
Abstract
Background:
Pyridoxine-dependent epilepsy (PDE) due to biallelic pathogenic variants in ALDH7A1 (PDE-ALDH7A1) is an metabolic disease of lysine catabolism. Current standard treatment includes pyridoxine, arginine, and lysine- or protein-restricted diet. Pyridoxine treats seizures. Arginine and lysine- or protein-restricted diet decrease elevated α-aminoadipic semialdehyde (α-AASA) and Δ1- piperideine-6-carboxylate (P6C) levels to improve neurodevelopmental outcomes. We previously reported abnormalities in tricarboxylic acid (TCA) cycle and electron transport chain in PDE-ALDH7A1. We report a new patient with PDE-ALDH7A1 who did not show any improvements in neurodevelopment on the current standard therapy. We hypothesized that triheptanoin will provide substrate to TCA cycle and improve abnormal energy metabolism leading to improvements in neurodevelopmental outcome.
Objective:
To treat this patient with triheptanoin to improve neurodevelopmental outcome.
Design:
Due to complex I deficiency and lack of response to the current standard therapy, we applied triheptanoin novel therapy.
Methods:
A 4-year-old male had compound heterozygous variants in ALDH7A1 and markedly elevated urine α-AASA. The goal dose of triheptanoin was 50% of the estimated energy requirement (EER). We assessed efficacy of triheptanoin using neuropsychological assessments. We measured 6-oxopipecolic acid using liquid chromatography tandem mass spectrometry.
Results:
Triheptanoin was started at 10 mL/day. There was nausea up to 3 weeks after each dose increase, which has improved allowing us to increase triheptanoin gradually. The maximum actual dose of triheptanoin was 40% of EER. Cognitive composite score improved from 16% to 63% on treatment. All chemistry and biochemical investigations were normal. 6-oxopipecolic acid levels did not normalize. Triheptanoin treatment seemed to be safe and tolerated well.
Conclusion:
Triheptanoin is an anaplerotic agent to provide substrates to the TCA cycle. This novel therapy improved neurodevelopmental outcome in our patient with PDE-ALDH7A1. We think that trihepatonoin should be the part of the current standard therapy to improve neurodevelopmental outcomes in patients with PDE-ALDH7A1.
Plain language summary
Pyridoxine-dependent epilepsy (PDE) is caused by genetic changes in the ALDH7A1 gene (PDE-ALDH7A1). This genetic condition involves breaking down lysine, a building block of proteins. The typical treatment for this condition requires giving pyridoxine (a vitamin), arginine and tryptophane (building blocks of proteins) and decreasing protein intake in daily diet.
We previously reported that people with PDE-ALDH7A1 have problems with energy production. A boy with PDE-ALDH7A1 had the typical treatment in the first year of life. Unfortunately, his walking, talking and understanding (cognitive abilities) did not get better on this typical treatment. We decided to try a new treatment using a special oil, if this new treatment will help his body to produce more energy, as we found in his muscle biopsy a slight energy deficiency.
We started a special oil therapy, called triheptanoin. The dose of this special oil was increased gradually as much as he was able to tolerate. We used structured tools to assess progress in his walking, talking and understanding every year. He started gaining new skills and understanding better. His cognitive abilities raised from 16% to 63% using structured tools.
In some people with PDE-ALDH7A1, the typical treatment may not work well. Adding special oil to the treatment may help to improve people’s walking, talking and understanding.
