
Abstract
Background:
Dentatorubral–pallidoluysian atrophy (DRPLA) is a rare, neurodegenerative disorder with no disease-modifying treatments. There is a dearth of information in the literature about the patient and caregiver experience living with DRPLA.
Objectives:
This study aimed to (1) understand symptoms experienced by adult- and juvenile-onset DRPLA populations and their impact on daily life and (2) explore patient and caregiver treatment goals and clinical trial participation preferences.
Design:
The study was a qualitative interview study.
Methods:
Interviews were conducted remotely with adult patients with DRPLA and caregivers. Participants described patient symptoms and the impact of those symptoms on daily life, and they discussed treatment goals and potential clinical trial participation. There were 18 patients described in the interviews with two patients and seven caregivers. Some participants were caregivers to multiple patients with DRPLA.
Results:
Interview transcripts were coded for themes, and reported symptoms were summarized with descriptive statistics. Adult-onset patients (N = 7) experienced difficulty with ataxia (100%), cognition (100%), fine motor skills (100%), gross motor skills (100%), speech (100%), personality changes (100%), and seizures (57%). Juvenile-onset patients (N = 11) experienced difficulty with ataxia (100%), sleep (100%), speech (100%), jerking/twitching (83%), behavior (82%), cognition (82%), fine motor skills (82%), gross motor skills (82%), sensory sensitivity (75%), and seizures (64%). When considering aspects of DRPLA to target for future treatment, patients prioritized ataxia/mobility (100%), juvenile-onset caregivers prioritized ataxia/mobility (60%) and independence (60%), and adult-onset caregivers prioritized personality (60%). Almost all patients (93%) would participate in a clinical trial if given the opportunity, but travel to a clinical site could pose a participation barrier for half.
Conclusion:
This study found that there are symptom domains that are relevant across the DRPLA population, but there is heterogeneity within each domain based on the age of symptom onset and disease stage, which has implications for clinical trial design.
Plain language summary
Keywords
Introduction
Dentatorubral–pallidoluysian atrophy (DRPLA) is a rare, neurodegenerative disorder, characterized by myoclonus, ataxia, epilepsy, cognitive impairment, and psychiatric symptoms.1,2 DRPLA is thought to occur most commonly in Japan, with an incidence of approximately 2–7 per million people3,4; however, the worldwide prevalence of DRPLA is currently unknown. Since DRPLA symptoms are associated with a broad differential diagnosis, it is often difficult to diagnose unless there is a known family history of the disease.2,3 DRPLA should be suspected in individuals that present with symptoms that are characteristic of DRPLA, have a brain magnetic resonance imaging test that shows cerebellar and brain stem atrophy and possibly white matter lesions, and have a family history that may suggest an autosomal dominant inheritance pattern. 5 A genetic test is needed to confirm the diagnosis. 5 The onset of DRPLA symptoms can occur at any age, but the median age at symptom onset is 31 years. 3 Patients are classified as either juvenile-onset (onset < 20 years) or adult-onset (onset ⩾ 20 years), and clinical presentation can vary with the age at symptom onset. 2 Juvenile-onset patients have a median age of onset of 15–19 years, and clinical features include epilepsy, rigidity, and intellectual disability. 2 Adult-onset patients have a median age of onset of 38–43 years, and clinical features include myoclonus, ataxia, choreoathetosis, and dementia. 2 It is more common for juvenile-onset patients to have seizures and more common for adult-onset patients to have chorea. 2 The median time from symptom onset to death is 15 years.2,3,6 The clinical management for patients with DRPLA is mostly supportive as there are currently no disease-modifying treatments for DRPLA.3,5
No clinical trials have been conducted in DRPLA, and there is a lack of information about the natural progression of the disease. Studying natural history is essential prior to drug development to understand disease progression and identify biomarkers. 7 While large-scale natural history studies among patients with DRPLA are lacking in the literature, 3 there are natural history studies that are currently in the planning stages. 2 For both natural history studies and clinical trials, the selection of clinical outcome assessments can be challenging in rare disease populations.8–11 The diversity of symptoms that characterize DRPLA and the lack of validated, disease-specific outcome measures can lead to using outcome measures developed for other diseases that may not reflect what is relevant or important to patients and caregivers. 12
Identifying the symptoms that are important to patients and caregivers is critical to ensure that clinical trials evaluate outcomes that impact participants’ daily lives in a real-world setting. Recent U.S. Food and Drug Administration (FDA) and European Medicines Agency guidance have recognized this point and have encouraged patient involvement throughout the entire drug research and development process.13–15 There is currently very little information in the literature about the patient and caregiver experience living with DRPLA. This qualitative interview study aimed to (1) understand symptoms experienced by adult- and juvenile-onset DRPLA populations and their impact on daily life and (2) explore patient and caregiver treatment goals and clinical trial participation preferences. The results of this study may provide needed information for the design of future clinical trials.
