
Abstract
SYNGAP1-related disorder (SRD) is a developmental and epileptic encephalopathy caused by a disruption of the SYNGAP1 gene. At the beginning of 2024, it is one of many rare monogenic brain disorders without disease-modifying treatments, but that is changing. This article chronicles the last 5 years, beginning when treatments for SRD were not publicly in development, to the start of 2024 when many SRD-specific treatments are advancing. We discuss the progress across many realms that have brought SRD to the forefront of drug development and highlight how Patient Advocacy Groups (PAGs) have had direct roles in accelerating the route to meaningful treatments for our children. We start with a summary of why SRD is an attractive pharmaceutical target. Second, we introduce the disease, the clinical features, and the number of patients. Next, we describe our PAG, our international partners and cite examples of the broad range of activities we believe are accelerating our pace toward treatments. We summarize the current SYNGAP1 pipeline and the status of each public project. Finally, we discuss two open questions that urgently need to be addressed in advance of clinical trials for SRD.
Plain language summary
SYNGAP1-related disorder (SRD) is a rare condition that is caused by accidental changes in the SYNGAP1 gene. It is one of many rare disorders that happen when a random change occurs in an essential gene, and one of many disorders with no gene-specific treatments. SRD causes multiple hardships for the people affected, including challenges from birth such as feeding and sleeping issues, a progression of seizure types that worsen over time, moving and thinking challenges, differences in sensing light, sound, touch, and motion, and communication difficulties. Our loved ones currently take many medicines that individually target different symptoms of SRD. Medicines are prescribed to reduce seizures, reduce anxiety and self-injurious behaviors, and improve sleep and allow for regular bowel movements. Even while taking many medicines, our kids have troubles in all these areas. In the year 2018, no treatments targeting the cause of SRD were publicly in development. In the beginning of 2024, many SRD-specific treatments are in development. New treatments that address the root cause of the disorder will mean less suffering for patients and their families. We hope to see reductions in seizures and improvements in sleep: improvements there may lead to more ease in communication and thinking, and possibly other symptoms too, like a reduction in sensory distress and anxiety. We hope to give fewer medicines to our kids, and for new therapies to have a more holistic healing effect than current options. This article serves to discuss the progress in many areas that have brought SYNGAP1 to the forefront of drug development, and to point out where SynGAP Research Fund and other advocacy groups have had direct roles in accelerating the journey on the road to meaningful treatments for our children.
Keywords
A compelling target for therapeutic development
SYNGAP1-related disorder (SRD) is a developmental and epileptic encephalopathy (DEE) caused by a disruption of the SYNGAP1 gene. While there are currently no disease-modifying treatments for the disorder, few monogenic ultrarare diseases have progressed as quickly as SRD on the path toward precision medicines. SRD presents a high value opportunity for industry given the clear roadmap to therapies, rapidly expanding assets and partners actively working on SRD.
SRD is caused by de novo mutations in one copy of the gene, resulting in a loss of fifty percent of the SYNGAP1 protein. One wild-type copy of the gene is in place, so finding a way to increase production or extend the utility of the protein is a compelling therapeutic strategy. Further, as we see in animal models, rescue is possible. 1 Multiple precision medicines are in development, and two repurposed drugs are in trials with patients.
Most importantly, there is an organized and effective patient community that is eager to partner with researchers and industry to accelerate disease-modifying therapies. Interestingly, this population, while already boasting impressive numbers, is significantly underdiagnosed for a variety of reasons, all of which are being addressed by researchers and industry in partnership with us. The size of this market, therefore, is expected to increase significantly.
We hope that this article will serve to encourage potential academic and industry partners, inform anyone interested in developing therapies for DEEs, and inspire other Rare Disease Leaders with the breadth of actions that can move the needle on reducing suffering.
SYNGAP1-related disorder
In 1998, the SYNGAP1 protein encoded by the SYNGAP1 gene was identified by two different research groups.2,3 The first linkage of SYNGAP1 heterozygous gene disruptions with intellectual disability (ID) in patients appeared in 2009 in Canada (n = 3). 4 Three key clinical features of SRD are ID, epilepsy, and autism spectrum disorder (ASD).5,6 In Figure 1 we compare the percentages of patients with various comorbidities reported in three recent publications describing SRD. All studies report six common comorbidities, including ID, epilepsy, features of ASD, severe behaviors, sleep problems, and gait problems. Vlaskamp et al. 7 (n = 57) extended the early clinical description to include eating problems, high pain threshold, tremor, hypotonia, and orthopedic abnormalities (Figure 1, green bars). In the study by Jimenez-Gomez et al. 8 (n = 15), only the main six features were reported (Figure 1, purple bars). The most recent McKee et al. 9 and largest study (n = 147; Figure 1, blue bars) evaluated four additional comorbidities, including sensory problems and anxiety, but did not evaluate hypotonia and orthopedic abnormalities reported in previous studies. In Figure 1, comorbidities are ordered to highlight their frequency and differences in reporting.

Clinical features of SYNGAP1-related disorder, not all symptoms are reported in all studies.
With a young patient population, most often diagnosed by pediatric neurologists, there is a need to characterize the clinical features of SRD adults to understand the disease’s progression. To date, Rong et al. 10 (n = 14) is the only dedicated publication of SRD adults, reporting:
Almost all adult patients depend on caregivers for many activities of daily living.
71% continue to have seizures.
Aggression and self-injurious behaviors are major management challenges.
50% of adults can ambulate with minimal or no assistance.
Publications related to the clinical features of SRD are summarized in Table 1, highlighting number of patients per study (n), and focus of clinical description.
Overview of publications reporting clinical features in SRD.
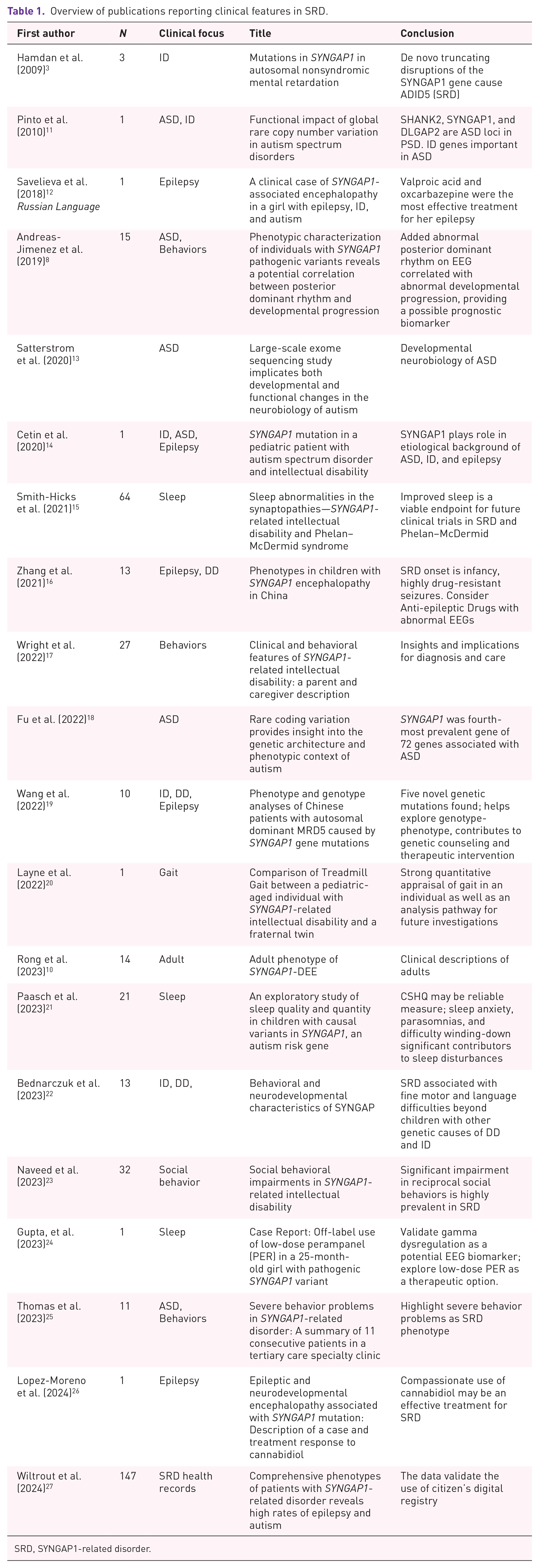
SRD, SYNGAP1-related disorder.