Keywords
Introduction
Pyridoxine-dependent epilepsy (PDE) due to biallelic pathogenic variants in ALDH7A1 (PDE-ALDH7A1) is a rare inherited metabolic disease of lysine catabolism. ALDH7A1 encodes α-aminoadipic semialdehyde (α-AASA) dehydrogenase, and its deficiency causes accumulation of α-AASA, Δ1- piperideine-6-carboxylate (P6C), and pipecolic acid (PA). P6C inactivates pyridoxal-5’-phosphate, which is involved as a cofactor in more than 70 enzymatic reactions in human metabolism.1–3
Since the first description of PDE in 1954, about 300 patients with PDE or PDE-ALDH7A1 were reported in the medical literature. 4 Neonatal or early infantile onset seizures, global developmental delay, and cognitive dysfunction are the common clinical features. 3 The seizure types range from focal seizures to generalized seizures, and patients can have more than one type of seizure.1,3,5 Other neurological features include abnormal tone (e.g., hypotonia or hypertonia), movement disorders (e.g., dystonia and tremors), and behavioral problems (e.g., autism spectrum disorder and anxiety).1,3,5,6 Non-neurological clinical features include poor feeding, vomiting, and abdominal distension. Abnormal biochemical investigations have been reported (e.g., hypoglycemia and lactic acidosis).1,3,5–14 All patients have elevated α-AASA/P6C in body fluids. Urine and/or plasma PA can be normal in some patients; however, it is elevated in cerebrospinal fluid (CSF) in all patients.1,3,5–14 The current standard therapy consists of supplementation of pyridoxine, arginine, and tryptophan and lysine- or protein-restricted diet with lysine-free medical formula.3,6,11,13,15–29 Pyridoxine monotherapy results in seizure freedom in about three-quarters of patients. 30 Despite seizure freedom, only about one-quarter of patients have normal neurocognitive functions with borderline impairments in various processing functions on pyridoxine monotherapy.13,15,30 Pyridoxine monotherapy does not improve accumulation of α-AASA/P6C, which are neurotoxic metabolites. Arginine and lysine- or protein-restricted diet with lysine-free medical formula are effective treatments to decrease accumulation of α-AASA/P6C to improve neurodevelopmental outcomes.6,13,15–29,31 Lysine-free medical formula is tryptophan-reduced, and patients with PDE-ALDH7A1 do not require tryptophan reduction. To prevent tryptophan deficiency, they are supplemented with tryptophane. Further improvements in seizures and neurodevelopment have been reported on arginine supplementation and/or lysine- or protein-restricted diet with or without arginine supplementation.3,6,13,15–29,31
We previously developed a knock-out aldh7a1 zebrafish model using CRISPR-Cas9 technology mimicking human PDE-ALDH7A1. 12 We reported impairments in energy metabolism pathways as a possible novel pathogenetic mechanism for PDE-ALDH7A1. 14 We reported low paired complex I+II and complex II+III and individual complex IV activities in muscle biopsy in one patient with PDE-ALDH7A1. 14 We reported elevated CSF citrate, succinate, isocitrate, and α-ketoglutarate in another patient with PDE-ALDH7A1. 14 Homozygous knock-out aldh7a1 zebrafish had significantly low tricarboxylic acid (TCA) cycle metabolites (citrate, malate, fumarate, isocitrate, and lactate) compared to the wildtype zebrafish. Decreased electron transport chain enzyme activities were identified in homozygous knock-out aldh7a1 zebrafish compared to wildtype zebrafish. 14 We listed the possible multifold reasons for the abnormal functions of TCA cycle and electron transport chain in PDE-ALDH7A1 in our previous study including (1) α-AASA dehydrogenase deficiency in lysine catabolism leading to decreased production of acetyl-CoA and citrate for TCA cycle; (2) Pyridoxal-5’-phosphate deficiency secondary to accumulation of P6C leading to decreased productions of succinate, oxaloacetate, α-ketoglutarate for TCA cycle; (3) Pyridoxal-5’-phosphate deficiency secondary to accumulation of P6C leading to decreased productions of reduced nicotinamide adenine dinucleotide (NAD), NAD-hydrogen (NADH), reduced flavin adenine dinucleotide (FADH2), and succinate that are substrates for complex II; and (4) Pyridoxal-5’-phosphate deficiency secondary to accumulation of P6C leading to decreased iron heme synthesis and iron-sulfur cluster synthesis that are the core redox centers of electron transport chain complexes I, II, and III. 14 Neurotoxicity in PDE-ALDH7A1 is likely secondary to accumulation of α-AASA, P6C, and PA; to deficiency of pyridoxal-5’-phosphate; and to abnormal energy production leading to neuronal cell death. Triheptanoin is a medium-chain triglyceride composed of three odd-chain, 7-carbon-length fatty acids (heptanoates).32–35 It serves as a source of odd-chain fatty acids to bypass long-chain fatty acid oxidation defects, acting as an anaplerotic agent to provide ketones and substrates for the TCA cycle.32,33 Triheptanoin is approved by Health Canada and Food Drug Administration (FDA) for the treatment of long-chain fatty acid oxidation defects.34,35
We report a new patient with PDE-ALDH7A1 who did not show any improvements in the neurodevelopment despite early treatment and good treatment compliance on the current standard therapy of PDE-ALDH7A1. This patient had mild complex I deficiency. We hypothesized that triheptanoin will improve neurodevelopmental outcome by providing ketones and substrates for the TCA cycle to improve abnormal energy production. Our study objectives were: (1) to treat this patient with triheptanoin as a novel therapy and (2) to assess the efficacy and safety of triheptanoin in a single patient with PDE-ALDH7A1 using neuropsychological assessments at baseline and throughout the therapy. We report the outcome of triheptanoin treatment in a single patient with PDE-ALDH7A1 in this study.