Methods
Study design and participants
This phenomenological qualitative interview study was designed and conducted collaboratively with contributions from advocacy groups CureDRPLA and Ataxia UK, and Emmes Endpoint Solutions, a division of Emmes, LLC (formerly Casimir LLC) specializing in developing assessments for rare diseases. English-speaking participants from the United States and the United Kingdom were referred by CureDRPLA using quota sampling to target recruitment of caregivers of children with DRPLA, caregivers of adults with DRPLA, and adult patients with DRPLA. Emmes Endpoint Solutions (formerly Casimir) study staff contacted interested potential participants through phone or email to set up a screening call. Eligibility criteria were broadly defined as any adult patient diagnosed with DRPLA or any caregiver of a child or adult diagnosed with DRPLA. Enrollment ceased once saturation of concepts was achieved. Saturation was defined as informational redundancy,16,17 where the same symptoms continued to be mentioned as more interviews were conducted. Participants were compensated for their participation, and no participants refused to participate or dropped out of the study.
Data collection
The development of the semistructured interview guide and selection of domains to discuss were informed by a literature review and an informal interview with a caregiver of a child with DRPLA. Interviews were conducted remotely and video/audio recorded through a web conferencing service. Two Emmes Endpoint Solutions (formerly Casimir) female researchers conducted the interviews using a semistructured interview guide. One of the interviewers (M.L.) has a Master of Education and was the founder and president of Casimir at the time of the study. The other interviewer has a PhD in Psychology and was the Director of Research at Casimir at the time of the study. Both researchers were trained in qualitative research methods and best practices in accordance with FDA Patient-Focused Drug Development Guidance 2. 15 Each interview lasted approximately 60–90 min. The interviewers did not have a relationship established with the interview participants prior to study commencement, and the participant did not know any personal details about the interviewers. The goals of the interview were stated in the introduction prior to starting the interview questions.
Prior to the start of the interview, the interviewer verified with participants that they were in a place where they could speak freely, without anyone else able to hear their responses. The study interview started with questions about the patient’s diagnosis story and early symptoms and moved into a discussion of current and previous symptoms for the following domains: gross motor skills, fine motor skills, communication, cognition, behavior, and activities of daily living/self-care. This discussion was followed by questions relating to the impact of the disease on daily life (patient, caregiver, and family). Lastly, the interview focused on treatment goals and potential clinical trial participation. If the participant was the caregiver to multiple patients with DRPLA, the interview guide topics were discussed for each patient under their care.
Data analysis
The interview audio was transcribed by an HIPAA-compliant transcription service and transcripts were compared to the audio/video to ensure accuracy. Transcripts were de-identified and assigned unique study identification numbers. Transcripts were coded by an analyst (R.W) trained in qualitative research. The analyst created a thematic codebook based on the interview guide and a review of transcripts. Codes were further refined with common themes or characteristics that emerged during the coding process. Key themes were summarized with representative quotes, and reported symptoms were summarized with descriptive statistics (n, %). All coding was conducted using NVivo 13 (QSR International).
Results
Patient/caregiver characteristics
There were 18 patients described in interviews with 2 patients and 7 caregivers (Figure 1). Seven adult-onset patients were described in interviews with two patients and six caregivers. There was one patient/caregiver pair who described the experiences of the same patient. Of those six caregivers describing adult-onset patients, three reported on observations of their husbands, one reported on observations of her uncle, one reported on observations of her father, and one reported on observations of his mother. Eleven juvenile-onset patients were described in interviews with five caregivers. Of those five caregivers describing juvenile-onset patients, one reported on observations of her son and daughter (n = 2), one reported on observations of her son (n = 1), one reported on observations of her daughter (n = 1), one reported on observations of her daughter and three sons (n = 4), and one reported on observations of her brother and two sisters (n = 3).

Detail on the patients described in each interview.
Both patients (100%) who were interviewed reside in the United States. Of the seven caregivers interviewed, four reside in the United States (57%), and three reside in the United Kingdom (43%). Of the 18 patients described in the interviews, 12 (67%) were male, and the patients ranged in age of symptom onset from 2 to 52 years, with a median age of symptom onset of 15 years. The patient-specific demographic information is provided in Table 1.
Patient-specific demographic information.
