
Abstract
The Koolen-de Vries Syndrome Foundation was founded in 2013 with the mission to educate, increase awareness, promote research and develop treatments for individuals living with Koolen-de Vries Syndrome (KdVS) and their families. With this aim, the foundation has focused on: developing scientific resources through patient cell and animal models, providing seed funding to basic and clinical researchers, establishing a natural history study of KdVS and increasing patient engagement. Projects have been prioritized across these areas of focus with an emphasis on expanding international research on KdVS, supporting translational research, establishing an international natural history study and conducting studies to assess patient priorities. With the incredible growth amongst our research and patient community in the last decade, our goal is to have our first clinical trial for KdVS in 2026.
Plain language summary
The Koolen-de Vries Syndrome Foundation (‘KdVSF’) was founded in 2013 with the mission to develop treatments for all individuals diagnosed with KdVS. With this aim, we have focused on several research priorities for our community: developing cell and animal models for KdVS for our researchers to utilize for experiments, providing research grants to KdVS basic and clinical researchers, establishing a natural history study of KdVS and increasing patient engagement and diversity. The KdVS research and patient community has expanded tremendously over the last decade, and there is growing excitement over the possible treatments currently being investigated amongst KdVS researchers. With our current focus on translational research and research aimed at identifying treatment strategies in KdVS patients, our goal is to have our first clinical trial for KdVS in late 2026.
Introduction
Koolen-de Vries Syndrome (KdVS) is a multi-system disorder due to a de novo, heterozygous 17q21.31 deletion or pathogenic variant in the KANSL1 gene.
1
KdVS is characterized by neonatal hypotonia, developmental delay, intellectual disability, speech difficulties,
2
epilepsy,3,4 congenital abnormalities of the heart and/or urogenital system, respiratory problems, ophthalmic issues, facial dysmorphism and musculoskeletal problems.5,6 Some children with KdVS are born with one or more congenital abnormalities,
5
and a large portion present with neonatal hypotonia and feeding problems. However, the diagnosis is often not recognized, as described by multiple parents recalling the start of their diagnostic odyssey: Our son, Max, was born with clubbed feet and an enlarged ventricle in his brain, but he was otherwise a healthy and happy baby. He was behind on milestones, but we attributed this to his leg braces for the clubbed feet but elected to do genetic testing on him just in case. Max was 18 months old and our family was dealing with our first bout of COVID-19. I was positive, wearing a mask at home and trying my best to keep my son from catching it. One day, Max slumped forward, hit his head on the ground, and began to have his first seizure in front of me. I called 911 and because I was COVID-positive, I couldn’t ride with him to the hospital. The doctors thought the seizure was fever-induced. Later that same day, we received a call with the genetic testing: Max had KdVS. In the span of a few hours, Max was both COVID-positive and KdVS-positive. A heart defect and brain abnormality was discovered at our 20 week ultrasound. We have weekly ultrasounds for the duration of the pregnancy and were told after open heart surgery at birth that would be it. At 2 weeks of age, we were admitted to the hospital for failure to thrive, jaundice, and feeding trouble. We were in and out of the hospital and on 15 medications round the clock. She had open heart surgery to repair 3 holes in her heart at 2 months old. She had trouble with anesthesia and her airway that required a lengthy PICU stay. Doctors noticed a sandal gap, wide set nipples, and an ear pit (things we didn’t even know existed). We were given a STAT full genomic sequencing test. A few weeks later, we met with the geneticist who told us that our daughter had Koolen-de Vries Syndrome and no one had ever heard of it. They gave us a pamphlet and walked us out the door.
These stories are reminiscent of the majority of the KdVS patient community and illustrate the basis upon which the Koolen-de Vries Syndrome Foundation was formed: to provide educational information, support and advocacy for families and their clinicians; to invest in research to better understand the clinical phenotypes to facilitate earlier diagnosis of KdVS; and to support research aimed at developing treatments for our patient community. The information below is a broad outline of the work we have done as a Foundation, the resources we have developed, the research priorities we have invested in and our roadmap for developing treatments for our children and young adults.
Developing a research roadmap
The Foundation invested in the development of a strategic research plan (SRP) conducted by COMBINEDBrain, a non-profit organization that advocates for rare, neurodevelopmental disorders. The project took approximately 5 months to complete and involved a team of translational neuroscientists to objectively review the current basic research, clinical and translational landscape of KdVS. This process involved interviews with current KdVS investigators and researchers, an in-depth assessment of the known and/or hypothesized molecular mechanism(s) of disease and a comprehensive review of existing disease model resources: patient samples, cell lines and animal models. A complete copy of the SRP complete along with the video presentation can be found on the KdVS Foundation website. 7
Our goal for developing a SRP was to clearly identify gaps in research where the Foundation could facilitate or recruit new or existing researchers to address those gaps.