Material and methods
Pyridoxine, arginine, tryptophan, and a lysine-restricted diet were started (Supplemental Table 1). Parents were very compliant with the current standard treatment. We did not see any improvements in developmental milestones during the first 10 months of the current standard treatment. There were declines in Bayley Scales of Infant and Toddler Development (Bayley-III) and Vineland Adaptive Behavior Scales-Third Edition (Vineland-3) scores across all domains. Declines in language, cognitive, fine motor, expressive language, adaptive behavior composites, communication, daily living skills, and socialization highlighted the severity of developmental declines on the current standard treatment (Supplemental Table 2).
A novel therapy using triheptanoin
Due to decreased electron transport chain enzyme activity and lack of response to the current standard therapy, we designed a novel therapy using triheptanoin and discussed with parents, while we continued the current standard therapy. This novel therapy study was discussed with Alberta Research Information Services (ARISE), and Clinical Research Dean at our institution. As there was a single patient and triheptanoin was provided as compassionate care treatment by Ultragenyx Pharmaceuticals, research ethics board approval was waived. Parents consented for this novel therapy study and signed a case report consent form to allow us to present their child’s de-identified information in the medical literature.
Goal dose of triheptanoin was designed as per the medium chain triglyceride (MCT) oil ketogenic diet treatment for the management of seizures, which is 40%–70% MCT oil. 36 As PDE-ALDH7A1 is one of the neurodevelopmental disorders and there were no seizures, we set the treatment goal to 50% of the estimated energy requirement (EER). The goal treatment duration was 2 years to assess efficacy of triheptanoin treatment using yearly neuropsychological assessments.
Assessment of the triheptanoin efficacy using neuropsychological assessments: We applied neuropsychological assessments using the Bayley-III and the Vineland-3 yearly and the Wechsler Preschool and Primary Scale of Intelligence-IV Edition (WPPSI-IV) at 4 years of age.
Assessment of the triheptanoin safety: We measured aspartate aminotransferase (AST), alanine aminotransferase (ALT), creatinine, urea, creatine kinase (CK), homocysteine, glucose, lactate, albumin, plasma amino acid analysis, PA in urine and plasma, and α-AASA in urine in clinical chemistry and biochemical genetic laboratories according to their methods.
Measurement of 6-oxopipecolic acid: Additionally, we developed a method for the measurement of 6-oxopipecolic acid in urine and plasma/serum samples in our biochemical genetics laboratory. Urine, plasma/serum samples were prepared for liquid chromatography tandem mass spectrometry (LC-MS/MS) analysis using a solid-phase extraction (SPE) method.37,38 In a 1.5-mL Eppendorf vial, 20 μL of calibrator, sample, or quality control (QC) material was combined with 20 μL of internal standard (pipecolic acid-d9 at 2.0114 μM and 6-oxopipecolic acid-d4 at 2.2151 μM) and 200 μL of acetonitrile containing 1% formic acid. The mixture was vortexed and transferred to a pre-washed and dried Agilent Captiva EMR-Lipid SPE cartridge. SPE was performed using a vacuum manifold set to −2.5 inHg to allow the sample to pass through the cartridge at a rate of 1 drop every 3–5 s. Once the solvent level was below the frit, 260 μL of a water:acetonitrile:formic acid solution (4:1, 1%) was added, the vacuum was maintained at the same flow rate. Following elution, a higher vacuum (−15 inHg) was applied to maximize sample recovery. The eluates were transferred to high-recovery 1.5 mL glass vials and dried under a nitrogen flow at 40°C. The dried residues were reconstituted in 75 μL of water containing 0.1% formic acid, vortexed thoroughly, and transferred to a 96-well plate. A 1 μL aliquot was injected for LC-MS/MS analysis.