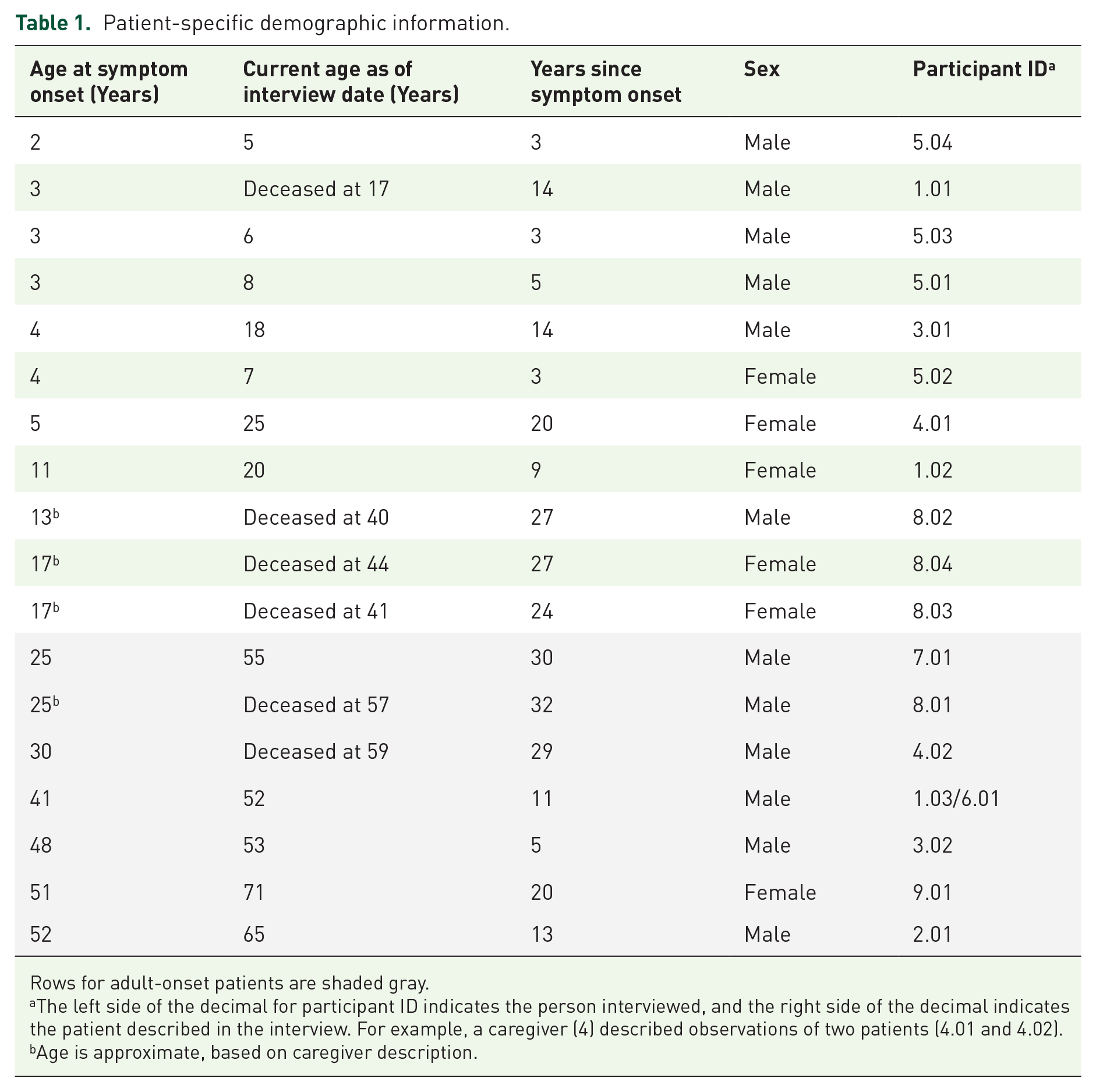
Rows for adult-onset patients are shaded gray.
The left side of the decimal for participant ID indicates the person interviewed, and the right side of the decimal indicates the patient described in the interview. For example, a caregiver (4) described observations of two patients (4.01 and 4.02).
Age is approximate, based on caregiver description.
Adult-onset DRPLA symptoms
Saturation of symptoms was achieved for adult-onset DRPLA participants through the interviews with six caregivers and two patients. The caregivers and patients described symptoms experienced previously and currently. Of the two patients who were interviewed, both (100%) reported experiencing difficulty with ataxia, cognitive ability, fine motor skills, gross motor skills, seizures, and speech (Table 2). Of the six patients described by caregivers, all (100%) experienced difficulty with ataxia, cognitive ability, fine motor skills, gross motor skills, speech, and personality changes. Three of six patients (50%) experienced seizures. The symptom domains described are not distinct and may be related to other domains (e.g., gross motor skills are related to ataxia, speech is related to cognition).
Adult-onset DRPLA symptoms experienced by patients, as reported by both patients and caregivers.