Figure 1 is a graphical representation of the KdVS communities’ existing resources as well as the current gaps in scientific/clinical research or community engagement with FDA/government officials. Items written in green font are strengths or resources currently available to the KdVS community, while items written in red font are limitations or unavailable resources (Figure 1). Overall, the community has an active patient registry and natural history study run by Drs. Koolen and de Vries 8 and has a number of international neuroscientists investigating the molecular consequences of 17q21.31 microdeletions or KANSL1 variants; however, there are still issues diagnosing patients within the first years of life and finding those individuals who are diagnosed (Figure 1). There is also a lack of inducible animal models – animals for which the Kansl1 variant can be ‘corrected’ or ‘induced’ at various developmental time points. These additional animal models are critical to understanding the optimal therapeutic window in KdVS. Another current limitation in the drug development process is the identification of biomarkers for KdVS: biomarkers in human biological samples, fluids or physiologic indicators of disease (Figure 1). Perhaps most importantly, there was a clear lack of translational research or research aimed at correcting the Kansl1 haploinsufficiency. There was also a lack of clinical research in terms of understanding patient and family priorities for symptoms and developing a standardized treatment protocol.

Resources and gaps along the KdVS drug development pipeline from diagnosis to clinical trials. Current available resources are outlined in green while missing components (gaps) are outlined in red. Additional opportunities in the future are outlined in blue.
Given the importance of basic research models for understanding and developing treatments for rare diseases, we outlined all existing basic research resources for KdVS (Tables 1 and 2) to articulate whether there were additional needs in these areas of in vivo and in vitro models. Having multiple models of disease are important for addressing different questions related to disease pathology and/or optimal therapeutic window. For instance, it was important to create cell and animal models which genetically resemble both conditions under which KdVS is diagnosed: 17q21.31 microdeletions and Kansl1 variants. Furthermore, it was important to understand whether different Kansl1 variants (i.e. missense versus nonsense variants) had different cellular and behavioral outcomes.
In vitro models of Koolen-de Vries Syndrome.

Point mutations in Kansl1 were introduced in some lines using clustered regularly interspaced short palindromic repeats (CRISPR) from a wild-type control (WTC) cell line.
Reporter lines with an in loci Kansl1 fluorescent label are currently under construction and will be available to the research community in late 2024.
Unaffected family members had donated wild-type (WT) control lines.
In vivo models of Koolen-de Vries Syndrome.

Colony at Emory is the same genetic model as those developed at IGMCB.
Developing treatments
Our SRP provided several recommendations for expediting the development of treatments in KdVS patients. Our highest two priorities among these recommendations were (1) to support translational research efforts with an aim to identify clinical treatments strategies, and (2) identify patient/caregiver priorities as a means to select relevant outcome measures. To address these priorities, we developed a research funding announcement (RFA) with a direct call for basic research experiments utilizing molecular approaches to treat KANSL1 haploinsufficiency. We developed a formal, objective review mechanism based upon the NIH scoring rubric and had select Scientific Advisors review and score each proposal. This process allowed us to collectively decide which proposals were worthy of funding (i.e. scientifically sound with high translational potential). There was an incredible response to our RFA – which has led to the Foundation supporting four investigators new to the field of KdVS, all utilizing different therapeutic strategies to increase KANSL1 expression. In addition, we are designing a high-throughput study with FDA-approved compounds as well as small molecules known to cross the blood–brain barrier as a means to identify compounds able to increase KANSL1 mRNA expression. In an effort to facilitate this work and the work of other KdVS investigators, we are developing fluorescent reporter lines to be able to quickly quantify changes in KANSL1 protein expression.
Establishing a natural history
As our Foundation continues to grow, we have developed a greater appreciation for the importance of patient and community engagement in the drug development process. As interest in the field of rare disease expands, there are increasing research and clinical trial opportunities for our community. While this is excellent news for the field of KdVS, we are sensitive to the burden we place on our community in terms of assessments, surveys, video interviews, physical exams, and more. We try to balance these requests with maintaining enthusiasm and motivation to participate in these research opportunities. To balance these individual research opportunities, our Foundation has been prioritizing and facilitating collaborative efforts across researchers, promoting sharing of de-identified patient data and encouraging data collection platforms which allow individual patients and caregivers to share and distribute their data to interested researchers.