LC-MS/MS analysis was performed on an Agilent 1290 Infinity UHPLC system equipped with an Agilent Poroshell 120 EC-C18 column (2.1 × 150 mm, 1.9 μm) coupled to an Agilent 6490 Triple Quadrupole mass spectrometer. Analytes were detected in multiple reaction monitoring (MRM) mode under positive electrospray ionization. Mobile phase and gradient conditions are summarized in Supplemental Table 3. PA was calibrated between 0.028 and 56.75 μM. 6-oxopipecolic acid was calibrated between 0.023 and 47.156 μM.
Statistical analysis
We used a combination of parametric and non-parametric statistical methods. Linear regression modeling was used to estimate the rate of change (slope) in scores as a function of age. Bootstrapping (9999 replicates) and 95% confidence intervals were applied to calculate bias-corrected and accelerated (BCa) for the slope. The Mann–Kendall non-parametric trend test was used to assess the statistical significance of monotonic trends in scores over time. The results were summarized using three key metrics: (1) the slope of change over time with its 95% BCa confidence interval; (2) quantification of the proportion of variance in scores explained by the age of the patient; and (3) the p-value from the Mann–Kendall test where p ⩽ 0.05 is significant. All statistical analyses were conducted in R (v. 4.4.0).
Results
Currently, he is 4 years old and has been seizure free since the initiation of pyridoxine treatment. He attends gymnastic and swimming classes as well as full time junior kindergarten. His developmental milestones were summarized in Supplemental Table 4. His growth parameters (weight at 53rd percentile; height at 40th percentile and head circumference at 94th percentile) and his physical examination were normal at the age of 4 years.
Triheptanoin treatment was started at 10 mL/day (10% of actual EER). Dose increases were every 2–3 weeks to reach the goal dose as per tolerance. The maximum actual dose of 40 mL/day (40% of actual EER) was at 2 months of treatment. Parents chose to mix arginine, tryptophan, lysine-free medical formula, and triheptanoin altogether and gave three times a day as per their preference and schedules. He had nausea and retching up to 3 weeks after each dose increase. There was no history of vomiting, abdominal discomfort, or diarrhea throughout the treatment. We summarize the goal and actual dose of triheptanoin in Table 1.
Triheptanoin novel therapy, its goal dose, and actual dose.

EER, estimated energy requirement.
Neuropsychological outcomes: All neuropsychological assessment results were summarized in Supplemental Tables 2 and 5. On the Bayley-III, the Motor Composite improved with a treatment-only trend slope of +0.62 [95% CI: +0.50 to +0.75; R2 = 0.987]. The Language Composite showed accelerated improvement with a treatment-only trend slope of +1.83 [95% CI: +2.00 to +3.42; R2 = 0.801]. The Cognitive Composite demonstrated notable gains with a treatment-only trend slope of +0.83 [95% CI: +0 – +0.83; R2 = 0.75]. Scaled scores for fine motor and gross motor skills showed slight improvements with slopes of +0.08 [95% CI: 0 to +0.25; R2 = 0.429] and +0.13 [95% CI: 0 to +0.25; R2 = 0.75], respectively. We depicted percentages of composite and scaled scores (25-75% normal population range) of Bayley-III assessments in Figure 1. His WPPSI-IV Full-Scale IQ was 82 (12th percentile). His visual spatial reasoning ability was above average (79th percentile) while his other intellectual abilities were in the low-average to very-low range (Supplemental Table 2 and 5).