Missing data for one caregiver due to the question about this topic not being asked.
DRPLA, Dentatorubral–pallidoluysian atrophy.
Ataxia: All the caregivers (100%) observed the patients having difficulty with balance. In addition to issues with balance, both patients (100%) described having poor spatial awareness: Well, I think the first thing that happened was when I was going through, you know, things like closet doors, or from room to room, I would gradually be hitting my head on the side of the door. And I realized that I’d never done that before . . . And that’s when I thought that I was having trouble just with judging where my body was. (ID 2.01, Patient)
Cognition: Both caregivers and patients described difficulty with cognitive ability, but they described it in different ways. Both patients described challenges with the clarity of their thoughts and changes in their ability to think: I was aware of the fact that this is one thing that DRPLA was taking from me, was having a clarity of insight. Yes, a certain clarity of intellectual engagement so I could develop an outline and then follow an outline to develop an argument. (ID 2.01, Patient)
The caregivers described the cognitive decline as slower processing speed: The processing speed. He hasn’t lost anything, and he’s very proud of that. So his memory is impeccable. He remembers things that I don’t remember. But the speed in which he processes things around him – if I’m trying to have a conversation, it’s very hard for me because I have to slow down . . . I have to look at him. I have to make sure he’s listening. I have to talk a little slower. I have to wait till he processes it, and make sure he hasn’t gone on to something else in his head. (ID 1.01, Caregiver)
Fine motor: All patients and caregivers (100%) described patients having difficulty using their hands: The first thing that I noticed, oh, quite a while ago was that I couldn’t button up shirts. I had a lot of trouble with those little dexterous muscles. (ID 2.01, Patient)
Gross motor: All patients and caregivers (100%) described patients having difficulty with gross motor skills that included challenges with walking, standing up, using stairs, getting dressed, and getting in and out of bed. A couple described stronger upper bodies than lower bodies: He’s completely immobile. He is hoisted from bed to chair, and hoisted into wheelchair, and that’s it. He doesn’t have any ability to use his legs. Still very strong in the upper part of the body, but . . . his legs, just completely immobile. (ID 7.01, Caregiver)
Seizures: Of the four patients (57%) who had experienced seizures, a patient reported having one grand mal seizure in his life and no need for medication, another patient reported about six to eight seizures in his life that are now controlled with medication, a caregiver reported on grand mal seizures that are now controlled with medication, and another caregiver reported on infrequent seizures that increased in frequency in the later stages of disease. Three of these patients currently have their seizures under control, and the other patient is deceased: No, he doesn’t have them anymore. They’ve been controlled with medication. He hasn’t had a seizure for about six years . . . But when he did have seizures, they were really bad, tonic-clonic, really, really bad seizures that lasted for a very long time. (ID 7.01, Caregiver)
Speech: Both patients (100%) described slurring words and having trouble with larger words, and all caregivers (100%) described that the patients had unclear and slurred speech: So there was kind of some, you know, weird speech . . . we’ve been refused service in bars. That’s . . . how it started with my husband. We were refused service in bars because they thought he was drunk. (ID 1.01, Caregiver)
Personality: All caregivers (100%) described patients having personality changes that included anger outbursts: He was very patient. Well he’s no longer patient, he gets riled up extremely quickly, he gets really angry with the children. It’s very, very difficult, because it’s like somebody changed my husband for a completely different one. (ID 3.02, Caregiver)
Patient ability to assess own condition
Three of the caregivers (50%) described that the adult patients they cared for would have difficulty assessing their own condition: [Describing her husband’s ability to assess functional change] Especially if it was in a positive, yes. In the negative, he tries to minimize, and – because obviously he doesn’t want to get worse. (ID 1.03, Caregiver) [Describing her husband’s ability to notice signs of DRPLA] No, we noticed it, he didn’t. He still struggles now to notice things. We noticed it, myself and my best friends. (3.02, Caregiver)
Juvenile-onset DRPLA symptoms
Saturation of symptoms was achieved for juvenile-onset DRPLA participants through the interviews with five caregivers. The caregivers described symptoms experienced previously and currently. Of the 11 juvenile-onset patients described by caregivers, all (100%) experienced difficulty with ataxia, sleep, and speech (Table 3). Five of six (83%) experienced hand tremors or twitching, and nine of eleven (82%) experienced behavioral issues, cognitive impairment, fine motor skill impairment, and gross motor skill impairment. Six of eight patients (75%) experienced sensory sensitivity to light or noise. Seven of eleven patients (64%) experienced seizures. The symptom domains described are not distinct and may be related to other domains (e.g. gross motor skills are related to ataxia, speech is related to cognition).
Juvenile-onset DRPLA symptoms, as observed by caregivers.

