
Abstract
Late-onset Pompe disease (LOPD) is a rare inherited disorder caused by deficiency of the lysosomal enzyme acid α-glucosidase (GAA), leading to an accumulation of lysosomal glycogen in tissues, profoundly affecting muscles. Patients with LOPD typically have some residual GAA activity but experience progressive skeletal muscle dysfunction resulting in muscle weakness and respiratory failure. Enzyme replacement therapy (ERT) with alglucosidase alfa, a recombinant human GAA (rhGAA), was the first disease-specific therapy for Pompe disease. Despite efficacy in the first years of use, many patients receiving alglucosidase alfa experience a decline in function over time. This may reflect the inherent challenges associated with rhGAA ERT, such as enzyme inactivation at the near-neutral pH of blood, inefficient target cell uptake, and a necessity for complete lysosomal processing once inside target cells. Cipaglucosidase alfa, a second-generation rhGAA, aims to address these challenges through natural enrichment with bis-mannose-6-phosphate-containing N-glycans to enhance cellular uptake while retaining capacity for complete postdelivery processing. Co-administration of cipaglucosidase alfa with the small molecule stabilizer miglustat (N-butyldeoxynojirimycin) enhances cipaglucosidase alfa stability in the bloodstream after infusion. We discuss published and new preclinical and clinical data on the efficacy and safety of miglustat in combination with cipaglucosidase alfa for treating LOPD. Studies in Pompe mouse models and patients with Pompe disease showed that stabilization by miglustat improved cipaglucosidase alfa exposure and availability for uptake into target tissues and was associated with improved functional outcomes and biomarker levels compared with cipaglucosidase alfa alone. In patients with Pompe disease, the once every 2 weeks dosing regimen of miglustat was well tolerated, with a low frequency of miglustat-related gastrointestinal events compared with daily miglustat regimens at higher doses used in the treatment of other diseases.
Plain language summary
In healthy cells, the enzyme acid α-glucosidase (GAA) works in parts of the cells called lysosomes to break down glycogen into glucose, an important source of energy for cells. In people with Pompe disease, a lack of working GAA means that glycogen cannot be broken down and builds up inside lysosomes. As muscles store large amounts of glycogen, they are especially impacted. Over time, excessive glycogen buildup leads to muscle damage that can cause weakness of the muscles that control movement (skeletal muscles) and breathing (respiratory muscles). The first treatment developed specifically for Pompe disease was an enzyme replacement therapy (ERT) called alglucosidase alfa. ERT for Pompe disease aims to replace the missing GAA enzyme in muscles but faces some challenges to be effective, such as reduced stability at the neutral pH of the blood as compared with the acidic environment found in lysosomes. After initial improvements, many people receiving alglucosidase alfa experience a worsening in muscle weakness and their health over time. Cipaglucosidase alfa is a next-generation ERT used with the enzyme stabilizer miglustat. Patients with late-onset Pompe disease take a low dose of miglustat once every 2 weeks, just before they receive cipaglucosidase alfa. In mice and in patients with Pompe disease, stabilization of cipaglucosidase alfa by miglustat in the bloodstream increased the amount of enzyme available to enter the muscles. Compared with cipaglucosidase alfa alone, combining miglustat with cipaglucosidase alfa improved functional outcomes in mice and reduced markers of glycogen buildup and muscle damage in human patients. Miglustat is also used to treat illnesses such as Gaucher disease and Niemann–Pick disease. For these illnesses, miglustat works differently and needs to be taken every day and at much higher doses than in Pompe disease. This can lead to high rates of side effects in the digestive system. In late-onset Pompe disease, the rates of side effects, such as diarrhea or nausea, were considerably lower compared with other uses of miglustat.
Introduction
Late-onset Pompe disease (LOPD) is a rare inherited disease caused by a deficiency of the lysosomal enzyme acid α-glucosidase (GAA). 1 GAA degrades glycogen to glucose in the lysosome; thus, reduced GAA enzyme activity leads to lysosomal glycogen accumulation in tissues, including skeletal, respiratory, and cardiac muscle. 2 While patients with LOPD typically have some residual GAA activity, they experience progressive dysfunction and degeneration of skeletal muscle, leading to muscle weakness and eventual respiratory failure.1,3
Enzyme replacement therapy (ERT) with alglucosidase alfa, a recombinant human GAA (rhGAA), was the first disease-specific therapy for Pompe disease 4 ; however, infusion of exogenous rhGAA has inherent challenges. 5 Less than 1% of infused alglucosidase alfa reaches muscle tissue due to clearance by nontarget tissues, such as the liver and spleen, and poor uptake into skeletal muscle cells.4,5 The uptake of rhGAA into cells and its subsequent delivery to lysosomes is driven by the cation-independent mannose-6-phosphate receptor (CI-MPR), which binds to mannose-6-phosphate (M6P) on newly synthesized lysosomal proteins. 4 CI-MPR can also cycle to the plasma membrane to facilitate receptor-mediated endocytosis, thus allowing cellular uptake of exogenous M6P-containing rhGAA. 4 The binding ability of rhGAA to CI-MPR is determined by its M6P and bis-phosphorylated N-glycans content, with bis-phosphorylated M6P (bis-M6P) having an approximately 3000-fold greater affinity for CI-MPR than monophosphorylated N-glycans. 4 Alglucosidase alfa has a low abundance of M6P, particularly bis-M6P, substantially reducing cellular uptake at low enzyme concentrations and, as such, limiting its effectiveness.4,6 Over time, many patients receiving alglucosidase alfa experience a plateau or decline in outcomes, such as lung function, muscle strength, and motor function, despite efficacy in the first years of use.7,8