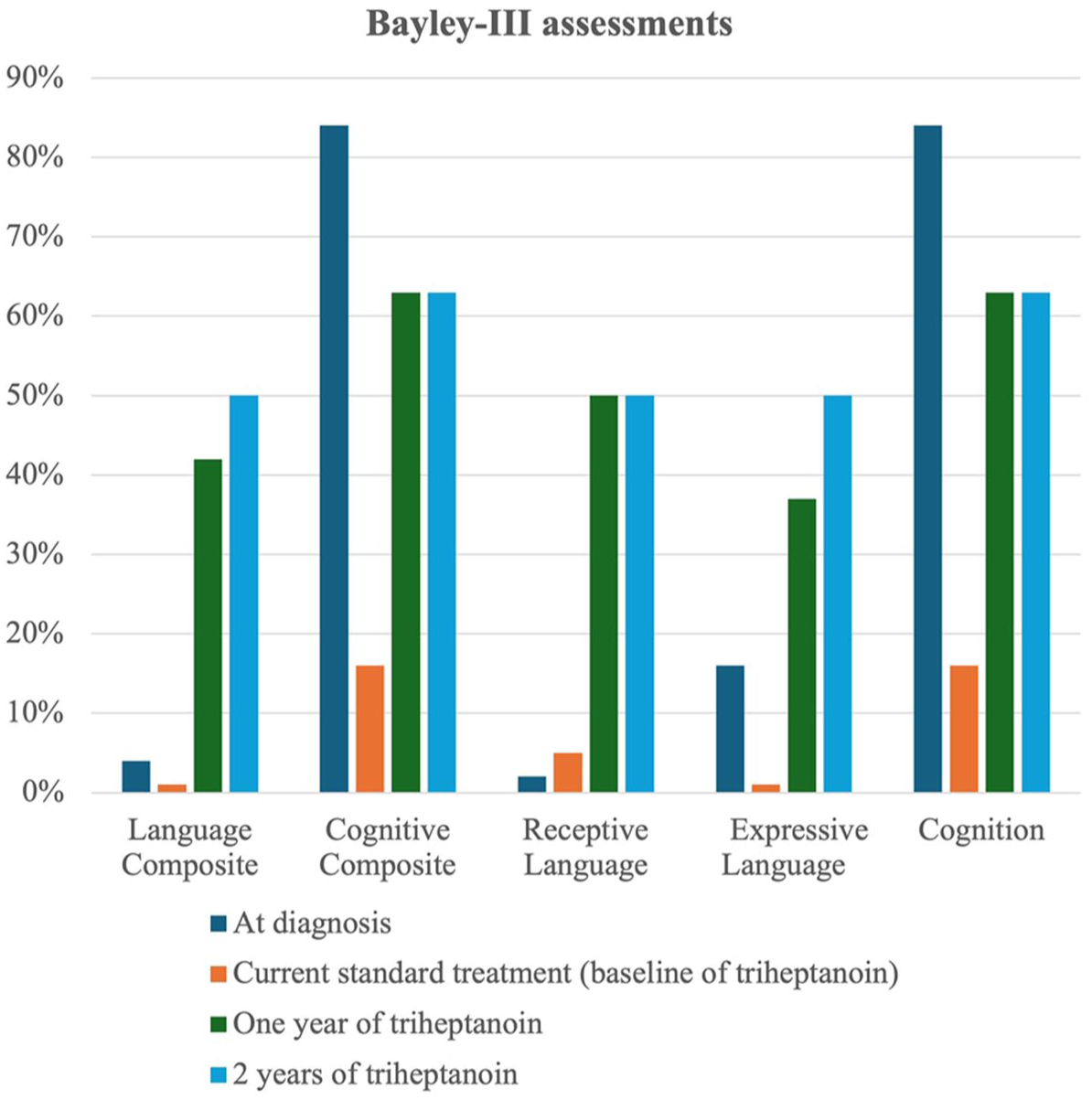
Percentages of composite and scaled scores of Bayley Scales of Infant and Toddler Development – III.
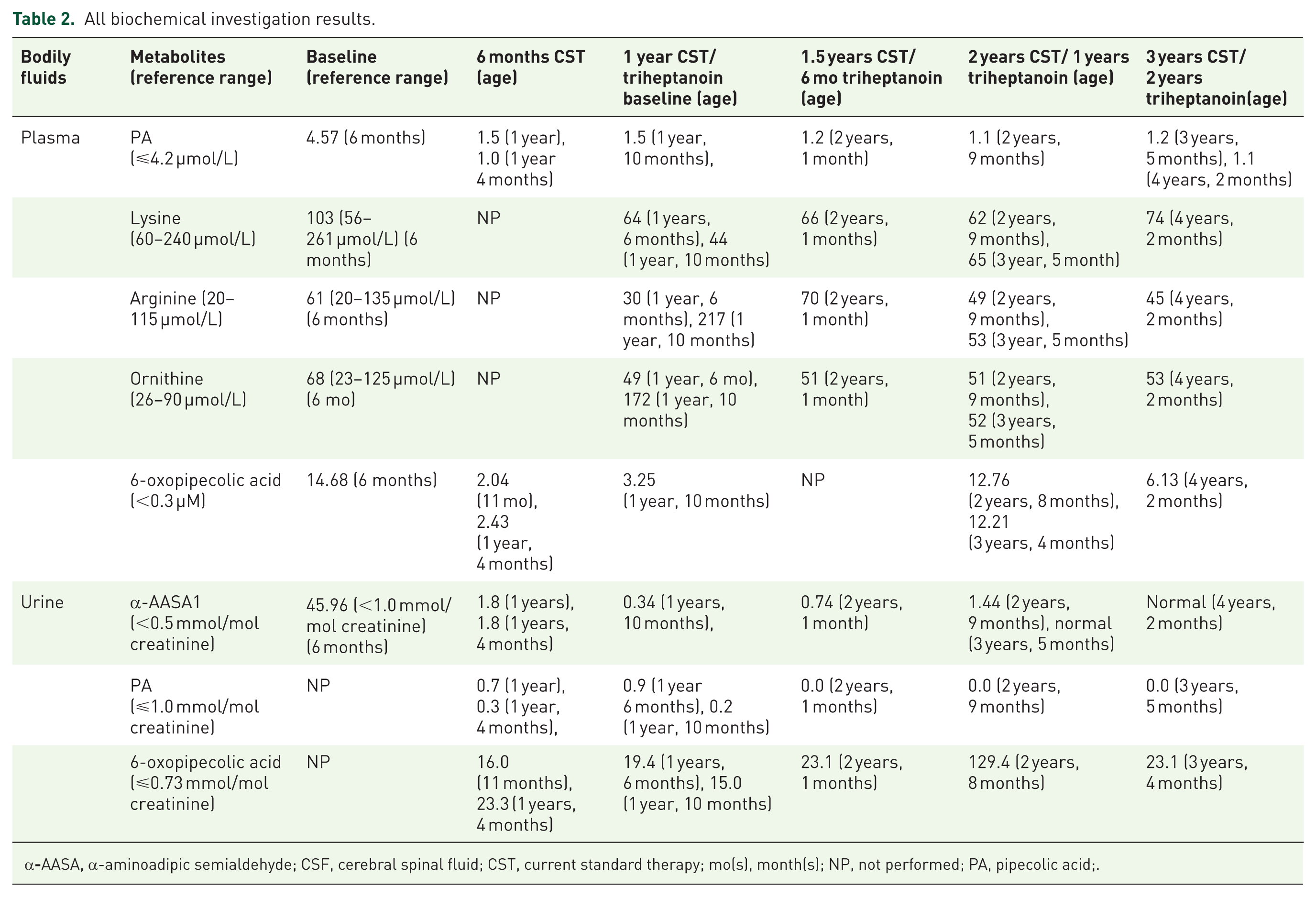
Safety assessments: All chemistry and biochemical investigations were normal. Results of the biomarkers of PDE-ALDH7A1 were summarized in Table 2. His plasma 6-oxopipecolic acid levels improved mildly, but urine 6-oxopipecolic acid levels remained elevated throughout the treatment.
All biochemical investigation results.
α
Triheptanoin treatment seemed to be safe and tolerated well. As there were progressive improvements in neuropsychological assessments compared to the current standard treatment, and triheptanoin was well tolerated, we did not discontinue the treatment after 2 years of this novel therapy.
Discussion
We report a new patient with PDE-ALDH7A1 who had poor response to current standard therapy despite early initiation and good treatment compliance. We previously reported impaired mitochondrial electron transport chain functions, and abnormal TCA cycle metabolites in single patients with PDE-ALDH7A1 and in knock-out aldh7a1 zebrafish. 14 Seizures and neurodevelopmental delays are likely due to several pathogenic mechanisms in PDE-ALDH7A1: (1) elevated α-AASA and P6C in the central nervous system due to α-AASA dehydrogenase deficiency; (2) pyridoxal-5’-phosphate deficiency which may not be completely replenished using pyridoxine for intracellular biochemical reactions in the human body; (3) gamma aminobutyric acid (GABA) metabolism abnormalities, the principal inhibitory neurotransmitter in the brain, due pyridoxal-5’-phosphate deficiency; (4) decreased production of acetyl-coenzyme A, the end product of lysine catabolism, due to α-AASA dehydrogenase deficiency; and (5) decreased production of succinate, the end product of GABA metabolism, due to pyridoxal-5’-phosphate deficiency.9,12,31,39 Lysine and GABA pathways provide substrates to the TCA cycle. Decreased substrate supply to the TCA cycle due to PDE-ALDH7A1 likely affect the main energy production pathways including TCA cycle and mitochondrial electron transport chain functions. The current standard therapy of PDE-ALDH7A1 partially decrease α-AASA and P6C levels in the central nervous system and likely partially replenish