The N refers to the number of patients reported on rather than the number of caregivers interviewed. There were five caregivers that reported on 11 patients.
Missing data for one patient due to the question about this topic not being asked.
Missing data for five patients due to the question about this topic not being asked.
Missing data for three patients due to the question about this topic not being asked.
DRPLA, Dentatorubral–pallidoluysian atrophy.
Ataxia: All of the caregivers (100%) observed the patients showing signs of ataxia and described falls, balance issues, and poor spatial awareness: The three-year-old at that time had significant balance issues and still does. . .any soft surface, any uneven surface, you know, playing on the grass or anything like that, he does not have the ability to balance. Like if he’s standing on a bed, when it gets soft and he starts to fall, he falls backwards, he doesn’t know how to catch that balance . . . It’s just like he just doesn’t have a sense of balance. . .I don’t really know how to describe it, other than he just doesn’t have a feeling for where he’s at in the world. (ID 5.03, Caregiver)
Behavioral: Caregivers of 6 of the 11 (55%) patients observed that the patients exhibited a lack of emotional control and tantrums: It seems like they don’t have very much . . . emotional control. So, when they get mad, they can’t just get mad and say ‘oh, I didn’t get a cookie, I’m upset.’ . . . they get mad, and they’re hitting, they’re kicking, they’re throwing fits, they’ll bite, anything they can do to show you how mad they are. (ID 5.01-04, Caregiver)
Caregivers of 5 of the 11 (45%) patients observed that the patients exhibited hyperactive behavior: He was a bull in a china closet. And . . . that continued until he couldn’t walk anymore . . . Maybe slept three or four hours a night. Just constantly going. You could not stop him. . .extreme, extreme hyperactivity. Just nonstop. Like, just – you know, the school just could not keep him. (ID 1.01, Caregiver)
Caregivers of 4 of 11 (36%) patients observed aggressive behavior, and caregivers of 3 of the 11 (27%) patients observed argumentative behavior: She’s very violent towards the other kids, towards anybody, really . . . She lashes out at them quite a bit. She attacks my oldest daughter pretty much every single morning, because she’s not getting what she wants . . . She’s pretty violent in regards to that . . . She says mean things to them. (ID 5.02, Caregiver)
Cognition: Caregivers of 8 of 11 (73%) patients described academic regression and intellectual decline: Just the academic piece, and just never progressing, and then starting to regress and stuff . . . I don’t think I ever left an IEP [Individualized Education Plan] for [ID 01.01] not crying. We would be called multiple times. No plan would ever actually work for him at school . . . He stopped going to school at age 12, 13. (ID 1.01, Caregiver)
Caregivers of 6 of 11 (55%) patients described deterioration of cognitive processing abilities: Her processing speed is super slow . . . she used to be able to follow commands no problem, and now it’s at the most two-step commands, and sometimes it’s, ‘[ID 01.02], turn around so you can sit on the toilet,’ and it takes two minutes for her to process to do that . . . there’s a lot of motor planning issues, so not only do I have to cognitively process what you just said, I have to try to tell my body to do that, which is also hard. (ID 1.02, Caregiver)
Fine Motor: Caregivers of 9 of 11 (82%) patients described worsening in the ability of patients to use their hands: When he was young, 5, 6 years old, he was doing better than he is now. He was able to write his name, he was able to hold a pencil, write a sentence, you know, do those kinds of things. Since that time, within the last year or so, he has definitely deteriorated with his fine motor skills. He is no longer able to write very well at all. He gets frustrated pretty quickly with it, because it takes so much effort just to write, you know, his name. And so, to write a sentence or anything, he just, he can’t do it. And his writing isn’t very legible anymore. (ID 5.01, Caregiver)
Gross Motor: Caregivers of 9 of 11 (82%) patients described patients having difficulty with gross motor skills that included worsening in ability to walk, climb stairs, and get dressed: It takes 2 people minimum to transfer him into the chair, and onto the toilet, or into his walker. He walks around in his walker to about three, four hundred steps, he can do obviously with us guiding it, because otherwise it would just go like a shopping trolley anywhere . . . His mobility is very, very limited . . . he can still walk up and down the stairs, but 2 of us have to maintain his balance on either side of him. (ID 3.01, Caregiver)
A couple of caregivers described motor planning and coordination challenges: Between five and a half and six years old, we noticed that some of the stuff that he was doing at the beginning of the school year, like playing on the monkey bars. . .he wasn’t able to do anymore. Where he used to be able to jump up and catch the bar, like on the monkey bars, you would jump up and catch it, and he could do it really well at the beginning of the year, and as the year progressed, that coordination went away, and he wasn’t able to catch it . . . Climbing up the steps, his feet wouldn’t cooperate to climb up the bars, or to climb up the steps. (ID 5.01, Caregiver)