Cipaglucosidase alfa is produced in a Chinese hamster ovary cell line that was selected for the generation of a rhGAA with optimal N-glycan profiles and high levels of bis-M6P.5,9,10 The natural enrichment of cipaglucosidase alfa with bis-M6P-containing N-glycans enhances cellular uptake by CI-MPR while retaining capacity for complete processing and GAA maturation, addressing some of the challenges associated with ERT. 5 Both proteolytic processing and N-glycan trimming within the endolysosomal pathway are required to attain the most efficient enzyme for glycogen hydrolysis. 11 While sufficient bis-M6P content of rhGAA is essential to maximize cellular uptake via CI-MPR, synthetic attachment of bis-M6P to rhGAA at unnatural linkages can prevent lysosomal processing and complete enzyme activation through inhibition of N-glycan trimming at critical enzyme recognition sites and subsequent steric hindrance of the active site by unnatural glycan residues.5,11,12 Cipaglucosidase alfa does not contain chemically conjugated bis-M6P; however, avalglucosidase alfa, another next-generation rhGAA, contains synthetic bis-M6P residues that are conjugated onto alglucosidase alfa N-glycans using oxime chemistry.4,5,13,14 The synthetic bis-M6P residues on avalglucosidase alfa did not appear to affect proteolytic processing of the enzyme in human Pompe fibroblasts.15,16 However, these assays were performed using samples treated with peptide:N-glycosidase F, which removes N-linked oligosaccharides from glycoproteins17,18 and can, therefore, obscure deficiencies in endolysosomal glycan trimming. Avalglucosidase alfa demonstrated improved glycogen clearance from the heart and skeletal muscle compared with alglucosidase alfa in Pompe mice.15,16 The efficacy and safety of avalglucosidase alfa (20 mg/kg) were directly compared with that of alglucosidase alfa (20 mg/kg) in patients with LOPD in the phase III COMET study (NCT02782741). 19 After 49 weeks of treatment, noninferiority but not statistical superiority versus alglucosidase alfa was met for the primary endpoint (percent predicted forced vital capacity). Avalglucosidase alfa was also associated with clinically meaningful improvements over alglucosidase alfa in secondary functional outcomes, including other respiratory function outcomes and 6-minute walk distance. 19 In an open-label extension study, patients who participated in COMET could continue receiving avalglucosidase alfa; data have been reported for up to 97 weeks of treatment. 20 Real-world data will be required to provide information from a larger number of patients and in the longer term.
While increasing the bis-M6P content of next-generation rhGAAs may improve cellular uptake in target muscle tissues, GAA is an acid hydrolase that is more stable under acidic conditions reflective of the lysosome, rather than at neutral pH of blood. Instability and inactivation at neutral pH compared with an acidic environment is a well-characterized aspect of all rhGAA ERTs,4,6,15,21–24 leaving them vulnerable to inactivation at the near-neutral pH of blood before reaching the muscles. 5 To address this challenge, cipaglucosidase alfa (20 mg/kg) is administered with the small molecule miglustat (260 mg for patients weighing ⩾50 kg or 195 mg patients weighing ⩾40 to <50 kg) for the treatment of adults with LOPD, to stabilize cipaglucosidase alfa in the bloodstream and increase bioavailability.5,9,10,25 Miglustat (N-butyldeoxynojirimycin; AT2221) was first approved as a competitive glucosylceramide synthase inhibitor 26 for patients with mild-to-moderate type 1 Gaucher disease (GD), who cannot be treated with glucocerebrosidase (GCase) ERT, and later for patients with Niemann–Pick type C (NPC).26–28 In GD, deficiency of the lysosomal enzyme GCase, which converts glucosylceramide into ceramide and glucose, results in the accumulation of glucosylceramide in various cell populations, predominantly macrophages.26,29 NPC is caused by a deficiency of lysosomal proteins (NPC1 or NPC2) involved in cholesterol efflux from lysosomes, leading to the accumulation of cholesterol and (secondarily) glycosphingolipids in various tissues, which results in cellular and organ damage.26,30,31 In these diseases, miglustat is used as a substrate reduction therapy (SRT) to decrease the biosynthesis of glycosphingolipids, reducing their lysosomal accumulation.26,29,30 In contrast, miglustat functions as an enzyme stabilizer when combined with cipaglucosidase alfa for treating LOPD.6,10,15,32 Miglustat has a similar structure to the terminal glucose on glycogen, the natural substrate for GAA. 6 Miglustat selectively binds to cipaglucosidase alfa in the blood during infusion, is internalized by muscle cells as a complex, and dissociates from cipaglucosidase alfa in the lysosome.32,33 Importantly, the posology of miglustat given as an enzyme stabilizer in LOPD differs significantly from its use in other indications. When miglustat is given as an SRT in GD and NPC, it is dosed three times per day to achieve steady-state concentrations for therapeutic effect. While other compounds are being evaluated as SRTs in Pompe disease, the enzyme stabilizer miglustat is dosed every 2 weeks, after a fasting period and just before the start of infusion of cipaglucosidase alfa. While miglustat use in patients with GD and NPC is associated with a high frequency of gastrointestinal side effects,27,28 the substantially lower and less frequent dosing in LOPD, along with a 4-hour fast (± 2 hours of dosing), helps to minimize the occurrence of gastrointestinal disturbances.32,33
As a treatment for Pompe disease, the co-administration of cipaglucosidase alfa with miglustat enhances the stability of a naturally highly phosphorylated enzyme at neutral pH with improved muscle-targeting properties. 6 This approach addresses the key challenges of using ERT for the treatment of Pompe disease without chemical conjugation of bis-M6P through synthetic linkers. This review presents published and new preclinical and clinical data in LOPD regarding the efficacy and safety of miglustat in combination with cipaglucosidase alfa.
Why use miglustat with cipaglucosidase alfa?