pyridoxal-5’-phosphate deficiency but does not help with the secondary deficiencies in the energy production pathways. Our patient had mild complex I (68% of normal) deficiency in muscle biopsy specimen. He did not have improvements in his developmental milestones. He showed declines in his neuropsychological assessments (language composite ratio of treatment to baseline 0.757; cognitive composite ratio of treatment to baseline 0.739; and adaptive behavior composite ratio of treatment to baseline 0.896) on the current standard therapy of PDE-ALDH7A1. As triheptanoin is an anaplerotic agent to provide ketones and substrates to the TCA cycle, we applied a novel therapy using triheptanoin in our patient to improve neurodevelopmental outcomes. Triheptanoin was well tolerated. There were accelerated improvements in cognitive composite (treatment-only slope +0.83), language composite (treatment-only slope +1.83), and steady improvements in motor composite (treatment-only slope +0.62). While adaptive functioning continued to decline, the rate of decline slowed down on triheptanoin therapy, particularly in adaptive behavior composite (−0.50 overall vs −0.33 post-treatment) and daily living skills (−0.70 overall vs −0.58 on triheptanoin). We aimed to increase triheptanoin to 50% of EER without changing caloric and protein intakes, but he was not able to tolerate the mixture of natural protein, medical protein, all supplements and triheptanoin. We depicted EER in Figure 2 throughout the therapy which was at or below 40% of EER (average 35%). If we would have reached our goal dose of triheptanoin, he might have higher Full-Scale IQ using WISC-IV during his third year of triheptanoin treatment. We report the first patient with PDE-ALDH7A1 who has been treated with triheptanoin which has resulted in improvements in neurodevelopmental outcomes and been well tolerated.

The goal and actual percentage of estimated energy requirement of triheptanoin treatment.