Based on SRD patient medical record data and healthcare claim data, the team led by Dr. Jillian McKee at Children’s Hospital of Philadelphia has presented the two largest cohorts of SRD patient data to date with a focus on epilepsy (n = 155) 28 and other clinical features (n = 246) 29 using novel data-driven approaches. Multiple new insights are presented in rich graphical summaries displayed in two posters at the 2023 American Epilepsy Society meeting. One example of an easily grasped visual description describes the onset of symptoms relative to age, based on clinical reports.
SYNGAP1-related disorder patient numbers
A critical question is, How many SRD patients are there?
The first insight comes from the answer to the question, How many patients can we count? The Syngap Global Network, a collection of collaborative SYNGAP1 Patient Advocacy Groups (PAGs) from around the world, publishes a census every quarter. The March 31, 2024 SYNGAP1 Census 30 identifies 1400 patients from 61 countries (Figure 2), but this number is known to be low, as only patients known to the PAGs are counted. From our own experiences as families, we know that our disease takes a long time to get diagnosed, can be misdiagnosed, is ultrarare, occurs in adults as well as children, and as a de novo heterozygous haploinsufficiency is sporadic, so it is spread over the globe. Despite the difficulties in obtaining an accurate count of patients, the steady growth of this number is notable.
Next, we consider the research that shows how many SRD patients occur in cohorts of people with common SRD symptoms. The data from cohort studies can be used to extrapolate the total patient population whether diagnosed or not, whereas counting does not address the undiagnosed population.Table 2 lists data showing that SRD patients are present in both ID and epilepsy patient cohorts. Based on these numbers, we expect many more SRD diagnoses than the 401 patients counted in the United States. It is easy to conclude that there are a large number of undiagnosed patients. Studies on cohorts of patients with ID from 2009 to 2020 found 1%–6% had a pathogenic variant of SYNGAP1. The aggregate among all studies is 1.7% (Table 2). These studies have led to the established belief that ‘SynGAP mutations are estimated to underlie approximately 1% of all intellectual disabilities and are associated with epileptic encephalopathies.’ 27
Incidence estimates for the 100 most common neurodevelopmental disorders (NDDs) caused by de novo variants are reported in the Supplementary Materials of a paper from Lopez-Rivera et al. 31 There, it is predicted that the incidence for SRD is 6.107 in 100,000 births. This prediction of pathogenic variant incidence is an aggregate for de novo protein truncating variants (at 0.787 in 100,000 live births) and de novo missense variants (at 5.320 in 100,000 births). From the clinical record data aggregated for SRD at Citizen Health, a secure online platform used to compile and store medical records (Table 3), we know that protein-truncating variants outnumber pathogenic missense variants for the currently diagnosed population. We see about 80% of the disease variants are considered haploinsufficient; ~12% are missense or small in-frame indels, requiring assays to understand the consequence of each individual variant. The 8:1 ratio of protein-truncating variants to missense variants seen in our patient population does not reflect the ratio of 1:6 ration of protein truncating variants to missense variants predicted in Lopez-Rivera et al.
Where might the rest of the predicted missense variants be? While a few patients with variants of uncertain significance (VUS) are in the Citizen Health cohort, the number of missense variants in de-identified lists of VUS from a few diagnostic labs (personal communication) is several times higher than all known US protein-truncating alleles, suggesting that even with current genetic testing rates, more patients will be identified with better VUS resolution tools.
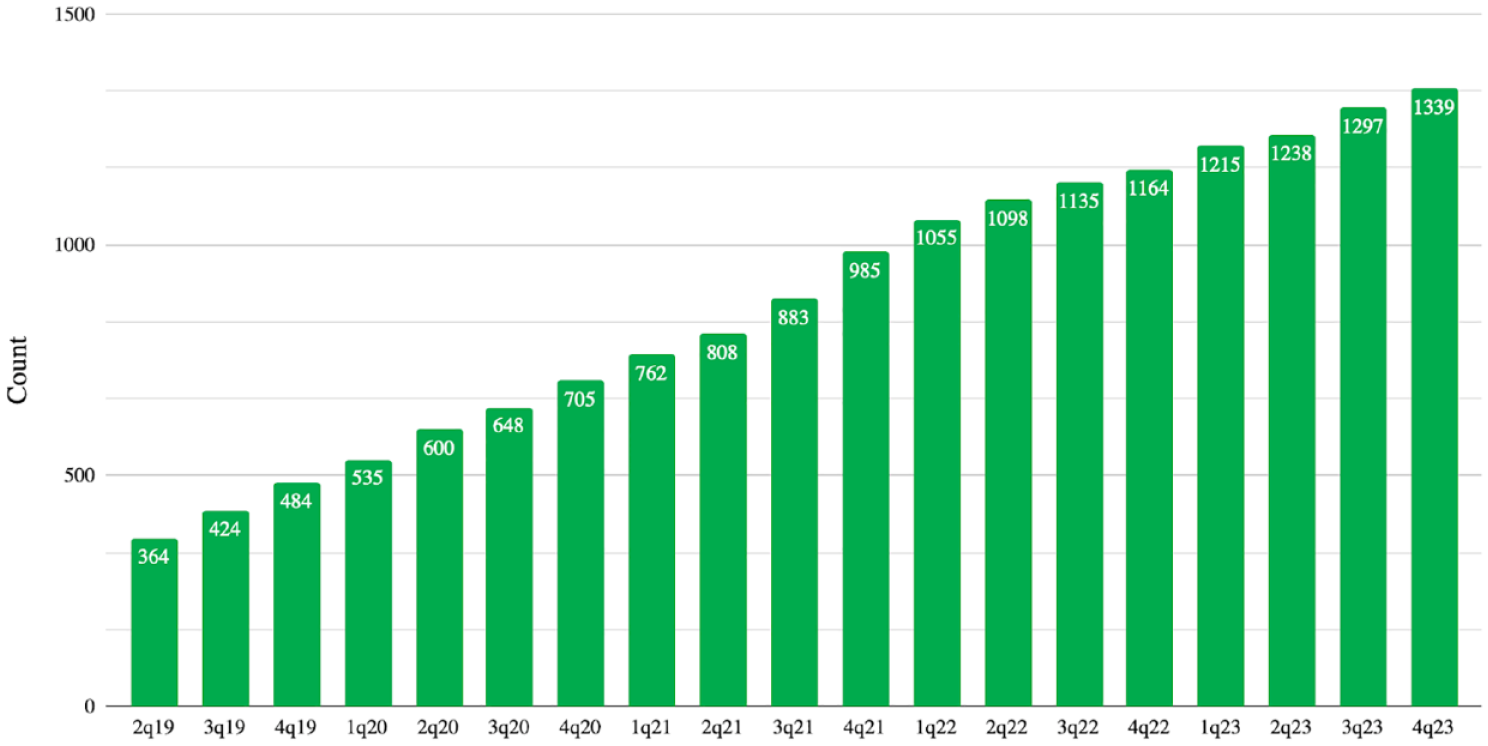
Patient counts from the SYNGAP1 census.
Incidence of SYNGAP1 pathogenic variants in ID and Epilepsy Cohort Studies.

NSID, nonsyndromic ID, **CNV, copy number variant.
Variant types in the first 195 SRD patients in the Citizen Health cohort.

PLP, Pathogenic or Likely Pathogenic; VUS, Variant of Uncertain Significance.
Why should we prioritize finding undiagnosed patients?
Expanded genetic testing for epilepsy, ID, and autism at all ages is required to find the patients. Identifying and observing more patients will aid in developing appropriate therapeutic drugs. Under current standards of care, expanded genetic testing is most likely to be initiated after one or more disease-modifying treatments for SRD are available and affordable, yet creating and approving those drugs requires more identified patients. Will treatments come before increased diagnosis, or will increased diagnosis come before disease-modifying treatments? We believe that equitable healthcare demands more genetic testing for underserved populations, including those with severe behaviors and ID. Increased diagnoses for all people with severe developmental disabilities will accelerate treatments for all.