Jerking/Twitching: Caregivers of five of six (83%) patients described that the patients twitched and had hand tremors: We noticed it from early age, when he started going to kindergarten, so probably three or four, when he would start holding a pencil or concentrating on holding a Lego piece or something, he would sort of – his hands would sort of have a little jerk and a tremor. (ID 3.01, Caregiver)
Seizures: Caregivers of 7 of 11 (64%) patients described that the patients experienced seizures, and the 4 patients who had not yet experienced seizures are early in their progression (3–5 years since symptom onset). All caregivers described frequent seizures, and most described challenges controlling them with medication: His seizures were on this really weird cycle of every three days he would have a cluster of 8 to 12 grand mal seizures, and then he would sleep for, like, 24 hours, and then he would be okay for, like, 48 hours . . . we never had control of his. So they started at age seven, and . . . we never had a med that we went, ‘oh, this is working, right?’ I mean, nothing ever worked . . . maybe we’d get a 10, 20 percent reduction initially, and then, you know, we’d lose that. And they just kept getting worse and worse and worse. (1.01, Caregiver) In general, we’ll start a med, and she’ll have, you know, maybe six months or even a year of pretty – you know, fairly good control with the new med, and then she’ll have an exacerbation, and then we’ll, you know, try something else, and then she’ll have some control for a while. So hers . . . Yeah, basically do respond to treatment, but . . . it’s been difficult, you know, more than the average just somebody with epilepsy. (1.02, Caregiver)
Sensory Sensitivity: Caregivers of two of eight (25%) patients described that the patients were sensitive to light, caregivers of four of eight (50%) patients described that the patients were sensitive to noise, and caregivers of two of eight (25%) patients described that they did not notice any sensory sensitivity.
Sleep: Caregivers of five of six (83%) patients asked about sleep described that the patients woke up during the night and got minimal amounts of sleep during the night, and the caregiver of one of the six (17%) patients asked about sleep described that the patient experienced dystonia.
And now the other kids are having sleeping issues too, so we actually have all four of them that are up and down during the middle of the night, not sleeping well. The doctors are trying to control that with the meds, but it’s definitely a challenge. . .[ID 05.01] gets probably, without his meds, he gets probably about two to three hours a night, and with the meds, we’re getting probably about five to six hours of sleep a night, which is a lot better. But still, you know, we’d like more. (ID 5.01-04, Caregiver)
Speech: The caregiver of only 1 of 11 (9%) patients described delays in speech, and the others described normal speech development early in life. Caregivers of three patients (27%) described slurred speech as an early sign of speech decline: It [speech clarity worsening] kind of went with the speech decline, with regression, started with the slurring of words, or difficulty in finding them, or just not understanding what they’re trying to say. You know, speech is a pretty intense motor planning activity, so it [speech clarity worsening] kind of went with the speech decline. (ID 1.01-02, Caregiver)
A caregiver of three patients (27%) early in their progression described that the patients alter their sentence structure in a way that can be confusing to understand: Sentence structure definitely is not there. You can tell with her, and actually the two younger boys also, they talk backwards a little bit. Like they can’t structure their sentences appropriately. And so, sometimes they’ll ask questions, but the words are rearranged . . . you have to really sit there and think about what it is that they’re asking. (ID 5.02-04, Caregiver)
Caregivers of seven patients (64%) described the patients using less and less words as they declined: My son never could really communicate past a three-year-old . . . between 11 and 13 it was progressively less words. I think the most we ever counted as far as his words was maybe 50 to 75 total words that he knew and could use. And somewhere between 11 and 13 he started regressing in those words, and by the time he was 14 he may have been able to mumble one or two words. (ID 1.01, Caregiver)
Treatment goals and meaningful change
Patients and caregivers described the aspects of DRPLA that they would most like to see improved with treatment (Table 4). Some patients and caregivers listed more than one treatment goal.
Aspects of DRPLA that patients and caregivers would most like to see improved with treatment.

Missing data for one patient
DRPLA, Dentatorubral–pallidoluysian atrophy.
Both adult-onset patients described that the aspect of DRPLA that they would most like to see improved with treatment was ataxia: I’m going to say if it can pull the ataxia, for me, that’d be the major thing. That keeps me from being normal. I feel that really keeps me from being normal. (ID 6.01, Patient)
Both adult-onset patients described that stopping the disease progression and having symptom stability would be a meaningful treatment impact.
Caregivers of 6 of 10 (60%) juvenile-onset patients described that the aspect of DRPLA that they would most like to see improved with treatment was independence.