Use of small molecule stabilizers in lysosomal diseases
Small molecules currently being used to treat rare lysosomal diseases can act in different ways to increase enzyme activity. 26 Chaperones bind and stabilize endogenous enzymes in the cell and help facilitate the correct folding process required for the progression of lysosomal enzymes from the endoplasmic reticulum through the Golgi to the lysosome, for example, migalastat in the treatment of Fabry disease. Stabilizers may be used in combination with ERT to stabilize exogenously administered enzymes in the circulation, for example, miglustat plus cipaglucosidase alfa in Pompe disease. Chaperones and stabilizers usually dissociate from their target enzymes in the acidic pH of the lysosome. 26 In Pompe disease, preliminary investigations of the use of duvoglustat hydrochloride 34 or miglustat 35 in combination with alglucosidase alfa found that small molecule stabilizers can increase the half-life of rhGAA and rhGAA enzymatic activity in plasma or dried blood spots of patients.
In vitro studies in fibroblasts from patients with Pompe disease showed that miglustat enhanced the effectiveness of alglucosidase alfa, leading to increased enzyme stability, improved delivery to lysosomes, and increased intracellular GAA enzyme activity. 36 These findings, along with in vivo studies in a mouse model of Pompe disease, 36 highlighted the therapeutic potential of combined therapy with ERT and an enzyme stabilizer. This therapeutic potential was further evaluated in a small clinical trial of ERT combined with N-butyldeoxynojirimycin (i.e., miglustat) in 13 patients with Pompe disease in Italy. 35 The trial demonstrated increased and prolonged GAA activity in dried blood spot samples obtained from patients after treatment with alglucosidase alfa (20–40 mg/kg) in combination with miglustat (four doses of 80 mg/m2, one on the evening before ERT and three on the day of the infusion) than in patients treated with alglucosidase alfa alone, highlighting the increased stability of the rhGAA alglucosidase alfa in plasma or blood cells in the presence of miglustat. However, the experiments did not provide clear evidence on whether miglustat improved the therapeutic efficacy of alglucosidase alfa in patients. A trend toward improvement in 6-minute walk distance was confounded by low patient numbers and a lack of statistical power. Percent predicted forced vital capacity and manual muscle test results remained stable, and biomarker levels (urine hexose tetrasaccharide [Hex4] and plasma creatine kinase [CK]) showed no significant changes. 35
Miglustat stabilizes cipaglucosidase alfa in vitro
In vitro studies have shown that all rhGAAs (alglucosidase alfa, avalglucosidase alfa, and cipaglucosidase alfa) are less stable at pH 7.4 (similar to the pH of blood) than at pH 5.2 (similar to the pH in the lysosome).6,15,21,22,24 Incubation of alglucosidase alfa, avalglucosidase alfa, or cipaglucosidase alfa in a neutral pH buffer (pH 7.4) at 37

Cipaglucosidase alfa and alglucosidase alfa have similar thermal stability at pH 7.4 and pH 5.2, and both enzymes are stabilized by miglustat at pH 7.4. Thermostability of cipaglucosidase alfa and alglucosidase alfa was measured in vitro in the absence and presence of miglustat (10, 30, or 100 μM) with a fluorescence-based thermal denaturation assay. 37 Unfolding/denaturation of rhGAA was monitored by the increase in SYPRO Orange fluorescence as a function of temperature. The Tm is the temperature at which 50% of the protein is denatured. A higher Tm indicates that the protein is more stable.
What is the effect of miglustat on cipaglucosidase alfa in preclinical studies?
Miglustat stabilizes ERT in Pompe mice and increases cipaglucosidase alfa exposure
Studies in Pompe mouse models (Gaa knockout mice) have shown that miglustat stabilizes ERT in vivo. In a Pompe mouse study reported alongside the first clinical trial of ERT with an enzyme stabilizer, co-administration of miglustat (6 mg/kg) with alglucosidase alfa (40 mg/kg) resulted in higher levels of GAA activity in the blood than with alglucosidase alfa administered alone. 35 The 40 mg/kg dose for mice is comparable with the dose recommended for use in patients with Pompe disease (20 mg/kg for alglucosidase alfa, though clinical trials using doses of 40 mg/kg are described in the prescribing information).38,39 However, doses used in mouse models may differ from those approved for use in human patients. A significant increase in GAA activity was also measured in the liver and muscles of the mice 48 h after administration of alglucosidase alfa with miglustat than with alglucosidase alfa alone. 35 These findings were consistent with those of an earlier study in Pompe mice, which showed increased GAA activity in the liver and muscle following co-administration of miglustat (4.3 mg/kg for 2 days) with alglucosidase alfa (40 mg/kg on the second day) than with alglucosidase alfa alone. 36 More recently, co-administration of a single dose of miglustat (10 mg/kg) with cipaglucosidase alfa (20 mg/kg) in Pompe mice increased cipaglucosidase alfa exposure (area under the curve [AUC]) by 6.8% compared with cipaglucosidase alfa alone (Supplemental Table 1). 21 In a different study, co-administration of miglustat with alglucosidase alfa or avalglucosidase alfa in Pompe mice had no significant impact on the serum exposure to these rhGAAs. 15
Cipaglucosidase alfa plus miglustat improves glycogen clearance and functional outcomes in Pompe mice
Pompe mouse models have also shown improved glycogen clearance and functional outcomes with cipaglucosidase alfa plus miglustat than with cipaglucosidase alfa alone.5,21 After four administrations of cipaglucosidase alfa (20 mg/kg) with or without miglustat (10 mg/kg) every 2 weeks, glycogen reduction in the quadriceps and triceps muscles was numerically greater with cipaglucosidase alfa plus miglustat than with cipaglucosidase alfa alone (Figure 2). 21 The combination of cipaglucosidase alfa plus miglustat reduced glycogen to levels statistically indistinguishable from those in wild-type mice in the quadriceps muscle, and to levels near those in wild-type mice in the triceps muscle (Figure 2). After eight administrations of cipaglucosidase alfa with or without miglustat every 2 weeks in the same study, muscle strength (assessed using grip strength) was also numerically greater with cipaglucosidase alfa plus miglustat than with cipaglucosidase alfa alone, with cipaglucosidase alfa plus miglustat improving muscle strength to levels similar (statistically indistinguishable) to that of wild-type mice (Figure 2). 21 Grip strength in Pompe mice treated with cipaglucosidase alfa plus miglustat improved over time, in contrast to the poor performance and progressive deterioration seen in untreated (vehicle-administered) Pompe mice (Figure 3). Grip strength with cipaglucosidase alfa plus miglustat was significantly improved compared with alglucosidase alfa after 2 months of treatment, but statistical comparisons of cipaglucosidase alfa with and without miglustat were not presented. 5 Importantly, these improvements were observed with a relatively small (~6.8%) improvement in enzyme exposure in mice, which suggests that the larger exposure benefits observed in patients with Pompe disease (~28.5%, discussed below) will also be functionally relevant. 21

Miglustat-induced increase in cipaglucosidase alfa exposure improved glycogen clearance in Gaa KO mice.