SynGAP Research Fund and the global SYNGAP1 patient community
SynGAP Research Fund has worked to ensure that the broad SYNGAP1 patient community is well organized, effectively engaged, and motivated to partner with industry to accelerate treatments for SRD.
Fundraising and grant making
The SynGAP Research Fund was founded by the parents of an SRD patient diagnosed in early 2018. The patient has an intronic mutation, 37 and the parents quickly realized the tremendous gaps in scientific understanding of how the SYNGAP1 gene works and how to meaningfully improve the lives of patients. As the name implies, the parents’ vision was that SynGAP Research Fund (SRF) would primarily fund scientific research. The story of SRF and its founders is described in various articles38 –41 and podcasts.42,43
SRF was incorporated on June 27, 2018, and granted 501(c)(3) status less than a year later. 44 At the end of 2023, after the first 5 years of work, SRF delivered quantifiable results 45 including having committed over $5M to research in 44 different grants, ranging from $5000 to $500,000.
SRF expanded the scale and scope of grant making from initial grants to develop a therapeutic treatment based on antisense oligomer technology to the broad array of topics required to deliver disease-modifying therapies to SRD patients. The categories are summarized in Table 4.
Categories of SRF Grantmaking.
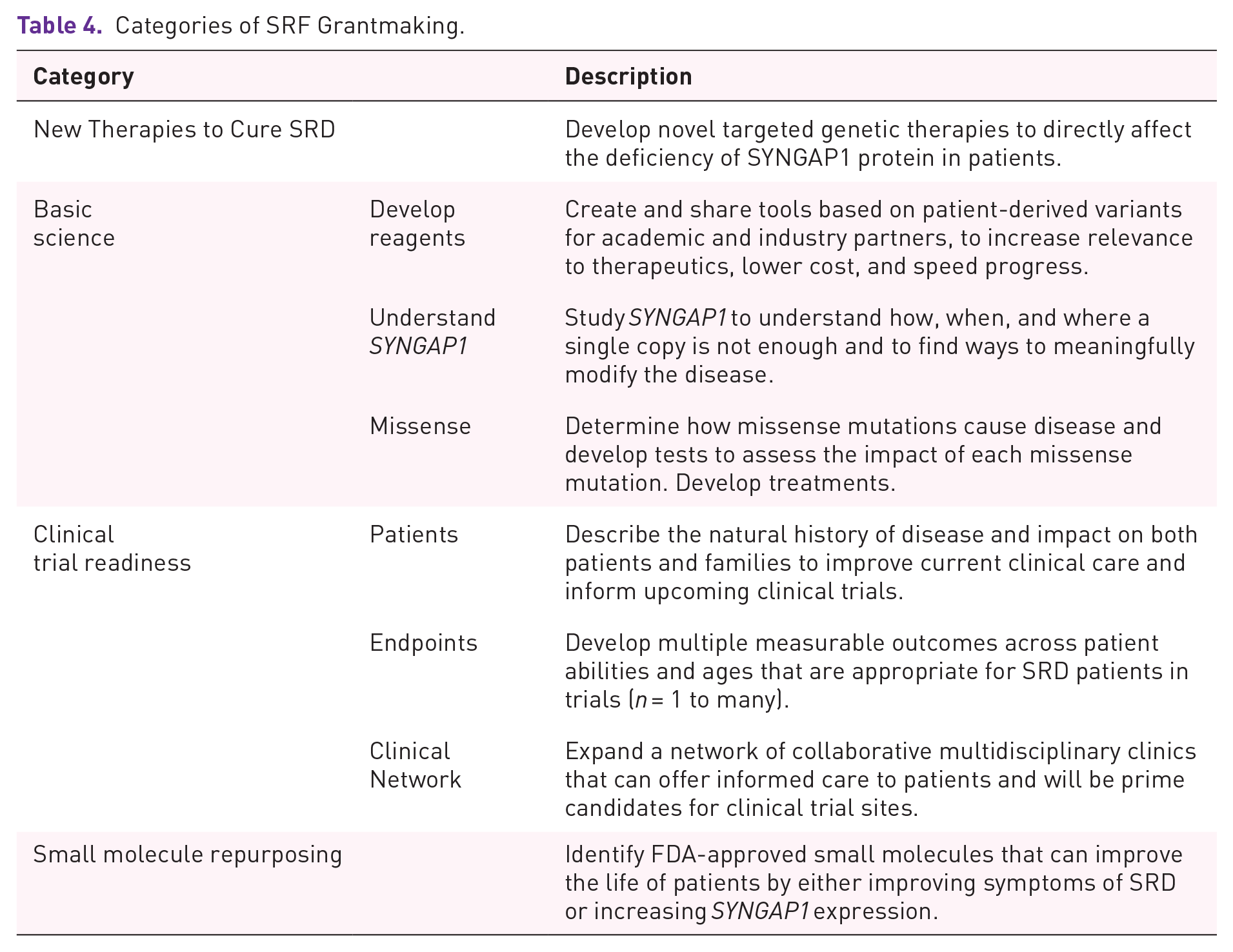
Table 5 lists specific grants funded by SRF within each category, by year initiated. This table is built on information that was presented at the 2023 SYNGAP1 Conference December 1, 2023 in Orlando, Florida.
Specific grants by category and year.

Txpta, CB, Transcripta Bio and COMBINEDBrain; **CH, Citizen HealthAAV, adeno-associated virus; ASO, antisense oligomer technology; BCH, Boston Children’s Hospital; CHOP NHS, Children’s Hosp. Philadelphia Natural History Study; EEG, electroencephalography; iPSC, induced Pluripotent Stem Cell lines; PPI, Protein–Protein Interactions; UCD, Univ. Calif. Davis; UCSF, Univ. Calif. San Francisco
Patient community activation
SRF was built on the initial premise that science was not moving fast enough to get disease-modifying treatments into our loved ones, so we would accelerate it by funding research. As we studied more-established PAGs for other rare DEEs, we learned about the crucial role advocacy groups have in addressing gaps in clinical science, and the regulatory challenges faced when developing a new, safe, effective, available treatment. As the SRF community grew and began connecting, we learned about our patient population from each other, recognized the gaps in our clinical care and considered new avenues for basic research. What began as an effort to raise funds and support a few labs already working on SYNGAP1 evolved into a coordinated PAG.
In addition to building standard PAG tools, including creating a website, 46 establishing groups on social media and developing a mailing list, gathering Scientific 47 and Clinical 48 Advisory Boards, SRF did a few things that were highly effective for activating the patient community.
Global Collaboration. SRF is a core member along with other collaborative SYNGAP1 organizations around the world that have formed an umbrella organization called the Syngap Global Network (SGN), which existed before SRF US was created in 2018.49,50
Danielle Williams originally founded SRF Australia in 2016; she allowed SRF US to use the same name when it was founded in 2018. Katrin Deckers founded SRF UK in 2020 and then Stichting SRF Europe in the Netherlands in 2022. Victoria Arteaga established Fondo de Investigación Syngap (Fondo) which was SRF Latin America in 2020 and secured formal legal status in Colombia in 2023. Each SRF organization is an independent legal entity that respects local governance practices. The groups chose to share a name, web-presence, and brand to signal the strong unified community to our partners and to make it easier for industry to engage with our community.
Census. With the invaluable support of other SGN members, SRF launched a global ‘SYNGAP1 Census’ to document current data on number and location of patients about whom the PAGs were aware. 30 The original article counted fewer than 400 worldwide, and after publishing it in 2019, 67 patients reached out to SRF to be counted. The effort has evolved into an ongoing project that is updated quarterly. The most recent count on March 31, 2024, was 1400. SRF has shared our census playbook with other PAGs, and they are developing similar efforts. 51
Electronic Health Records, for Natural History Data. SRF was the first Rare Disease PAG to launch with citizen in 2020, 52 when ciitizen (now Citizen Health) expanded beyond its founding purpose helping cancer patients. Citizen Health offers a retrospective natural history platform which collects, standardizes, normalizes, and then shares back data about patient populations. 53 Importantly, the platform also gives patients a dashboard with their own information and facilitates sharing the same. This is all done in full compliance with the patient privacy protections outlined in the Federal Health Insurance Portability and Accountability Act. SRF has recruited over half of the patients about whom it is aware in the United States to Citizen Health. The work of engaging patients – one by one – to this novel platform catalyzed significant connectivity within the community.