Independent living, like being able to care for yourself . . . it’s huge, because, you know, if you can’t care for yourself, then you are a burden on somebody else, which takes, you know, quality of life for two people. (ID 1.02, Caregiver)
Caregivers of 6 of 10 juvenile-onset patients (60%) described that they would like to see patient mobility improved with treatment: The physical stuff is a big thing, you know, I want to keep them physically active and able to move around, and being able to do things on their own. (ID 5.01-04, Caregiver)
Caregivers of three of five adult-onset patients (60%) described that they would most like to see patient personality return to pre-symptom onset or remain intact without further decline with an effective treatment: But when there’s nothing left of that person’s personality in the end, when you lose them over a 10, 15-year period, to the point where there’s nothing left of their mind, then I don’t know that controlling any other symptom would be worthwhile. (ID 8.01, Caregiver)
Caregivers of 14 of 15 patients (93%) described that stopping the disease progression and having symptom stability would be a meaningful treatment impact, and one caregiver described that symptom improvement was the goal.
Clinical trial participation
Both adult-onset patients (100%) reported that they would participate in a clinical trial for DRPLA treatment if given the opportunity. One of the patients mentioned that a barrier to participation could be the time commitment of the trial. The caregivers of 13 of 14 patients (93%) asked about participation reported that the patient would participate in a clinical trial for DRPLA treatment if given the opportunity. The caregiver of the patient who would likely not participate in a clinical trial would choose not to participate due to a fear that unsafe drugs could exacerbate DRPLA symptoms.
The factors considered for clinical trial participation depended on the patient’s current level of function. Caregivers of those that were not as progressed were more concerned about the risk of potentially worsening patient function: I would be scared of what that [new drug] would do to the speed of the progression if it was not positive response . . . she kind of is somewhat stable all the time. You know, she only ends up in the hospital maybe once every other year, and those are, you know, maybe over– just one overnight stay or something. So it’s just – you don’t want to rock that boat type thing. (ID 1.01-03, Caregiver)
Caregivers of patients that were more progressed in their disease expressed more concern about the patient dying than potential negative consequences of a clinical trial treatment: I . . . absolutely would jump at an opportunity, because there isn’t anything available for his illness, and I know that drugs bring a risk with them, but he is dying versus a risk, surely we’re going in the right direction, and I understand the morality behind it, and the questions, but I would rather he tried something than was just left to die, so absolutely would jump at an opportunity. (ID 3.01, Caregiver)
Caregivers of 7 of 14 patients (50%) reported that travel to a distant clinical site could pose a barrier to clinical trial participation: Traveling with [ID 03.01] is difficult, and also it’s difficult for him to travel. It makes his epilepsy worse, and it tires him out . . . so, not having to travel too far would be a definite bonus. But, I mean, otherwise we would do anything. (ID 3.01, Caregiver)
Discussion
Through qualitative interviews, patients with DRPLA and caregivers described the symptoms patients experienced and their impact on the lives of the patients, caregivers, and other family members. This study found that there are symptom domains that are relevant across the DRPLA population, but there is heterogeneity within each domain based on age of symptom onset and disease stage. Patients and caregivers both prioritized ataxia/mobility and independence/self-care as aspects of DRPLA to target for future treatment. Almost all patients would participate in a clinical trial for DRPLA if given the opportunity, but travel to a distant clinical site could pose a barrier to clinical trial participation.
While other studies have reported on heterogeneity in the clinical presentation of DRPLA from patient to patient,2,18 this study found similarities in the symptom domains experienced by patients. Within the adult-onset population, patients and caregivers described similar experiences with ataxia, cognition, fine motor skills, gross motor skills, speech, and personality changes, but they described varying experiences within the domain of seizures. Only about half of adult-onset patients experienced seizures, and the majority have their seizures under control. Research in a large Japanese population similarly found that epilepsy was present in 54% of DRPLA patients, but the study population included both juvenile and adult-onset patients. 6 This study found that there was more variability within domain discussed in the juvenile-onset population, and some of that variability may have been due to differing ages of symptom onset. For example, academic and gross motor achievements are vastly different when comparing patients with symptom onsets at age 3 versus age 11. Within the juvenile-onset population, caregivers described patients eventually having similar experiences with ataxia, cognition, fine motor skills, gross motor skills, jerking/twitching, seizures, and sleep. The caregivers described varying experiences within the domains of behavior, sensory sensitivity, and speech. While the majority of caregivers stated that patients experienced behavioral issues, the behavioral issues varied from hyperactivity to issues with self-regulation. While all caregivers described deterioration of patient speech, the experiences differed, including delayed speech, slurred speech, altered sentence structure, and loss of words. Sensory sensitivity was present in three-quarters of patients and differed between light and noise.