Grip strength with cipaglucosidase alfa plus miglustat approached levels in Gaa KO mice similar to those of wild-type mice after 4 months of treatment.
What is the effect of miglustat on cipaglucosidase alfa in patients with LOPD?
Pharmacokinetic data from Pompe mice were used to estimate the appropriate dose of miglustat in combination with cipaglucosidase alfa for clinical trials in human patients
The target dose and timing of miglustat for use with cipaglucosidase alfa in clinical trials with human patients were first determined using nonclinical pharmacokinetic (PK) data from Pompe mice. Concentration–response curves were evaluated to predict the optimal miglustat dose to bind and stabilize cipaglucosidase alfa in the blood while minimizing miglustat exposure in the muscles and thus prevent any potential unwanted postdelivery inhibition of GAA activity in the lysosome. 5 Evaluation of a range of miglustat doses (1–30 mg/kg) revealed that co-administration of a 10 mg/kg dose of miglustat in Pompe mice (corresponding to a dose of ~260 mg in humans) with a 20 mg/kg dose of cipaglucosidase alfa optimized the stability of GAA activity in plasma and maximized glycogen reduction in the quadriceps (Supplemental Figure 1). 5 Lower doses of miglustat (1, 3, or 5 mg/kg) were associated with suboptimal GAA stabilization, while the highest dose of miglustat (30 mg/kg) was associated with a lower degree of glycogen reduction, likely due to excessive miglustat exposure in the muscles, which could cause postdelivery inhibition of GAA activity in the lysosome (Supplemental Figure 1). 5
The clinical dose of miglustat in combination with cipaglucosidase alfa was confirmed using PK data from patients with LOPD
In patients with Pompe disease, population PK modeling was performed on pooled data from two clinical studies: the open-label, phase I/II, first-in-human ATB200-02 study of cipaglucosidase alfa plus miglustat in adults with Pompe disease (NCT02675465), 40 and the pivotal randomized, phase III PROPEL study of cipaglucosidase alfa plus miglustat versus alglucosidase alfa in adults with LOPD (NCT03729362). 41 Using PK modeling, it was predicted that a miglustat dose of 260 mg, administered orally 1 hour prior to cipaglucosidase alfa (20 mg/kg), would result in a molar excess of miglustat during and shortly after the 4-hour cipaglucosidase alfa infusion period (Supplemental Figure 2). It was also estimated that this dose and timing would provide >90% occupancy of cipaglucosidase alfa with miglustat for up to 12 hours following infusion (Supplemental Figure 2), meaning >90% of cipaglucosidase alfa molecules would be in complex with miglustat in the bloodstream. Hence, the 260 mg dose was proposed to provide effective rhGAA stabilization in the blood during biodistribution, while minimizing excessive miglustat exposure to prevent enzymatic inhibition once inside the lysosome. 5 The lysosomal half-life of rhGAA after uptake in cultured cells is estimated to be 8 to 10 days, considerably longer than the ~6-hour terminal elimination half-life of miglustat in plasma.32,33,42,43 Hence, miglustat is absent from the blood for most of the treatment period in patients with Pompe disease.
Compared with cipaglucosidase alfa alone, co-administration of cipaglucosidase alfa (20 mg/kg) with 260 mg of miglustat in adult patients with Pompe disease increased cipaglucosidase alfa exposure (AUC) by 28.5% (Figure 4), 21 increased the half-life of cipaglucosidase alfa by 46.7% (Figure 4), and increased the exposure partial AUC of cipaglucosidase alfa in the bloodstream from the period spanning peak plasma levels to 24 hours postdose by 43.6%, consistent with stabilization of cipaglucosidase alfa in plasma (Supplemental Table 2). 5 The greater increase in cipaglucosidase alfa exposure in the presence of miglustat in patients with Pompe disease (Figure 4) than in Pompe mice (6.8%; Supplemental Table 1) may be due to the different infusion methods used for human versus animal studies (4-hour infusion versus bolus injection).

In patients with LOPD, co-administration of miglustat (260 mg) with cipaglucosidase alfa (20 mg/kg) increased cipaglucosidase alfa exposure (AUC) in plasma by 28.5% compared with cipaglucosidase alfa alone.
Miglustat dose adjustments for pediatric patients are derived from simulations and mathematical modeling
Cipaglucosidase alfa plus miglustat is currently approved for use in adult patients with LOPD.9,10,32,33 In ongoing pediatric trials in LOPD and infantile-onset Pompe disease (IOPD),44,45 adjusted doses of cipaglucosidase alfa and miglustat based on modeling and simulations, body weight, and differences in clearance rates from adults are being evaluated. In an expanded access program, cipaglucosidase alfa at doses up to 37 mg/kg every week plus miglustat at doses of 30–115 mg showed a positive clinical response in a pediatric patient with IOPD. 46 Alglucosidase alfa and avalglucosidase alfa have been administered to patients with Pompe disease, including pediatric patients, at doses of up to 40 mg/kg.47–51 While there are no clinical trial data evaluating cipaglucosidase alfa 40 mg/kg alone or with miglustat, cipaglucosidase alfa at doses of 20 mg/kg and 40 mg/kg with a miglustat dose of 260 mg for an adult weighing at least 50 kg were simulated and assessed in an occupancy binding mathematical model. Plasma concentration ratios of miglustat remained in molar excess to cipaglucosidase alfa until at least 24 hours after the infusion, indicating that dose adjustment of miglustat would not be necessary in a 40 mg/kg cipaglucosidase alfa dosing scenario (not currently labeled).