Podcasts. In March 2021, SRF launched a podcast called SYNGAP10 54 to update the community and stakeholders about the work of SRF. This has proven to be a powerful tool for patient engagement and support which has been copied by multiple other PAGs.55 –57 In 2023, SRF expanded its use of this tool to include two interview series, SYNGAP1 Stories 58 and for our Spanish-speaking community, Café SYNGAP1, 59 all available on a SYNGAP1 channel 60 on Apple Podcasts, as well as on other major podcast platforms.
Webinars. In order to educate ourselves, families, and researchers, we have hosted 89 webinars as of June 1, 2024, on topics ranging from basic science, treatments in development, clinical insights, and more. 61
Conferences. Looking for inspiration from the Dravet Syndrome Foundation and their annual Roundtable the evening before the annual American Epilepsy Society meeting, SRF began with a breakfast the morning of the Dravet Roundtable in 2019. 62 In 2020, the SRF Roundtable 63 was online due to COVID-19. Similarly, the 2021 Roundtable 64 was also virtual but done in partnership with the STXBP1 Foundation and cureSHANK. The ‘SRF Roundtable’ grew to a full 2 day conference in 2022 65 which continued into 2023. 66 Most of the talks are available on the SRF YouTube channel and website.
Fondo, SRF-Latin America, has held two virtual meetings where hundreds of clinicians from the Americas joined for presentations and discussions around SRD. These events took place in 2021 67 and 2023. 68
Advocacy
SRF has supported multiple families as they have had to advocate for appropriate medical care and education for their loved ones. Patient privacy prevents us from sharing detailed stories, but a few anonymized examples of SRFs work include:
A document written specifically for advocacy in education around the impact of SRD on student learning, behaviors, and development, is available: SYNGAP1: Background, Development, and the Impact on Children 69
Social Security Administration: Effective 8/11/21 SRD qualifies for automatic disability under the US Social Security Administration 70
When a preschool student with SRD was confined to limited services and instruction as a result of his behaviors, SRF advocates coordinated with state disability organizations, a university law center, and a private attorney to defend the child’s right to Free Appropriate Public Education and Least Restrictive Environment. Jackie Kancir, SRF Patient Advocacy Director, provided a written report that used existing scientific research on SRD to support the student’s legal right to appropriate services and accommodations. The document was used to reach a favorable conclusion of the case for the family. Excerpt: Our position is that behaviors must be managed with appropriate behavior intervention plans and accommodations and these behaviors cannot be a reason for limiting services or education time, as they are a symptom of the disability... .After reviewing all information provided, no challenges experienced to date appear outside the realm of SYNGAP1, and thus all challenges are believed to be symptoms of his disability. Individuals with symptoms of disability are protected from discrimination under ADA and required to be provided accommodations and supplementary aids and services under IDEA (ADA, Americans with Disabilities Act; IDEA, Individuals with Disabilities Education Act)
Supporting a NJ Family has involved patient advocacy through various aspects of healthcare, emotional support, living arrangements, and medical treatments. This has been helpful for families in need, as they are facing challenges such as financial hardship, lack of access to quality care, and emotional stress. Support has been successful by providing guidance and resources to help the family overcome these obstacles.
Jackie Kancir, Tennessee parent of a daughter with SRD, successfully advocated for development of Intellectual and Developmental Disabilities (IDD) Emergency Department Stabilization Protocols, 71 establishing a more dignified, appropriate, and holistic experience for patients with IDD. The protocols were first adopted by Vanderbilt University Medical Center 72 and since have been adopted by other hospitals across the nation, as well as the National Center for START (Simple Triage and Rapid Treatment) Services. 73
Jackie Kancir continues to advocate for improved crisis systems for patients with SRD and other NDDs. Her daughter was chosen as a pilot participant for the Tennessee Simple Triage and Rapid Treatment Assessment and Stabilization Teams (TN START AST) program. 74 TN START AST aims to reduce hospitalizations and police interactions with personalized crisis support teams and care coordination.
SRF Leadership provides comprehensive Individualized Education Plan (IEP) reviews and attends IEP meetings with families virtually upon request.
SRF has provided travel assistance for studies and conferences to ensure that families are able to participate, contribute, and benefit from these events despite financial constraints ($8076, in 2023).
Clinical advocacy
In addition to these efforts, SRF has advocated for ICD codes and a clinical guidance document.
Through the leadership and perseverance of Dr. Hans Schlecht, there now exist both an ICD-10 code from the US Centers for Disease Control, (CDC) effective 10/1/21 75 and an ICD-11 code from the World Health Organization (WHO) in 2023 (Table 6). Both of these codes required sustained advocacy and engagement with the relevant organizations. The impact of the existence of these codes is hard to overstate. Already in theUnited States, having a code has facilitated both patient counting and the use of billing data to understand medication trends. Further, in securing an ICD-11 code at the WHO level, SRF has seeded the code for international use, and as countries adopt ICD-11, they will be able to begin counting patients and understanding the resources they consume across the world. But more importantly, when precision medicines are available for SRD, the healthcare systems will have a diagnosis code to bill against. While this may sound painfully obvious, it is remarkable (and discouraging) to note how much resistance SRF and other rare disease PAGs have met seeking to secure an ICD-10 code from CDC, despite resounding expert support.
When a doctor sees a patient with SRD, it is often the first SRD patient they have seen. With the leadership of Dr. Marta Dahiya, Clinical Director, SRF created a Medical Considerations Guide for clinicians to help families partner with their practitioners to improve care for loved ones with SRD. 76
To further improve current medical care for people with SRD, SRF has worked to establish a Clinical Network so that people with SRD can be seen by doctors who know about DEEs and SYNGAP1. In addition, we are working to develop a Standard of Care, by asking individual clinicians to write case studies, and by asking clinicians to build on the SRF Clinical Considerations recommendations by creating a peer reviewed Standard of Care in the literature.
One of the most impactful acts of clinical advocacy, which has literally built our community, is getting SYNGAP1 sequencing onto multiple genetic testing panels at multiple testing companies. The phone-calling efforts directly to testing companies, by Aaron Harding, Critical Operations Director and others, have led to many people being diagnosed earlier, or at all. Besides Whole Exome and Whole Genome sequencing, SYNGAP1 can be found on panels for epilepsy, ID, autism, and combinations of these indications.
ICD codes for SYNGAP1-related disorders.

Diversity, equity, and inclusion
We believe that inclusivity and representation are crucial in understanding the full spectrum of this condition and key in the development of effective treatments as well as recognizing the importance of supporting families and caregivers who are navigating the challenges of SRD. Fondo is a strong advocate of Diversity Equity and Inclusion within the SRF community and the SRF Board of Trustees is invested in ensuring inclusion in our community, research studies, and clinical trials.
Global partners support the #SyngapCensus and recruiting
In addition to the international collective of SRF groups, there are a number of other national European organizations which have made significant contributions to our community. SYNGAP Elternhilfe in Germany, 77 Overcôme in France, 78 SYNGAP1 España, SYNGAP1 Switzerland, Famiglie Syngap1 in Italy, and SYNGAP1 Poland. 79 These organizations along with SRF are part of the SGN. 80 All of these groups contribute to the quarterly census effort.
In addition to active collaboration – on the census, various social media efforts and the annual SYNGAP1 Awareness Day, #Splash4Syngap, on June 21 each year – these groups work together to constantly recruit for studies. Some studies, such as questionnaires or digital assessments can enroll participants from multiple geographies and the SRF and SGN community collaborates to ensure we maximize participation. Thanks to joint effort of all SGN members, there is a medication survey result, which has data of 228 families from all around the world. 80 Similarly, for the Adult Phenotyping study led by Dr. Andrade, we are recruiting on three continents, North America, Europe, and Latin America with sites in Canada and the Netherlands.