Platforms that fall in line with our Foundation goals to minimize burden on patient families include Simons Searchlight, RARE-X, Ciitizen and COMBINEDBrain. All platforms have either objective mechanisms by which to share de-identified data and/or have the ability for patients and caregivers to download and share their own data. These platforms are designed for longitudinal data collection, albeit the response rate amongst the community has been modest. As a means to develop a formal natural history of KdVS, in January 2024, we provided seed funding to Drs. David Koolen and Bert de Vries, who will establish a protocol at their local institute. The goal is to recruit 200 individuals with KdVS over 2 years and to share the developed protocol with additional, international KdVS clinicians to establish a robust cohort of KdVS patients and a large, longitudinal natural history study.
Patient identified priorities and engagement
The KdVS Foundation has increased its community base significantly since the launch of the Foundation in 2013 as highighted by the recent family turnout at the KdVS Foundation Family & Scientific Summit in 2023 which brought together approximately 100 KdVS families. The Foundation has launched a patient registry where they are able to collect basic demographic information and location information for members of the KdVS community. This registry allows for participants to indicate their willingness to provide their information to researchers and with other families. This is one of the primary mechanisms by which the community is informed about existing and new research opportunities.
We have also recently launched a monthly webinar series hosted by the Foundation and moderated by our Chief Scientific Officer. These monthly meetings are broadcast live on a variety of social media platforms as well as recorded and uploaded to our website. 9 The goal of these webinars is to review new clinical research opportunities and provide useful clinical information to our community.
In an attempt to better understand our community priorities, we have initiated two projects geared toward understanding the aspects of KdVS which are most burdensome on families/caregivers/patients. The first was an online survey developed by our Foundation to better understand symptoms of KdVS – as well as those which impact the caregivers and patients lives more significantly. In addition to prompting participants to select symptoms present in their child or young adult, we asked participants, ‘If you had a pill that could fix 3 symptoms [in the individual with KdVS], what would they be?’ Participants ranked symptoms in terms of most impactful to least. Over 90 caregivers completed this survey. A complete review of our findings from this survey will be published; however, briefly, we found that improvements in communication and GI symptoms would drastically improve quality of life for patients and caregivers.
A preliminary literature review for KdVS symptoms was done as part of the SRP completed in 2023. Figure 2 gives a general overview of the symptoms currently reported in the literature. This preliminary model outlines categories of symptoms present in KdVS using 12 ‘domains’: cognition, communication, neurological, behavioral, emotional, motor, musculoskeletal, sleep, gastrointestinal, vision, skin and ‘other’. Within these domains, specific symptoms are reported with the ‘size’ of the pie chart segment indicative