Miglustat is associated with improved cipaglucosidase alfa-induced reductions in Hex4 and CK in patients with Pompe disease
Muscle damage caused by the accumulation of lysosomal glycogen in Pompe disease can be detected by evaluating the biomarkers Hex4 and CK. Hex4, a breakdown product of glycogen, is detected in urine and acts as a biomarker for glycogen storage metabolism; higher urine levels of Hex4 typically indicate higher levels of glycogen buildup52,53 and correlate with skeletal muscle glycogen content. 53 CK, which leaks into the bloodstream from injured muscle tissue, is detected in serum and may act as a biomarker for muscle damage. 54 However, CK levels may also decrease with advanced muscle damage, for example, in muscular dystrophy, when muscle tissue is replaced by fibrotic and adipose tissue. 55 Although the number of patients was low, data showed that co-administration of cipaglucosidase alfa with miglustat numerically improved (i.e., decreased) levels of urine Hex4 and serum CK with a miglustat dose-dependent trend in ERT-experienced patients with Pompe disease (Figure 5), suggesting a reduced glycogen burden over time. Clinical doses of miglustat (260 mg) led to a further numerical decrease in Hex4 and CK compared with cipaglucosidase alfa (20 mg/kg) alone or with subclinical doses of miglustat (130 mg; Figure 5), consistent with a pharmacodynamic benefit from enzyme stabilization in plasma. The changes from baseline in absolute values of urine Hex4 and serum CK are shown in Supplemental Figure 3.

Treatment with cipaglucosidase alfa plus miglustat improved levels of urine Hex4 and serum CK in patients with LOPD.
Cipaglucosidase alfa plus miglustat improves functional outcomes in patients with Pompe disease
Whether the positive impact of miglustat on cipaglucosidase alfa, seen in vitro and in vivo, translated into clinical improvements in people living with Pompe disease was investigated in the pivotal phase III PROPEL trial.41,56 PROPEL was a head-to-head study of cipaglucosidase alfa (20 mg/kg) plus miglustat (260 mg, or 195 mg in patients weighing 40 to <50 kg) versus standard of care (alglucosidase alfa 20 mg/kg), conducted over 52 weeks in 123 adults with LOPD. 41 While PROPEL did not meet its primary endpoint (superiority in change from baseline to week 52 in 6-minute walk distance with cipaglucosidase alfa plus miglustat versus alglucosidase alfa), the outcomes showed stability in respiratory function and improvement in motor function and biomarkers (Hex4 and CK) after 52 weeks of treatment with cipaglucosidase alfa plus miglustat versus alglucosidase alfa in both the overall study population and in ERT-experienced patients previously treated with alglucosidase alfa, 41 which were considered to be clinically meaningful. 57 These findings were supported by a within-group effect size analysis of patients who switched from alglucosidase alfa to cipaglucosidase alfa or remained on alglucosidase alfa in the PROPEL study. This post hoc analysis showed that patients who switched treatment from alglucosidase alfa to cipaglucosidase alfa plus miglustat mostly showed within-group improvement or stability from baseline to week 52, with the largest effect sizes and significant improvements in various measures of motor function, muscle strength, and biomarker levels. However, patients remaining on alglucosidase alfa mainly showed worsening or stability from baseline to week 52, with significant worsening in many lung function assessments and biomarker levels. 58
All patients who completed PROPEL were eligible to enroll in an open-label, single-arm extension study (NCT04138277) to evaluate the long-term safety and efficacy of cipaglucosidase alfa plus miglustat (at the same dosage used in PROPEL). 56 Patients treated with cipaglucosidase alfa plus miglustat for a total of 2 years maintained any improvements (or stability) in motor and respiratory function and biomarker levels gained during the initial 52 weeks of treatment in PROPEL, indicating durable long-term benefits from cipaglucosidase alfa plus miglustat. 56 Patients who switched from alglucosidase alfa to cipaglucosidase alfa plus miglustat at the start of the extension study showed stabilization or improvement in motor function, respiratory function, and muscle strength over their first year of treatment with cipaglucosidase alfa plus miglustat, accompanied by a rapid improvement in biomarker levels. 56 Reduction in urine Hex4 and serum CK in patients with LOPD in PROPEL who received cipaglucosidase alfa plus miglustat suggests enhanced rhGAA uptake and GAA activity within muscle cells, resulting in increased glycogen clearance and reduced muscle damage, translating to beneficial outcomes for patients. 56 Results from PROPEL and the open-label extension support the therapeutic rationale and clinical effectiveness of combining cipaglucosidase alfa with miglustat.
What is the safety profile of cipaglucosidase alfa plus miglustat?