Patients can mobilize significant resources
SRF has raised and committed approximately 5.75 million US dollars to SYNGAP1-related research at the time of writing, and some of these grants have been in partnership with SRF EU, UK, and Overcôme. But other groups have also been active in mobilizing resources: specifically SRF-Australia and Elternhilfe.
SRF-Australia was able to secure AUD$500,000 for research at the Florey in 2018. 81
More recently, the German patient organization Syngap Elternhilfe e.V. (Germany) initiated the EURAS project 82 (EUropean network for neurodevelopmental RASopathies), which focuses on neurodevelopmental diseases including CFC, Costello, Noonan, and SRD. EURAS involves 16 teams (researchers, clinicians, and patient representatives) from 8 countries and is funded by the EU and UK Research and Innovation with €8.45 million, effective June 2023. The 4-year project aims to improve disease tracking with a patient registry, create disease models, identify biomarkers, conduct drug screening, develop brain-targeted delivery technologies, and validate treatments through preclinical trials. With an emphasis on patient involvement, EURAS includes a Patient Board representing 25 European patient organizations and support groups, to ensure a strong collaboration between researchers and the patient community to promote accurate diagnosis and effective treatments.
How we navigate our journey: The Roadmap
SRF was founded with three core values, urgency, collaboration, and transparency, which infuse every decision (Figure 3(a)). Each person involved with SRF feels a strong sense of urgency to reduce suffering in our patients and families. Time is passing our children by without properly growing up their brains. Years without enough SYNGAP1 protein leads to deficits in brain development. This is why we say: Time is Brain. Our personal and collective sense of urgency helps us identify productive actions and to quickly pivot or evolve when necessary. Our commitment to collaboration is born from seeing how much there is to do, and how many others have similar goals. We have never wanted to build from scratch, we prefer to join umbrella organizations and share our homework with other PAGs. Our reliance on collaboration fuels our informal and formal partnerships, with people with similar goals in other PAGs, researchers in both academia and industry, and advocates. Our desire to operate in a way that is above reproach, and therefore, attractive to industry and academia, led to our commitment to transparency. Our evolving strategies and tactics can be found in every blog, podcast, newsletter, conference, and webinar.

SynGAP Research Fund Roadmap. (a) Founding values. (b) Selected specific actions by year, including goals for the next 2 years. (c) Principles to navigate by and our desired actions as a PAG. (d) The finish line.
Over the last 5 years, significant progress has been made on multiple fronts. Some of our less expected achievements are highlighted in the Roadmap, along with a few of our guiding philosophies (Figure 3(b)). Of all the possible actions that can be carried out by PAGs, we have two guiding principles to help us choose what to do when. First, we do our best to take advantage of opportunities. We are not afraid to introduce ourselves, ask questions, and follow-up. Second, we look for places to add a modest amount of our precious donated dollars to a project or lab that is already funded, so that our dollars leverage other monies. Examples include funding a 2-year postdoc position in a lab that has state-of-the-art core facilities and mentorship, and following up frequently with industry partners who are considering a technology that could be used as a measurable outcome for many DEEs, so that our population is part of the development of the tool. Both are predicated on relationships and amplify the power of our funds.
SRF focuses on the actions that accelerate the availability of disease-modifying treatments (Figure 3(c)). This is a long list: fund research and development, create partnerships with researchers, industry, other rare disease groups, advocates, and others. It is especially gratifying to facilitate the exchange of ideas and disease model assets, important accomplishments on the road to therapies. We prioritize patient engagement – in our case, engagement with caregivers of those affected with SRD – and it often leads to family friendships. Community is formed around the unusual and challenging issues that tend to leave us lonely and isolated in our local communities. SRF promotes awareness of SRD amongst scientists, clinicians, educators, and undiagnosed families. SRF’s Clinical Considerations document, blogs, Conferences, podcasts, and webinar series serve as communication and education around SRD, not just for professionals but importantly also for families, so that families can bring trusted information to their child’s own clinicians, teachers, and even local lawmakers and first responders. The result is a higher level of care, right now, for people living with SRD. Furthermore, engaged clinicians are key to finding institutions willing to host preliminary observational studies of repurposed drugs or supplements.
The Finish Line may seem obvious but notice the breadth: the equitable availability of affordable, safe, disease-modifying treatments for all people living with SRD (Figure 3(d)). That is a broad ask, and includes better diagnosis, resolution of VUS, safety, and regulatory wins, along with the many different therapeutic strategies in development.
Remarkable scientific progress continues on multiple fronts
Strong body of literature in place
The scientific literature on SYNGAP1 is robust and expanding. The Huganir Lab wrote a summary of the first 20 years published in 2020—this is an ideal introduction to SYNGAP1. 83 The rate of new articles is growing especially fast recently, with 152 articles from 1998 to 2019 per PubMed.gov and 117 from 2020 to 2023, with 44 in 2023 alone (see Figure 4).

284 Papers Published Related to ‘SYNGAP1’ from 1998 to mid-2024.
Labs working on SYNGAP1
When SRF was created in 2018, we found only a few labs working on SYNGAP1: Dr. Huganir, Dr. Rumbaugh, and Dr. Holder. SRF gave grants to all three. While there must have been other labs engaged in SYNGAP1 research, they were not public enough to find easily. Five years later, the number of labs and clinics working on SYNGAP1 is hard to keep up with, but many are working on SYNGAP1 with, if not because of, SRF support (see Table 7). Most of the labs listed have public webinars 84 discussing their work.
Labs working on SYNGAP1 or SYNGAP1-related disorder.

SIBD has multiple labs working on SYNGAP1 under the leadership of Prof. Peter Kind, PhD, and is supported by the Simons Foundation.
ENDD has multiple labs working on SYNGAP1 under the leadership of Dr. Ben Prosser, PhD, and is supported via an anonymous grant. 86
Support in partnership with U of Penn, Orphan Disease Center, Million Dollar Bike Ride (MDBR).
Research assets in place
A remarkable collection of assets with SYNGAP1 pathogenic variants are in place to inform research. Multiple patient-derived cell lines are available in multiple locations, a strong organoid collection is under study at the Coba and Quadrato Labs at USC, and a growing number of animal models from multiple species are showing new insights. We also have a biobank of patient biosamples available via our collaboration with COMBINEDBrain.
Induced Pluripotent Stem Cell lines (iPSCs). SRF maintains a public list of iPSCs and patient controls on our website. 87 We have learned that moving cell lines between labs is sometimes challenging and moving lines from labs to industry can be an onerous if not impossible task. As such, SRF has invested in a set of patient-derived iPSCs with same-sex family controls at the CRO Transcripta Bio (formerly known as Rarebase). These have been shared with multiple companies and labs for standard fees and are still available, see Table 8.
Organoids. SRF supported work in the Coba Lab 88 to explore SRD patient-derived organoids. Dr. Coba collaborated with the Quadrato Lab who has recently published a detailed study based on one organoid. 89 SRF has also given a grant to the Quadrato Lab to expand this work on three additional lines. We refer industry partners looking for expertise on iPSCs and organoids to USC to work with these labs.
Animal Models: Mice. At the time of writing, there are six mice on the Jackson Laboratory site resulting from a search for SYNGAP1. Three of these are experimental model mice (not patient-derived models) from the Huganir
90
and Rumbaugh91,92 labs, but the other three are recent and important additions to the collection. • Two mice have been developed and characterized
93
in the Huganir lab with patient-derived mutations c.3583-9G>A
94
and L813RfsX22.
95
This work was supported, in-part, with the first grant SRF gave in 2018. • The Prosser Lab created a SYNGAP1-humanized mouse
96
that is also available. This is useful for therapies that act on parts of the human SYNGAP1 gene which are not expressed in the murine version of SYNGAP1. • SRF has recently developed a third patient-derived mutant mouse with the Jackson Labs, the nonsense variant SYNGAP1-Q503X, and it is currently being characterized with SRF support. It will be the seventh SYNGAP1 mouse at Jackson Labs, and will be available later in 2024.
