo DLBCL, 32 (80%) patients died with a median follow-up of 18 months. 23 The median follow-up for the 8 surviving patients was 34.5 months. In another case series of 29 individuals, 14 (48%) patients died, 13 (45%) survived, and 2 (7%) were lost to follow-up. 24 The median progression-free survival was 11.5 months, and the median overall survival was 46.7 months. In addition to lymphoma involvement in the bone marrow and peripheral blood, many of these patients also manifested with other sites of extra-nodal disease, including spleen, lung, liver, bone, cerebral spinal fluid, bowel, kidney, pancreas, adrenal gland, and testes.9,17,19,22,24 The diversity of organ involvement may be explained by the high tumor burden associated with circulating malignant cells and the differential expressions of adhesion molecules on cell surfaces. 25 The imbalance of adhesion molecule expression in tumor cells leading to peripheral blood involvement is well described in chronic lymphocytic leukemia and intravascular B-cell lymphoma. 25
Case studies and case series of de novo or relapsed DLBCL presenting in the leukemic phase.

Cases were presumed de novo if the article did not mention de novo versus relapsed disease AND if the patient’s past medical history did not include a prior diagnosis of lymphoma.
Case series with more than 1 patient.
Abbreviations: ARDS, acute respiratory distress syndrome; ASCT, autologous stem cell transplant; CHOPE, cyclophosphamide, doxorubicin, vincristine, prednisone, etoposide; C-MOPP, cyclophosphamide, vincristine, sulfate, procarbazine, prednisone; CNS, central nervous system; CR, complete remission; CSF, cerebral spinal fluid; DA-EPOCH, dose-adjusted etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin; DHAP, dexamethasone, cytosine arabinoside, cisplatin; EBRT, external beam radiotherapy; hyper-CVAD/MA, hyper-CVAD alternating with high-dose methotrexate and cytarabine (MA); IPI, International Prognostic Index; IV, intravenous; LDH, lactic dehydrogenase; NR, not recorded; NS, not specified; OS, overall survival; O-ICE, ofatumumab, ifosfamide, carboplatin, etoposide; PD, persistent disease; PFS, progression-free survival; PO, per os, by mouth, oral; PR, partial remission; R, rituximab; R-BFM-90, rituximab-Berlin-Frankfurt-Münster-90 protocol; R-CHOP, rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone; R-COMP, rituximab, cyclophosphamide, vincristine, non-pegylated liposomal doxorubicin, prednisone; R-ICE, rituximab, ifosfamide, carboplatin, etoposide; R-hyper-CVAD, rituximab and hyper-fractionated cyclophosphamide, vincristine, doxorubicin, dexamethasone; SCT, stem cell transplant; TECAM, thiotepa, etoposide, cyclophosphamide, cytosine arabinoside, melphalan.
TP53 is a tumor suppressor gene that is responsible for DNA repair, apoptosis, and cell cycle arrest. It plays an integral role in maintaining genetic stability and eliminating cells with pathogenic mutations. For patients with DLBCL, the presence of TP53 mutations, especially in the TP53 DNA-binding domains, is an independent negative prognostic factor for survival.26-29 The methylation or mutation of the TP53 promoter, followed by the loss of an intact gene allele, is one proposed mechanism that may explain tumorigenesis and the malignant transformation of DLBCL. 30 TP53 gene mutations are present in approximately 20% to 25% of DLBCL cases.26,31,32 Clinically, DLBCL patients with TP53 mutations have higher rIPI scores, higher serum LDH levels, the presence of B symptoms, and a lower likelihood of achieving complete remission. 31 Patients with DLBCL and TP53 mutations have lower life expectancies compared to those without such mutations. 31
Although an elevated serum lactic acid level is often associated with sepsis as a result of tissue hypoperfusion and hypoxia, it can rarely also serve as a negative prognostic factor for survival in DLBCL through a phenomenon known as the Warburg effect. During malignant conversion, cancer cells undergo accelerated glycolysis and produce large amounts of lactic acid, even in the presence of oxygen. The imbalance between tumor lactic acid production and hepatic lactic acid breakdown causes a type B lactic acidosis that is occasionally associated with hypoglycemia.33-36 This form of lactic acidosis is more frequently associated with hematologic rather than solid malignancies.
Our patient’s elevated lactic acid level in the setting of a leukemic manifestation of relapsed DLBCL and dysregulation of the TP53 tumor suppression gene foreshadowed his particularly ominous prognosis. Only a limited number of cases of lactic acidosis in DLBCL have been reported in the medical literature.37-44 The vast majority of these patients did not receive treatment for their lymphoma because of the fulminant nature of their disease. The few patients who survived generally responded to rapid initiation of chemotherapy. Other therapies have included sodium bicarbonate and steroids, but the benefits have been inconsistent or uncertain.38,41,44 Thiamine and riboflavin deficiencies have been hypothesized to play a role in the development of a type B lactic acidosis, although the exact mechanism is not fully understood.45-47 Given the high mortality associated with an elevated serum lactic acid level in patients with DLBCL, timely diagnosis of this rare complication of aggressive lymphoma and close monitoring for timing of salvage chemotherapy is essential.
Our case report and literature review have limitations. Despite highlighting 3 important factors associated with negative prognoses in DLBCL, we ultimately are unable to establish causality. In addition, the generalizability of our study is limited, given that this fatal triad is not well-described in the scientific literature. Only a small number of retrospective case reports and case series of DLBCL presenting in the leukemic phase recount the presence or absence of TP53 mutations and elevated serum lactic acid levels, therefore limiting our ability to ascertain a representative population for causal inference.
Our report also stimulates further research questions. Efforts to elucidate possible predictive factors for the leukemic conversion of DLBCL may aid in the early recognition of this disease and improve clinical outcomes. Moreover, the recent approval of tafasitamab-cxix, lenalidomide, selinexor, and chimeric antigen receptor T-cell (CAR-T) therapies by the United States Food and Drug Administration are examples of the rapidly expanding array of salvage treatments options available to patients with DLBCL. Further research and development of targeted and salvage chemotherapy regimens, in addition to clinical and epidemiological studies to determine whether these therapies improve survival, will improve cancer care and guide clinicians in managing this disease.
Conclusion
The clinical triad of a leukemic presentation of recrudescent DLBCL, dysregulation of the TP53 tumor suppressor gene, and an elevated serum lactic acid level are uniquely ominous prognostic factors that portend short survival. A better understanding of the mechanisms by which these risk factors lead to an aggressive clinical course, as well as broadening the spectrum of therapeutic options available to these patients, is important in optimizing medical management and goals of care discussions.
Footnotes
Acknowledgements
We thank Virginia M. Green, PhD, for her editing expertise and assistance with manuscript preparation. We also thank the patient and his family for generously allowing us to share his story with the medical community.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
CSH, DGH, and DMA wrote the manuscript. CSH was the resident physician who cared for this patient, DGH performed the pathology review, and DMA was the primary oncologist for this case. All authors reviewed and approved the final manuscript.
Ethics Approval
Our institution does not require IRB approval for case studies.
Informed Consent
This patient is deceased, and we therefore obtained written informed consent for the publication of his case information and clinical images from his wife.