A pooled safety analysis from three clinical trials was performed, which included 151 adults with LOPD who were treated with cipaglucosidase alfa plus miglustat in the ATB200-02 study (n = 29), 40 the PROPEL study (n = 85), 41 or who were switched to cipaglucosidase alfa plus miglustat in the PROPEL open-label extension (n = 37). 56 The total median duration of treatment with cipaglucosidase alfa plus miglustat was 21 months; 120 patients had been treated for more than 12 months. 10 The most common treatment-related adverse events (AEs) with cipaglucosidase alfa plus miglustat reported in the pooled safety analysis were headache, diarrhea, fatigue, nausea, abdominal pain, and pyrexia, all occurring in ⩾5% of patients. 10 In the PROPEL study, the most common AEs in patients treated with cipaglucosidase alfa plus miglustat, regardless of relatedness to treatment, were falls (29%), headache (24%), nasopharyngitis (22%), myalgia (16%), and arthralgia (15%). 41 The incidences of these AEs were generally similar to the alglucosidase alfa plus placebo group in this trial (falls [39%], headache [24%], nasopharyngitis [8%], myalgia [13%], arthralgia [13%]). Similarly, falls, nasopharyngitis, and headache were the most frequently reported AEs in the pivotal placebo-controlled trial of alglucosidase alfa. 59 The exact incidence has not been reported, but was described to be similar to the placebo group. Overall, the three approved ERTs, alglucosidase alfa, avalglucosidase alfa, and cipaglucosidase alfa plus miglustat, had similar safety profiles. 60 To evaluate how miglustat affected the overall safety profile of cipaglucosidase alfa plus miglustat combination therapy in LOPD, we assessed the incidence of AEs that were considered by the investigators to be related to miglustat only in the pooled safety analysis. Overall, 21 of 151 patients (13.9%) experienced a total of 68 AEs that were related to miglustat only. The most frequent miglustat-related AEs were gastrointestinal (diarrhea: 15 events in eight [5.3%] patients; and nausea: 14 events in five [3.3%] patients) (Figure 6). All other miglustat-related AEs occurred in only one or two patients and were mainly gastrointestinal (Table 1).

Miglustat is administered with fasting and at a lower dose and less frequently in patients living with LOPD than in other approved indications and has less frequent GI side effects.
Of 151 patients with LOPD treated with cipaglucosidase alfa (20 mg/kg) plus miglustat (260 mg) in three clinical trials, 13.9% had AEs related to miglustat only, none of which were serious or led to study drug discontinuation.
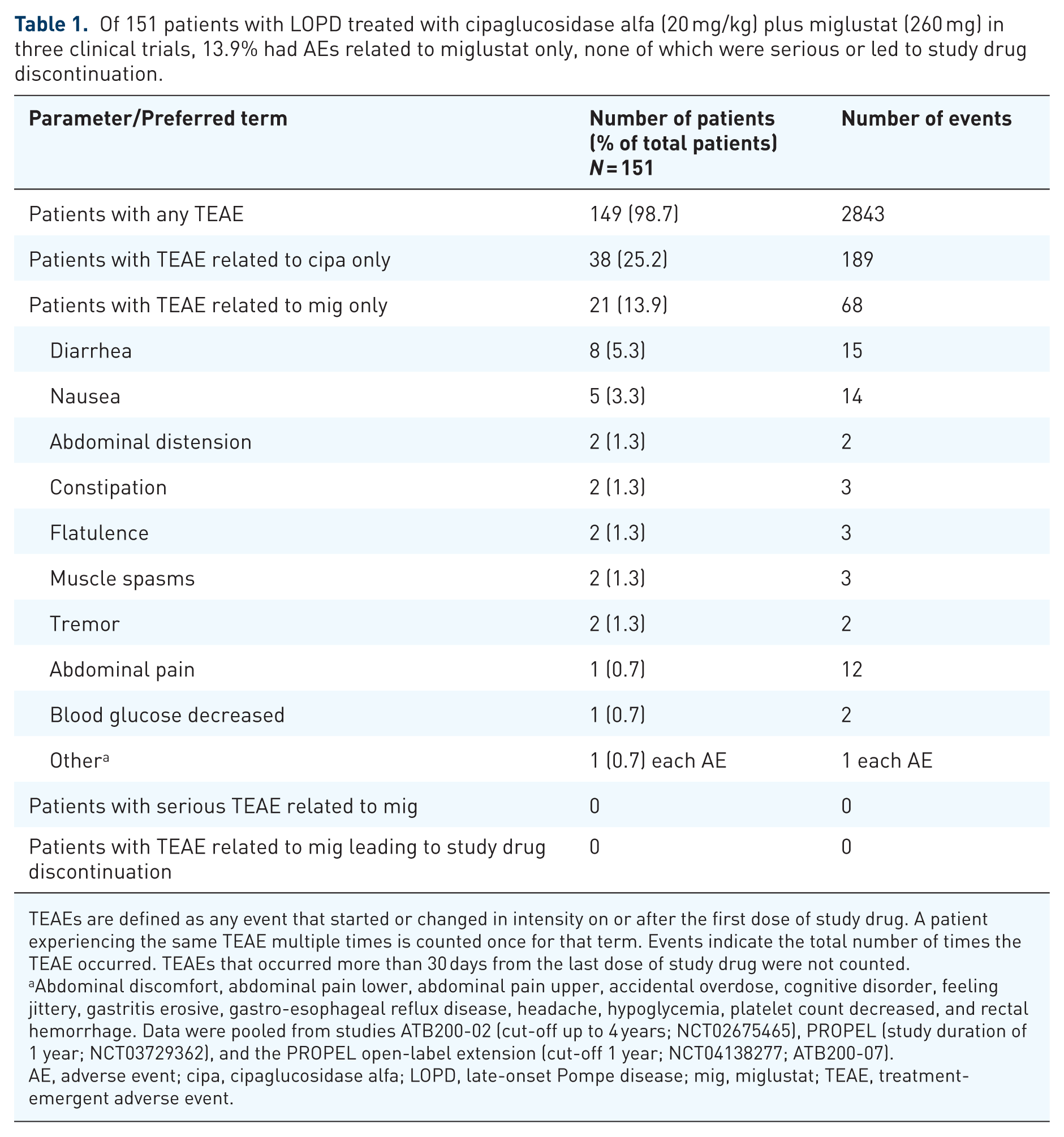
TEAEs are defined as any event that started or changed in intensity on or after the first dose of study drug. A patient experiencing the same TEAE multiple times is counted once for that term. Events indicate the total number of times the TEAE occurred. TEAEs that occurred more than 30 days from the last dose of study drug were not counted.