Animal Models: Rats. There are multiple rat models of SRD as well. The majority are at the SIDB at U of Edinburgh, where the rat is the preferred model of study due to its detailed brain anatomy, EEGs, 97 and a broad range of behavioral assays (e.g., social hierarchy.) 98 Additionally, the Kennedy Lab collaborated on a model with SIDB, 99 and the Willsey and Kastner Labs at University of California San Francisco (UCSF), funded by SRF, have begun a 2-year project on SYNGAP1 protein haploinsufficiency, widening the focus from PSD localization in brain tissue to include other cellular localizations (for instance, cilia) and other tissues (e.g., respiratory tract, gut, and kidney).
Animal Assays: Zebrafish. The Baraban Lab developed a zebrafish SYNGAP1b model of epilepsy 100 that is available for collaborators. The Dallman Lab both published101,102 and has forthcoming work 103 on zebrafish models including SYNGAP1a/1b, with assays for excitability and gut motility.
Animal Assays: Frogs. The Willsey Lab presented findings at the SYNGAP1 Conference 2023 that we expect to be published later in 2024, and SRF has recently begun supporting work on frogs in this lab. Earlier work by Dr. Willsey 104 showed that Xenopus tropicalis embryos can be used as powerful, fast, high-throughput models and assays. In addition to studying SYNGAP1 biology, the frogs may hold the key to an assay that could segregate missense and VUS variants into categories by function.
iPSC lines from SYNGAP1-related disorder patients available at Transcripta Bio. 87

For protein-truncating variants, the (partial length protein is not expected to be expressed, resulting in a haploinsufficiency model of disease. For single nucleotide changes the full-length mutant protein is expected to be expressed, and the disease model is uncertain.
Clinical research
A number of clinics serve patients with NDDs and many of these are building expertise with SRD and have a collaborative relationship with SRF and/or an international SYNGAP1 PAG. In Table 9, we highlight clinics with SRD clinical research studies.
Clinical research sites – US unless other two-letter country code (Head Researchers).

Support in partnership with the American Epilepsy Society (AES).
Adult study collaboration.
To demonstrate whether a treatment meaningfully modifies a disease, the progression of the untreated disease must be understood. Case Studies, Clinical Descriptions, and Natural History Studies build on each other, detailing the abilities and challenges of patients over time. The SRD community has three published clinical descriptions of patients, and four Natural History Studies currently in process.
Clinical descriptions of SRD
Michaud’s group first linked the SYNGAP1 gene to a disorder (Autosomal Dominant ID Type 5) in 2009 with a description and sequencing from three pediatric patients. 4 In 2018, Dr. Kluger’s group in Germany, with support from SYNGAP ELTERNHILFE e.V., reported a new type of reflexive seizure found in pediatric patients. 105 In 2019 a group led by Jimenez-Gomez published a retrospective clinical data analysis of 15 patients in which they described a possible biomarker found in an abnormal posterior dominant rhythm on EEG. 8 Also in 2019, a group led by Ingrid Scheffer published a description of the epileptology of 57 patients with SRD, 7 including reporting a new type of seizure specific to SRD, a reflex seizure during eating. Together these three seminal papers give strong insights into the patient journey.
Natural history studies in progress
Currently, there are several Natural History Studies in progress. The first, run by Citizen Health, formerly owned by Invitae, is a retrospective study mining data from the life-long Electronic Health Records of patients. The dataset now contains 218 patients, is updated multiple times a year (as patients age and receive more medical care), and additional patients can be added to the cohort. The data are already being used by four academic researchers and three industry partners (Figure 5).

Citizen Health and SynGAP Research Fund partner to present standardized, normalized, and harmonized data based on to researchers in academia and industry.
A traditional Natural History Study requires multiple examinations and tests over time. The Center for Epilepsy and Neurodevelopmental Disorders (ENDD) at Children’s Hospital of Philadelphia (CHOP) has enrolled and had the initial exams with 70 SRD patients, since July 2023. Each patient sees five separate specialties all in 1 day to reduce the burden on families and patients. The goal is to enroll 100 patients, and the longitudinal study begins in July 2024 by having the same 100 patients return for follow-up visits.
The EURAS project: The German patient organization Syngap Elternhilfe e.V 77 (Germany) initiated the EURAS project, which focuses on neurodevelopmental diseases such as Costello, Noonan, and SRD. 106 EURAS involves 16 teams (researchers, clinicians, and patient representatives) from 8 countries and is funded by the EU and UK Research and Innovation with €8.45 million from June 2023. The 4-year project aims to improve disease tracking with a patient registry, create disease models, identify biomarkers, conduct drug screening, develop brain-targeted delivery technologies, and validate treatments through preclinical trials. With an emphasis on patient involvement, EURAS includes a Patient Board representing 25 European patient organizations and support groups, to ensure a strong collaboration between researchers and the patient community to promote accurate diagnosis and effective treatments. 82
Finally, Professor Ingrid Scheffer has been developing the DEE Natural History Study over many years. This builds on 30 years of enrolment of families into the Genetic Basis of Epilepsy study, which has led to the discovery of many epilepsy genes, including SYNGAP1. She delineated the phenotype of SYNGAP1-related disorder in a research study which has led to the diagnosis of epilepsy in many individuals with SRD. Approximately 2000 patients with DEEs are currently involved in the study, as well as >10,000 individuals with epilepsy but not developmental challenges. The natural history study will be open to international patients from mid-2024. The study combines sophisticated data input with family and clinician input. It will foster collaboration with researchers on specific genetic epilepsies. This clinical research will delineate the natural history study of many specific genetic epilepsies, providing vital information to determine the efficacy of new therapies, including gene therapies.
Biomarkers and endpoints
Physiological. SRF is collecting bio samples via the COMBINED Brain Roadshow. The samples will be analyzed for ‘-omics’ discovery with COMBINED Brain partner studies and in projects directed by SRF.
EEG
The seizure progression in SRD starts out subtle and intensifies through escalating seizure types over years, as described in presentations at the 2023 SYNGAP1 Conference by Dr. Levin and Dr. Sulser. 84 SRF continues to advocate by speaking to researchers, connecting researchers, funding projects, and identifying partners to help describe our patients and determine appropriate endpoints for trials. For example, Beacon Biosignal’s work analyzing EEG data for SCN2A encephalopathy is an analysis we want to see replicated for SRD. 107
Validated scales
People with SRD often score at the bottom of the widely used measures of communication, cognition, and activities of daily living. To measure an improvement meaningful to our patients and families and to be able to detect and show a worsening, validated scales with appropriate functional ranges must be available.
ORCA
The Observer-Reported Communication Ability measure was developed to get detailed and nuanced descriptions of communication ability when communication is especially impacted. SRF helped recruit families to participate in interviews and validation studies to expand the ORCA from Angelman Syndrome to other DEEs. 108
NET
The Neurobehavioral Evaluation Tool was developed to capture a range of neurobehavioral abilities for DEEs. SRF supported Frazier to add SRD to the NET. 109 SRF broke records for recruitment to the study, as can be seen when comparing participants with different DEEs.110,111 In 2023, SRF has extended the collaboration by funding the validation of NET for SRD. 112
SRD pipeline
When SRF was formed, there were no therapeutic targets, no companies publicly working on SRD and no small molecules identified or in trials. Today, as seen in Figure 6, the reality is much different. Two small molecules are in trials, one in preclinical and another in discovery. At least three different repurposing screens are in progress. Three companies and ENDD have SYNGAP1 anti-sense oligonucleotides (ASOs) listed in public facing pipelines, two companies and ENDD have publicly declared adeno-associated virus- (AAV-) based therapies, and we are aware of other companies with SYNGAP1 programs. There is also a hematopoietic stem cell therapy project at University California Davis.