Previous studies have reported improvements in neurodevelopmental outcomes and biomarkers on the current standard therapy.3,6,13,15–29 An observational study reported that lysine restriction was well tolerated and led to decreases in neurotoxic biomarkers, although improvements in age-appropriate skills were observed in only four out of five patients, where one patient stopped the lysine-restricted diet after 6 months due to no significant improvement of speech and the patients desire for animal protein. 13 The neurodevelopmental outcome of early versus late treated siblings reported better neurodevelopmental outcomes, although there was no statistically significant difference. 29 Lysine- or protein-restricted diet resulted in significant increase in objective developmental assessments if started in the first 6 months of life. 40 Arginine supplementation resulted in improvements in neurocognitive functions and biomarkers in a patient with PDE-ALDH7A1 who was treated at the age of 11 years for 1 year. 17 One study reported age-appropriate language development after 10 months of current standard therapy. 24 Our group reported milder phenotypes if the current standard therapy was started within the first 7 months of age. 6 Previous studies reported improvements in developmental milestones within 12 months of therapy.3,5,16,18,40 Unfortunately, our patient had cognitive composite score of 84% prior to treatment start which deteriorated to 16% (5.25-fold decrease) on the current standard therapy. To the best of our knowledge, we report the first patient with PDE-ALDH7A1 who had unfavorable short-term outcome on the current standard therapy started at the age of 8 months.
Lysine- or protein-restricted diet have shown to reduce or normalize α-AASA levels in plasma, or urine previously.5,6,18 Arginine supplementation therapy led to a 57% decrease in CSF α-AASA level at 12 months of therapy. 17 CSF α-AASA level remained elevated in a patient on the current standard therapy. 31 Elevated levels of 6-oxopipecolic acid, formed by oxidation of 6-hydroxy-pipecolic acid as a novel biomarker in the blood, urine, and CSF, were reported in patients with PDE-ALDH7A1.7,41–45 6-oxopipecolic acid is relatively stable at room temperature, making it suitable for newborn screening.42,45 We monitored plasma and urine 6-oxopipecolic acid levels throughout the therapy that remained elevated in our patient similar to previous reports.41–44 As lumbar punctures are invasive and urine α-AASA level normalizes on the current standard therapy, blood dot spot 6-oxopipecolic acid might be a better biomarker for the monitoring of therapy in PDE-ALDH7A1.