Abdominal discomfort, abdominal pain lower, abdominal pain upper, accidental overdose, cognitive disorder, feeling jittery, gastritis erosive, gastro-esophageal reflux disease, headache, hypoglycemia, platelet count decreased, and rectal hemorrhage. Data were pooled from studies ATB200-02 (cut-off up to 4 years; NCT02675465), PROPEL (study duration of 1 year; NCT03729362), and the PROPEL open-label extension (cut‑off 1 year; NCT04138277; ATB200-07).
AE, adverse event; cipa, cipaglucosidase alfa; LOPD, late-onset Pompe disease; mig, miglustat; TEAE, treatment-emergent adverse event.
Gastrointestinal side effects have also been described with miglustat treatment in GD and NPC, at a considerably higher rate than seen with miglustat treatment in LOPD.27,28,61,62 Gastrointestinal effects, mainly diarrhea, happen in more than 80% of patients with GD 28 and can occur at the onset of miglustat treatment or intermittently later during treatment. 27 Miglustat inhibits intestinal disaccharidases in the gastrointestinal tract and reduces the absorption of dietary disaccharides in the small intestine, causing osmotic diarrhea.27,28 In clinical practice, miglustat-induced diarrhea in patients with GD or NPC may respond to changes to patients’ diet (reduction of carbohydrate intake) and to taking miglustat between meals.27,28 Here, it is important to note the marked differences in the cumulative miglustat doses between the indications. The recommended miglustat dose for adults with NPC is 200 mg, taken three times a day, that is, a total dose of 600 mg/day. For adults with GD, the recommended starting dose is 100 mg, also taken three times per day, that is, 300 mg/day.27,28 In contrast, patients with LOPD receiving cipaglucosidase alfa co-administered with miglustat take a single 260 mg dose of miglustat (or 195 mg for patients weighing ⩾40 to <50 kg) every 2 weeks.32,33 Hence, the cumulative miglustat dose over 2 weeks is 8400 mg in NPC and 4200 mg in GD, compared with 260 mg in LOPD (Figure 6). In addition, the single-dose posology of miglustat in the cipaglucosidase alfa plus miglustat regimen for LOPD allows for a 2-hour fasting period before and after taking miglustat,32,33 which may further reduce gastrointestinal side effects. Such a fasting period would not be feasible in the treatment of GD or NPC, where miglustat is taken three times per day. In the 52-week PROPEL study, diarrhea occurred with similar overall frequency in patients treated with cipaglucosidase alfa plus miglustat and those treated with alglucosidase alfa (13% vs 11%). 41 In the pooled cipaglucosidase alfa plus miglustat safety analysis, no cases of miglustat-related diarrhea were serious or led to treatment discontinuations (Table 1). Of the 15 events in eight patients, 13 were mild and two were moderate in intensity, and all recovered. Overall, the considerably lower dose of miglustat in LOPD compared with the doses in GD and NPC, and the fasting period before and after taking miglustat, led to an improved side effect profile that appears to be well tolerated by patients with LOPD (Figure 6).
Discussion
In order to be effective, treatment of LOPD with ERT has had to overcome several challenges encountered by rhGAA following infusion. These include the vulnerability of infused rhGAA to inactivation at the near-neutral pH of blood that was not observed in the acidic pH of the lysosome, inefficient uptake of exogenous rhGAA by target cells at low enzyme concentrations, and the need for complete endolysosomal processing (proteolytic processing and removal of N-glycans) for GAA enzyme maturation once inside target cells. 5 Cipaglucosidase alfa plus miglustat was designed to address these three challenges. Initial investigation of potential small molecule enzyme stabilizers, such as duvoglustat hydrochloride, 34 highlighted that increasing the bioavailability of alglucosidase alfa, which has a low abundance of bis-M6P,4,6 only has negligible effects on improving treatment efficacy with this ERT in Pompe disease. The rhGAA cipaglucosidase alfa is naturally bis-M6P enriched (with an average of 1.1–1.3 moles of bis-phosphorylated N-glycans per mole of rhGAA) to facilitate uptake by target cells while retaining the capacity for complete GAA processing and maturation. 5 However, this exogenous enzyme still encounters the challenge of instability at the near-neutral pH of blood following infusion. Co-administration of cipaglucosidase alfa with the enzyme stabilizer miglustat aims to overcome this challenge. Miglustat binds to and stabilizes cipaglucosidase alfa following infusion, minimizing the loss of enzyme activity while in the circulation.6,15,32 This stabilization reduces the nonproductive clearance of denatured enzyme, increasing both distribution half-life and overall exposure. 21 Importantly, the role of miglustat as a small molecule enzyme stabilizer for cipaglucosidase alfa in the treatment of LOPD, given at a low dose just before ERT as indicated in the product label,32,33 differs from that of small molecule pharmacological chaperone therapies, for example, migalastat for the treatment of Fabry disease, which is administered regularly as monotherapy and acts to correct misfolded endogenous enzymes produced within the target cell. 26