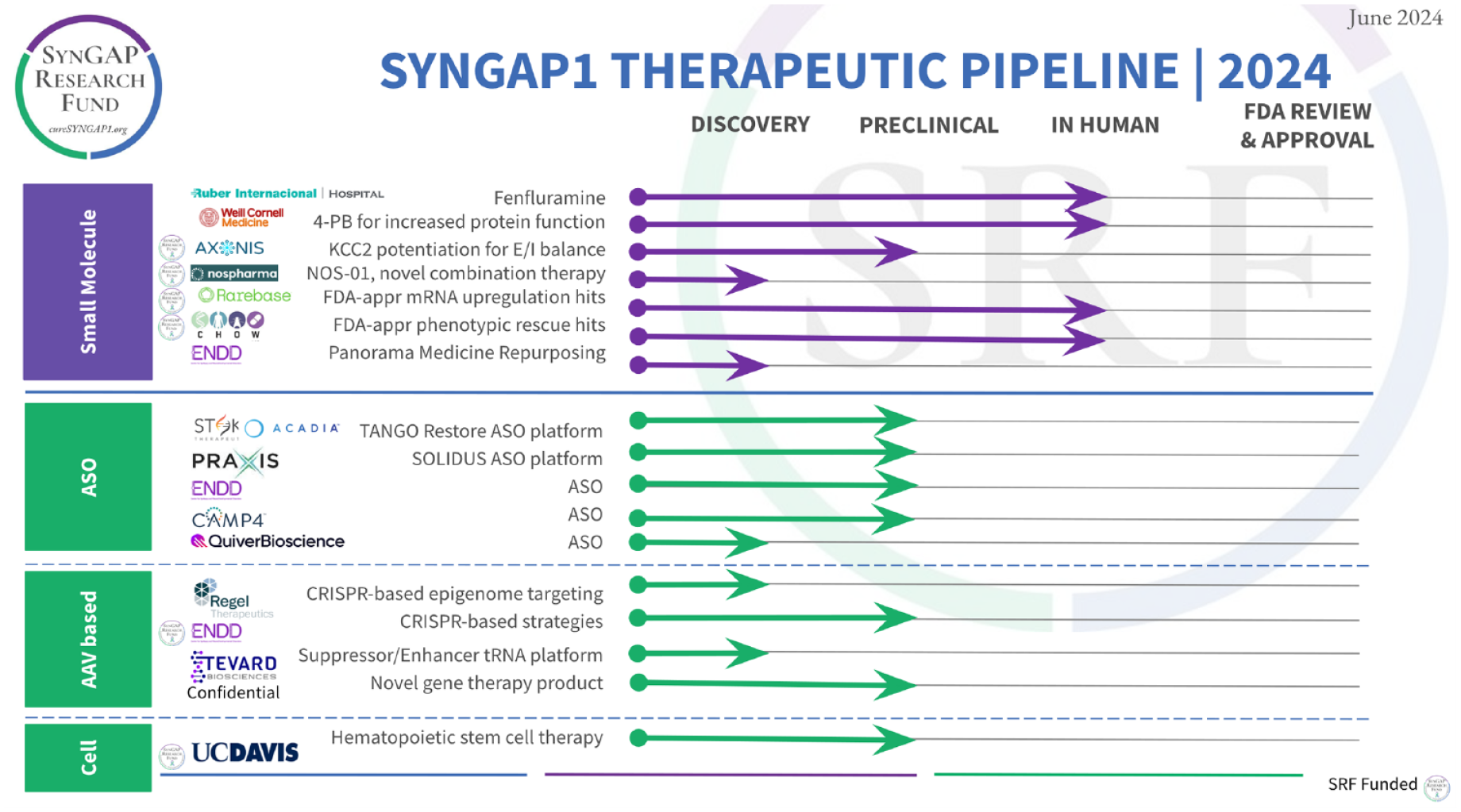
SYNGAP1 therapeutic pipeline for 2024.
Small molecule overview
SRF is currently tracking four specific molecules or combinations thereof to see if they can either ameliorate the symptoms of SRD or upregulate the protein. Two of these are in human trials already, the SRF-supported effort is in preclinical 113 and another in development. 114 Additionally, we are working with three broad screens looking for targets, two of which are entirely SRF-funded. These trials are important not only for the ability to serve patients in the near term but also as they could inform trial design for precision genetic therapies.
A trial at Weill Cornell Medical Center is exploring the possibility that 4-phenylbutyrate can help SRD patients. Building on promising results from a repurposing trial 115 with STXBP1 and SLC6A1, Dr. Grinspan has secured product and support to test 4-phenylbutyrate in other diseases. Based on research from Burre 116 and ongoing engagement with SRF, at least ten SRD patients have enrolled in the trial at the time of writing. SRF is not currently supporting this work financially, but we are working with Dr. Grinspan to support recruitment.
Broad drug repurposing efforts
SRF has invested in broad repurposing efforts to identify small molecules that may directly address the deficiency of SynGAP protein in our patients. While we are committed to the precision therapies mentioned above, they face a lengthy and uncertain development and regulatory process that promises to take more time than our families can accept. If we can find help for our patients with small molecules sooner, then we must. SRF has found two strong partners we are funding in this work: the Chow Lab and Transcripta Bio described below. Additionally, the ENDD program – who we partner with closely – has a drug screening program. 117
We are aware that the Rumbaugh lab secured an NIH grant in 2017118,119 and again in 2021 120 for a small molecule screen and subsequently announced a partnership with Praxis Precision Medicines in 2022 121 on the same effort. To date, we are not aware of any hits from this work nor the status of the partnership.
Chow Lab at University of Utah. With funding from SRF, the Chow lab has identified a robust assay for identifying the rescue of a SYNGAP1 haploinsufficiency model in Drosophila, finished screening a library of safe drugs, 122 and is currently following up on a validated hit.
Transcripta Bio, patient-derived cell lines in Palo Alto, California. We funded a multiyear research collaboration agreement with Transcripta Bio (formerly Rarebase) 123 to establish their Function drug discovery platform 124 and look for drugs that upregulate SYNGAP1 gene expression. This work involved a small molecule drug screen in wild-type glutamatergic neurons with RNA sequencing as the primary readout. This research has yielded several candidate molecules, including Food and Drug Administration- (FDA-) approved drugs, that upregulate SYNGAP1 gene expression and are under further study. As part of this agreement, Transcripta Bio established multiple patient peripheral blood mononuclear cells-(PBMC-) derived iPSC lines representing a series of different SYNGAP1 variants and made them available to the broader research community. The eight best-performing FDA-approved active molecules from the compound screening efforts were tested in concentration response mode in differentiated glutamatergic neurons from each of four patient-derived cell lines. In addition, Transcripta Bio has developed several orthogonal assays for SYNGAP1-mutant glutamatergic neurons, including confocal microscopy-based assays for calcium signaling, and an MEA electrophysiology assay for neuronal signaling. We are currently assessing the path forward for a small number of FDA-approved small molecule drugs that can more quickly advance to clinical testing.
ASO overview
SRD is a haploinsufficiency, meaning only one copy of the gene is working, and that is not enough for typical functioning. Strategies that may upregulate SYNGAP1 expression include ASOs designed to knock down a naturally expressed antisense RNA (SYNGAP1 AS1) or knock-down the naturally occurring splice variants that incorporate poison exons. Before January 2022, there were no companies working on SRD, and the only ASO we were aware of was the one being generated by the Huganir Lab with SRF funding, employing a knock-down of the natural anti-sense RNA. The collaborative design of a mouse with a humanized SYNGAP1 gene was initiated with this type of project in mind. Then in a presentation 125 Stoke Therapeutics announced that SYNGAP1 and Rett syndrome were both in their pipeline in partnership with Acadia. Three months later, Praxis Precision Medicine announced SYNGAP1 was on their pipeline in an April 2022 Presentation, 126 potentially based on this patent 127 from their Chief Scientific Officer, Dr. Steven Petrou.
ENDD in collaboration with IONIS was working on ASOs for SYNGAP1 per Dr. Prosser’s presentation to SRF in September 2022. 128 This work is based on previous work describing the binding and action of polypyrimidine tract binding protein 2, and splice variants that lead to nonsense-mediated decay. The work was published in Nature in May 2023 by Dr. Prosser 129 and described in this patent application. 130 The same insight was published by the Zhang Lab at U Chicago, who found it independently, in Cell. 131 Quiver Biosciences also notes SYNGAP1 in their pipeline. 132
AAV-based therapies
Regel Therapeutics announced their T3 Platform in January 2023, 133 and SYNGAP1 is third in their pipeline. 134 The Regel platform ‘combines AAV delivery with two proprietary technologies: a programmable dCas coupled with a modulator to epigenetically tune abnormal gene expression back to healthy levels, and a regulatory element (RE) that restricts the dCas intervention to the appropriate cells.’