Limitations
Our study had several limitations including: (1) we have a single patient on triheptanoin treatment and we are not certain if all patients will improve in their neurodevelopmental outcomes; (2) we did not measure CSF metabolites on triheptanoin treatment or prior to onset of current standard therapy to compare if triheptanoin improved accumulation of neurotoxic metabolites in CSF; (3) we did not measure TCA cycle metabolites or electron transport chain complexes on triheptanoin treatment to see if any improvements as these were invasive investigations and parents did not consent for these investigations; and (4) we had different neuropsychological assessments due to young age of our patient, which did not allow us to compare outcomes of those assessments statistically as we used WPPSI-IV during the last assessment at the third year of triheptanoin treatment, and we did not have any comparison. Despite these limitations, we think that our study is valuable to improve neurodevelopmental outcomes of patients with PDE-ALDH7A1 as a novel therapy using triheptanoin.
Conclusions
We report a new patient with PDE-ALDH7A1 on a novel triheptanoin treatment and his objective improvements in neurodevelopmental outcomes who did not respond to the short-term current standard therapy of PDE-ALDH7A1. We found a mild complex I (68% of normal) deficiency in muscle biopsy specimen which shows that PDE-ALDH7A1 causes secondary mitochondrial electron transport chain dysfunction. The current standard therapy focuses on the elevated α-AASA and P6C levels and pyridoxal-5’-phosphate deficiency but does not address the secondary deficiencies in the energy production pathways. As triheptanoin is an anaplerotic agent to provide ketones and substrates to the TCA cycle, trihepatonoin should be the part of the current standard therapy to improve neurodevelopmental outcomes of patients with PDE-ALDH7A1.
Supplemental Material
sj-pdf-1-trd-10.1177_26330040261427020 – Supplemental material for A novel therapy for pyridoxine-dependent epilepsy due to biallelic pathogenic variants in ALDH7A1: secondary mitochondrial energy deficiency and improvements of neurodevelopmental outcomes on triheptanoin treatment
Supplemental material, sj-pdf-1-trd-10.1177_26330040261427020 for A novel therapy for pyridoxine-dependent epilepsy due to biallelic pathogenic variants in ALDH7A1: secondary mitochondrial energy deficiency and improvements of neurodevelopmental outcomes on triheptanoin treatment by Anastasia Ambrose, Morganne McCabe, Shalini Bahl, David Sinasac, Thomas Snyder and Saadet Mercimek-Andrews in Therapeutic Advances in Rare Disease
Supplemental Material
sj-pdf-2-trd-10.1177_26330040261427020 – Supplemental material for A novel therapy for pyridoxine-dependent epilepsy due to biallelic pathogenic variants in ALDH7A1: secondary mitochondrial energy deficiency and improvements of neurodevelopmental outcomes on triheptanoin treatment
Supplemental material, sj-pdf-2-trd-10.1177_26330040261427020 for A novel therapy for pyridoxine-dependent epilepsy due to biallelic pathogenic variants in ALDH7A1: secondary mitochondrial energy deficiency and improvements of neurodevelopmental outcomes on triheptanoin treatment by Anastasia Ambrose, Morganne McCabe, Shalini Bahl, David Sinasac, Thomas Snyder and Saadet Mercimek-Andrews in Therapeutic Advances in Rare Disease
Supplemental Material
sj-pdf-3-trd-10.1177_26330040261427020 – Supplemental material for A novel therapy for pyridoxine-dependent epilepsy due to biallelic pathogenic variants in ALDH7A1: secondary mitochondrial energy deficiency and improvements of neurodevelopmental outcomes on triheptanoin treatment
Supplemental material, sj-pdf-3-trd-10.1177_26330040261427020 for A novel therapy for pyridoxine-dependent epilepsy due to biallelic pathogenic variants in ALDH7A1: secondary mitochondrial energy deficiency and improvements of neurodevelopmental outcomes on triheptanoin treatment by Anastasia Ambrose, Morganne McCabe, Shalini Bahl, David Sinasac, Thomas Snyder and Saadet Mercimek-Andrews in Therapeutic Advances in Rare Disease
Footnotes
Acknowledgements
We would like to thank the parents for allowing us the publish their child’s information and their excellent care. We would like to thank the metabolic physicians, metabolic dietitians, metabolic nurses, genetic assistants and genetic counselors for their excellent patient care. We would like to thank all biochemical genetics laboratory scientists and technicians (North and South Alberta) for developing 6-oxopipecolic acid measurements in urine and blood and for organizing biochemical investigations. We would like to thank Stollery Children’s Hospital health care team for their excellent patient care. We would like to thank Ultragenyx Pharmaceuticals for providing triheptanoin for compassionate care treatment.
Declarations
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.