In patients with LOPD, stabilizing cipaglucosidase alfa with miglustat translated into durable improvement or stabilization of clinical outcomes versus alglucosidase alfa at 52 weeks in the phase III PROPEL trial and relative to baseline at 104 weeks in the PROPEL extension study.41,56 However, the potential benefit of combining miglustat with other rhGAAs aside from cipaglucosidase alfa has yielded varying results in vitro and in vivo. While the co-administration of miglustat alongside alglucosidase alfa has been shown to increase GAA activity in fibroblasts and dried blood spots from patients with Pompe disease and to increase GAA activity in the muscles of Pompe mice,35,36 a recent study reported no added benefit of miglustat co-administration with alglucosidase alfa or avalglucosidase alfa on glycogen clearance in Pompe mice.15,16 Alglucosidase alfa and cipaglucosidase alfa have similar stability in vitro.6,21,22,24 The stability of alglucosidase alfa and avalglucosidase alfa is also similar, 15 suggesting that all three rhGAA have similar instability at neutral pH compared with acidic pH. It has been postulated that the lack of benefit of miglustat on PK parameters, such as AUC and glycogen clearance, with alglucosidase alfa and avalglucosidase alfa in Pompe mice described in a recent publication 15 may be due to a weekly dosing scheme rather than the every-2-weeks dosing that is used in the clinical regimen. 21 In mice, this may obscure the benefits of enzyme stabilization by saturating muscle cells with rhGAA. 21 Differences in glycan processing between avalglucosidase alfa and cipaglucosidase alfa could also obscure a potential benefit of coadministration of miglustat with avalglucosidase alfa. If miglustat increased the lysosomal delivery of avalglucosidase alfa, this may not be translated into increased activity if the enzyme could not be processed into its most active form 11 once it has reached the lysosome. In addition, it has been described previously that the increase in the cipaglucosidase alfa AUC with miglustat was considerably higher in human patients than in Pompe mice.5,15 Clinical trials of miglustat in combination with alglucosidase alfa or avalglucosidase alfa have not been conducted. Therefore, the contribution of enzyme stabilization of these ERTs with miglustat in patients with Pompe disease is unknown. Nevertheless, the durable positive clinical outcomes with cipaglucosidase alfa plus miglustat support the idea that an improved mechanism of action, including increased bis-M6P and enzyme stabilization, leads to clinical benefits. 5
A concern with the clinical use of miglustat is the high frequency of gastrointestinal side effects seen when used as an SRT in patients with GD and NPC. Such side effects, mainly presenting as diarrhea, are seen in over 80% of patients with GD and NPC receiving miglustat (Figure 6).27,28,62 However, in a pooled safety analysis, gastrointestinal side effects occurred in <10% of patients treated with miglustat in combination with cipaglucosidase alfa for LOPD. 32 The treatment regimen of miglustat when used as an enzyme stabilizer for cipaglucosidase alfa in patients with LOPD requires a substantially lower and less frequent miglustat dose (260 mg once every 2 weeks during a 4-hour fasting period) than when used as an SRT in patients with GD and NPC who receive miglustat three times per day with a cumulative dose of 4200 mg and 8400 mg, respectively, over the same 14-day period.27,28,32 Lower dosing, along with administration of miglustat during a short period of fasting (which is not feasible with the frequent GD and NPC treatment regimen), reduces the potential for gastrointestinal events in LOPD.
Limitations
There are several limitations to the available data examining the impacts of ERT with and without stabilizers for LOPD, primarily the lack of head-to-head trials of cipaglucosidase alfa plus miglustat versus avalglucosidase alfa, 5 cipaglucosidase alfa alone (without miglustat) versus alglucosidase alfa or avalglucosidase alfa, or miglustat in combination with alglucosidase alfa or avalglucosidase alfa. More real-world evidence and clinical experience are needed to elucidate the relative benefit of approved ERTs in Pompe disease. As expected for a rare, heterogeneous disease such as Pompe disease, the sample sizes in studies ATB200-02, PROPEL, and the PROPEL open-label extension were relatively small, and heterogeneity between patients may have introduced variability into the small datasets.40,41,56 Furthermore, the unblinded format of study ATB200-02 40 and the PROPEL open-label extension study 56 may have impacted safety findings. Another important limitation is that data for cipaglucosidase alfa plus miglustat in IOPD and pediatric studies in LOPD are not available yet, although studies are ongoing.44,45 Lastly, approved ERTs in Pompe disease do not cross the blood–brain barrier. Development of GAAs that can cross the blood–brain barrier is ongoing, 63 and future studies are needed to evaluate the potential effect of miglustat, which can cross the blood–brain barrier, 62 on such treatments.
Conclusion
Studies in mice and humans showed that stabilization of cipaglucosidase alfa by miglustat in the circulation improved cipaglucosidase alfa exposure and availability for uptake into the intended target tissue. This was associated with improved functional outcomes (in mice) and biomarker outcomes (in humans) compared with cipaglucosidase alfa alone. In a phase III clinical study in patients with LOPD, cipaglucosidase alfa plus miglustat led to clinically meaningful improvements. Miglustat was well tolerated in patients with LOPD, with a low frequency of gastrointestinal events considered to be related to miglustat alone.
Supplemental Material
sj-docx-1-trd-10.1177_26330040261425686 – Supplemental material for Miglustat: a first-in-class enzyme stabilizer for cipaglucosidase alfa for the treatment of late-onset Pompe disease
Supplemental material, sj-docx-1-trd-10.1177_26330040261425686 for Miglustat: a first-in-class enzyme stabilizer for cipaglucosidase alfa for the treatment of late-onset Pompe disease by Robert J. Hopkin, Barry J. Byrne, Mazen M. Dimachkie, Priya S. Kishnani, Tahseen Mozaffar, Mark Roberts, Benedikt Schoser, Nadine A. M. E. van der Beek, Ans T. van der Ploeg, Stephan Wenninger, Jon Brudvig, Brian Fox, Fred Holdbrook, Vipul Jain, Franklin Johnson, Jennifer Zhang and Giancarlo Parenti in Therapeutic Advances in Rare Disease
Footnotes
Acknowledgements
The authors thank the patients, their families, Pompe disease patient organizations, and the ATB200-02, PROPEL, and PROPEL open-label extension study investigators. Medical writing assistance was provided by Alyson Bexfield, PhD, and Simone Boldt, PhD, at Amiculum, under the direction of the authors in accordance with Good Publication Practice guidelines and was funded by Amicus Therapeutics, Inc.
Declarations
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.