Tevard Biosciences is using AAV-delivered RNA therapies to target haploinsufficiency. In their CEO’s presentation at the recent SYNGAP1 Conference, 135 he shared that Tevard is looking at SYNGAP1 as a potential target for at least one of their technologies.
The ENDD team at U Penn is also developing AAV-based therapies both via gene replacement under Dr. Davidson 136 and CRISPRa under Dr. Heller, 137 who began her SYNGAP1 research with SRF support. 138
Hematopoietic stem cell therapy
SRF funded 139 Dr. Joe Anderson of UC Davis in 2022, to explore using hematopoietic stem cell therapy for SRD as he had done for Angelman syndrome. 140 SRF has since funded follow-on work that is not yet formally announced.
Additional efforts in early stages
In addition to what is shown on the 2024 SYNGAP1 Therapeutic Pipeline update SRF is funding three researchers in the hopes their work progresses toward a therapeutic. Prof. Cobb at University of Edinburgh is working on a replacement gene therapy using AAV9 to deliver the gene, 141 Prof. Coller 142 is working on novel RNA therapies, and Prof. Zempleni is exploring exosomes as a delivery vehicle for SRD therapies. 143
Current challenges and open questions
As the goal of having safe, disease-modifying treatments for SRD moves closer, there are two areas of SYNGAP1 biology that pose important and urgent challenges for translational medicine.
Isoforms: Function and expression
There are many fascinating issues being addressed around SYNGAP1, but one of the most pressing from a translational perspective is isoforms. Some genetic/precision medicine approaches seek to upregulate production by the wild-type allele and as such leave the question of which isoforms to express to the existing regulatory mechanisms found in each cell type. Other approaches, however, may select a particular isoform to add back or increase and this forces the question, ‘Which one?’ Gross 144 explains the importance of SYNGAP1 isoforms while commenting on results from Kilinc, et al., 145 which shows ‘upregulation of α1/2 expression improved learning and increased seizure latency.’
The length of the SYNGAP1 gene pushes the limit for viral vectors, so finding ways to minimize the gene without losing function is paramount. Since there is compelling evidence that rescue with the major isoform is possible in rodents, 1 the next question is whether replacing only one isoform of SYNGAP1 will be helpful to SRD patients.
Like most human genes, SYNGAP1 has multiple isoforms. The ends have optional start and finish sites with five known choices at the N-terminus (A1, A2, B, C, and D) and four choices at the C-terminus (α1, α2, β, and γ). Most of the research to date has investigated the major isoform, A1α, encoding 1343 amino acids (Uniprot Q96PV0).
When and where are the different isoforms expressed? It is clear that ‘α1 isoforms are found enriched in the postsynaptic density,’ consistent with a major role in synaptic plasticity, learning, and epilepsy, whereas α2 isoforms have a developmental shift from a nonsynaptic to a mostly postsynaptic density localization with age, and β isoforms have been found enriched in nonsynaptic locations. The differential expression and subcellular distribution of SYNGAP1 isoforms may contribute to isoform-specific regulation of small GTPases, explaining SYNGAP1 pleiotropy. 146
Which SYNGAP1 isoforms have overlapping functions? Do any have an antagonistic function? Is protein localization, affinity for protein partners, or other activity altered due to differential N- or C-terminal exon choices? While we realize that meaningful treatments do not require a full mechanistic understanding, it is imperative to rigorously reduce the risk of harm when designing precision medicines. Understanding more about the roles of different isoforms is crucial.
Missense activity/VUS resolution
The second question is simple to ask but frustratingly complex to answer. ‘What happens to SYNGAP1 protein activity when a single amino acid is changed?’ Since SRD is a haploinsufficiency, the pathogenic variant is typically categorized as nonfunctional, or null, which is true for approximately 80% of currently diagnosed patients: those with a protein truncating variant (nonsense, frameshift, intronic) or large disruption of the gene (see Table 3). Missense variants, which make up 13% of the first 195 SYNGAP1 variants in Citizen Health, don’t follow the same simple disease model. In fact, for each missense variant, there are multiple possible categories of activity of SYNGAP1 protein ranging from benign to more severe than the haploinsufficiency (dominant negative). It is expected that any null and hypomorph variants can be grouped with the protein-truncating variants. However, missense variants could show hypermorphic, neomorphic, or dominant negative activities, which may have a different mechanism of disease.
The complications in determining the disease model for each missense variant have two consequences for SRD patients. First, patients with missense variants might be worsened by the therapies that upregulate SYNGAP1 gene expression, because the mutant variant will be upregulated along with the wild type. Therefore, for each missense variant SYNGAP1 protein stability and activity over multiple assays should be tested to determine which precision therapies are appropriate for each. Second, the inability to categorize variants with single amino acid changes into pathogenic and benign results in a shocking percentage of variants in SYNGAP1 labeled ‘uncertain significance.’ Categorization requires knowledge about the clinical presentation and insights into the structure and function of the changed protein. While it is unlikely that all people with a VUS in SYNGAP1 have SRD, it is important to note that the VUS population seems to be several times the size of the currently diagnosed population, so even a modest rate of SRD among VUS could significantly increase the patient population with the current rate of genetic testing.
Missense/variant of uncertain significance studies
A detailed set of assays in 57 patient-derived HEK cell lines was an early indication that the questions around missense variants are imperative to understand. 147
A multisite federally funded program called EpiMVP 148 is looking at all SYNGAP1 missense and VUS, as one of four nonchannelopathy monogenic diseases under study, to assess protein structures and functions and to increase the ability to predict the function of altered proteins. In addition to being part of the EpiMVP team, Dr. Carvill has been awarded funding via the Million Dollar Bike Ride (MDBR/UPenn) to expand on EpiMVP work on SYNGAP1. 149
In Finland, two separate missense projects are underway. Dr. Courtney 150 uses an AAV vector to express different SYNGAP1 missense variants in cells. He uses multiple assays including high-power imaging to describe in detail the effects of different single amino acid changes. The second project is a collaboration between Dr. Postilla and Dr. Pentikainen. 151 Their expertise in structural bioinformatics applied to SYNGAP1 will yield ‘The SynGAP Missense Server – A Resource Providing in Silico Predictions of SynGAP Variant Functionality’. These projects build on early support for the work from Leon and Friends, 152 a PAG based in Austria.
The structure of SYNGAP1 protein is currently based on three partial structures that include the C2 and RasGAP domains, and the coiled-coil domain. Over 50% of the protein structure is either not known or may be flexible, for example, in the large Domain of Unknown Function. Dr. Underbakke at Iowa State University looking at how missense variants alter stability and structure. 153 He has at his disposal powerful techniques to determine complex and flexible structures, including quantitative cross-linking mass spectrometry and hydrogen deuterium exchange mass spectrometry.154,155
Conclusion
We are encouraged by the speed at which disease-modifying therapies are advancing toward our loved ones with SRD. Still, every day our children living with SRD are building and living in a brain and body without enough SYNGAP1 protein. They are suffering, and so are we as parents and caregivers. We need collaboration. We feel urgency. Time is brain.
Footnotes
Acknowledgements
The authors would like to thank the following people for help with this manuscript:
For assistance with the content and design of Figures and Tables, we thank Suzanne Jones, Pavel Gerovich, Yulia V. Surovtseva, and Lindsay Wieczorek. We thank leaders of International Syngap PAGs for sharing their data summaries, information, and suggestions: Verena Schmeder and Marcos Mengual Hinojosa of SYNGAP Elternhilfe e.V., Krefeld, Germany; Katrien A. Deckers of Stitching Syngap Research Fund EU; Beata Tarasiuk of SYNGAP1 Poland; Victoria Arteaga of Fondo de Investigación Syngap, Columbia; and Danielle Williams of Syngap Research Fund, Australia. For assistance with the content and references in the sections on advocacy we thank Jackie Kancir. We thank Lindsay Wieczorek for comprehensive and detailed editing of the entire manuscript
The authors acknowledge their affected and unaffected children, who rise to challenges every day, and who have inspired and motivated all our work.