When considering aspects of DRPLA to target for future treatment, adult-onset patients prioritized ataxia/mobility, caregivers of juvenile-onset patients prioritized ataxia/mobility and independence, and caregivers of adult-onset patients prioritized personality. Similarly, an Externally-Led Patient Focused Drug Development (EL-PFDD) meeting for polyglutamine spinocerebellar ataxias (SCA) and DRPLA found that balance was one of the top three most troubling symptoms for individuals with DRPLA and losing independence was the highest ranked worry among individuals with DRPLA. 18 Another study found that among ataxia patients and caregivers, fitness/recreation, mobility, and independence were the biggest challenges or areas they desired improvement. 19 Similar to the findings from this qualitative interview study, the EL-PFDD meeting found that individuals with DRPLA are willing to participate in clinical trials, 18 but travel to a distant clinical site could pose a barrier to half of the patients described in this study.
The findings from this study highlight some methodological challenges that need to be addressed in the design of clinical trials. While all patients may eventually experience difficulty with the symptom domains described in this study, they may not experience difficulty with a specific symptom domain at the time they present for participation in a clinical trial, since participants will be entering a clinical trial at varying stages of disease progression within each domain. This variation may make it challenging to draw conclusions about the absence of disease progression during a clinical trial. Additionally, while adult-onset patients begin their disease progression from peak development, juvenile-onset patients begin their disease progression from varying stages of development, depending on the age of symptom onset. Clinical outcome assessments will need to take into account the wide range in function that could be seen within each symptom domain, and there may be some domains that are more relevant than others depending on the patient’s disease stage. As is the case in many rare diseases, there may not be one outcome that is applicable across an entire patient population at differing disease stages at the time of trial participation, and multiple clinical endpoints may be needed to determine patient benefit. 8 In this study, caregivers also highlighted that adult patients with DRPLA may have difficulty assessing their own condition accurately, and the lack of awareness of symptoms, impairments, and performance has similarly been found in patients with neuropsychiatric conditions. 20 This lack of patient ability to assess their own condition should be taken into consideration when considering the use of patient-reported outcome measures. 9 In addition to measurement challenges, clinical trials may experience difficulty with recruitment. Due to the rare nature of DRPLA, it is likely that patients will be geographically dispersed and need to travel long distances to clinical sites for trials. DRPLA is an autosomal dominant disorder and multiple family members may be affected, which can further complicate family travel. Since half of the patients in this study would view travel to distant clinical sites as a potential barrier to participation, clinical trials may need to consider the use of remote assessments to lessen the travel burden for patients and families.11,21
This study had several limitations. First, the patients described in the interviews were at different stages of their disease progression at the time of the interview, and the results are not able to take into account the symptoms that patients may eventually develop. Second, some of the caregivers who were interviewed were asked to recall observations and experiences from decades ago, and it is possible there could have been issues with recalling accurate and specific information. Third, the study had a small sample size and only included English-speaking participants; the participants in this study may not be representative of all patient and caregiver experiences with DRPLA.
Conclusion
While the symptoms that characterize DRPLA are diverse, patients and caregivers described being impacted by similar symptom domains. Within each symptom domain, it will be essential to factor in age of onset and disease stage. The symptoms identified in this study impact the daily life of patients and caregivers, and these findings can be used to inform the outcome measure selection for future research. As clinical trials are designed, it will be important to continue to engage patients and caregivers in the process to ensure that trials are assessing outcomes of importance to patients.
Supplemental Material
sj-docx-1-trd-10.1177_26330040241252447 – Supplemental material for Understanding dentatorubral-pallidoluysian atrophy (DRPLA) symptoms and impacts on daily life: a qualitative interview study with patients and caregivers
Supplemental material, sj-docx-1-trd-10.1177_26330040241252447 for Understanding dentatorubral-pallidoluysian atrophy (DRPLA) symptoms and impacts on daily life: a qualitative interview study with patients and caregivers by Marielle G. Contesse, Rebecca J. Woods, Mindy Leffler, Silvia Prades, Julie Greenfield, Andrea Compton and Jeffrey B. Carroll in Therapeutic Advances in Rare Disease
Supplemental Material
sj-docx-2-trd-10.1177_26330040241252447 – Supplemental material for Understanding dentatorubral-pallidoluysian atrophy (DRPLA) symptoms and impacts on daily life: a qualitative interview study with patients and caregivers
Supplemental material, sj-docx-2-trd-10.1177_26330040241252447 for Understanding dentatorubral-pallidoluysian atrophy (DRPLA) symptoms and impacts on daily life: a qualitative interview study with patients and caregivers by Marielle G. Contesse, Rebecca J. Woods, Mindy Leffler, Silvia Prades, Julie Greenfield, Andrea Compton and Jeffrey B. Carroll in Therapeutic Advances in Rare Disease
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.