of the frequency at which these symptoms are reported in the literature. This draft conceptual model for KdVS confirms the presence of several symptoms which have yet to be fully interrogated by clinical researchers in terms of prevalence and potential impact on clinical care. For instance, the model highlights the multitude of communication deficits in KdVS. Furthermore, Figure 2 outlines the variety of musculoskeletal problems, including congenital defects in several organ systems which impact standard of care for a significant number of KdVS patients.
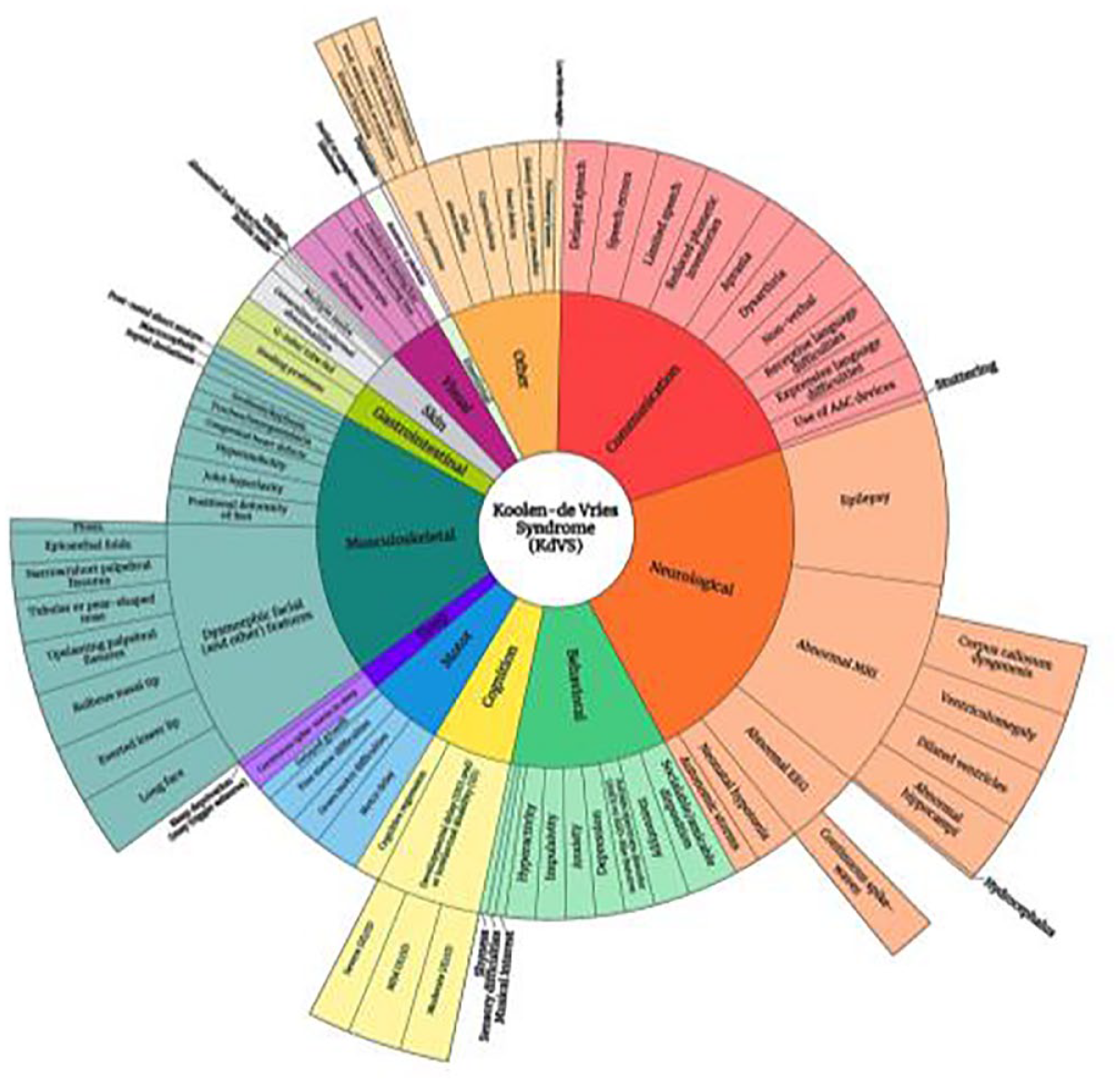
A preliminary conceptual model for KdVS based upon current published literature outlining symptoms in KdVS. Symptoms reported are categorized into 12 domains: cognition, communication, neurological, behavioral, emotional, motor, musculoskeletal, sleep, gastrointestinal, vision, skin and ‘other’. Specific symptoms within those domains are reported as a third or fourth tier to the pie chart. The size of each domain ‘wedge’ reflects the frequency at which those symptoms are reported in the literature.
A more formal study of KdVS symptoms is currently being done through the genetic counselor program at Medical College of Wisconsin. This study involves semi-quantitative interviews with caregivers, clinicians, educators, and therapists who have experience with KdVS. These interviews will develop an extensive landscape of the symptoms and disease burdens of KdVS from which to select and/or develop outcome measures for clinical trials. This often requires between 10 and 20 interviews with key opinion leaders. Select participants should represent different genetic conditions, clinical severities and ages of KdVS. These findings will be published and leveraged as the Foundation collaborates with researchers and clinicians to select relevant outcome measures for the first round of clinical trials for KdVS. These interviews will begin in late 2024.
Takeways
After over a decade of advocacy on behalf of KdVS children and their families, there are several prominent themes which continue to appear in the success stories of other rare diseases further along in the drug development pipeline. These themes primarily focus around transparency, advocacy and collaboration. Transparency and collaboration amongst our researchers to build upon each other’s work and potentially validate initial findings in additional disease models. Collaboration is amongst the clinicians who are novel to the field of KdVS with those clinicians with expertise in the of treatment a multi-system, that is a rare neurodevelopmental disorder. Collaboration amongst basic and clinical researchers to push the pace at which basic research findings are safely translated into clinical treatments. These messages and themes have been a focus of our work within the KdVS Foundation and our broader patient and scientific community. In addition to our work within our own disease community, we understand the importance of learning from communities further along in the drug development process and have developed collaborations with other advocacy groups, and last but not the least, a powerful base of patient advocates, who engage the patient and research community, promote diversity amongst basic and clinical research and consistently advocate for their children and young adults in a world that is primarily designed for those without cognitive, emotional or physical impairments.